
Oxidative DNA Damage and Repair Dynamics in Multiple Sclerosis: Insights from Comet Assay Kinetics, Base Excision Repair Gene Expression, and Genotype Analysis
 , , , , ,
, , , , ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Groups
2.2. Sample Collection and Preparation
2.3. Alkaline Comet Assay
2.4. RT-PCR for mRNA Expression Analysis
2.5. Genotyping of SNPs
2.6. Statistical Analysis
3. Results
3.1. Characteristics of the Study Population
3.2. Differences in DNA Repair Between MS Patients and Controls
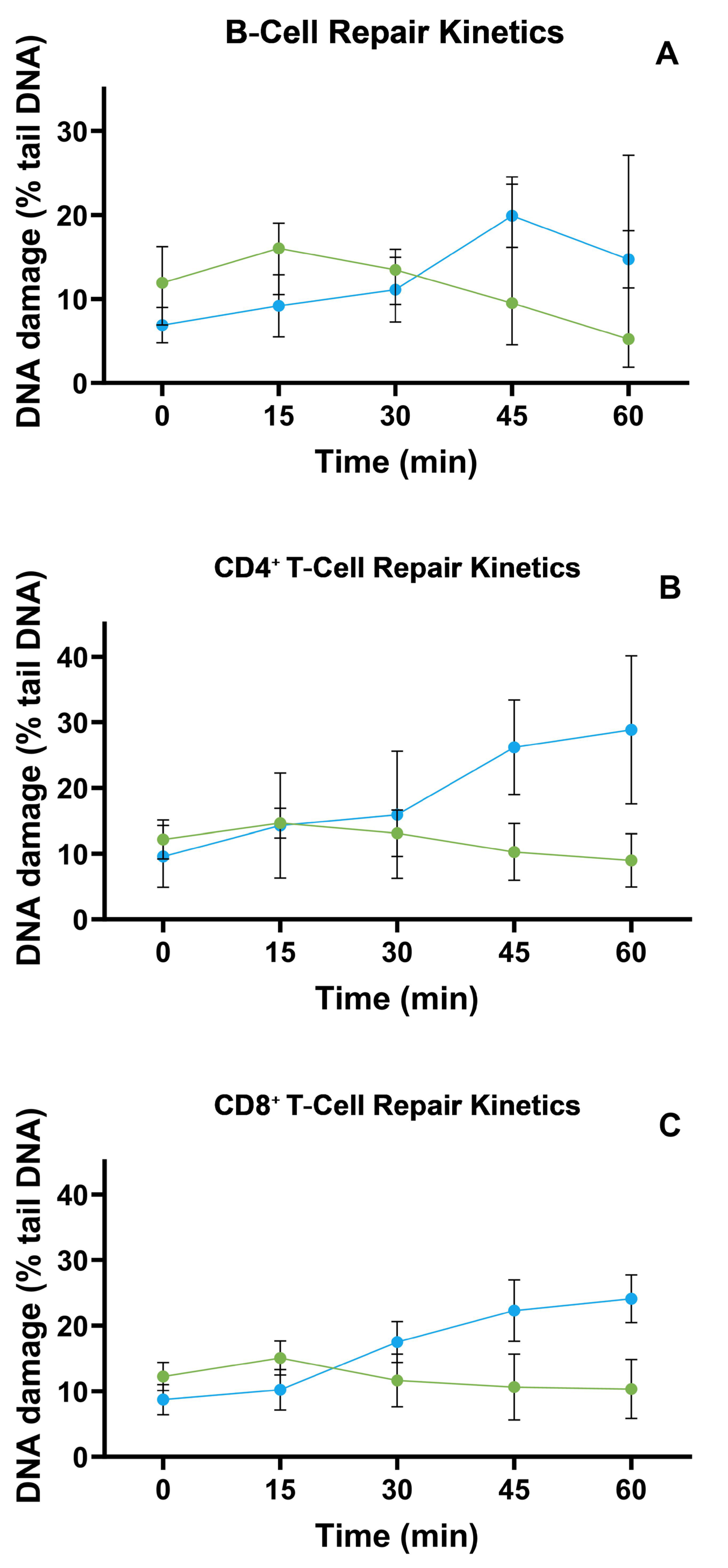
3.3. TBH-Induced DNA Repair Kinetics in B, CD4, and CD8 Cells
3.4. Differences in BER mRNA Expression Levels Between MS Patients and Controls
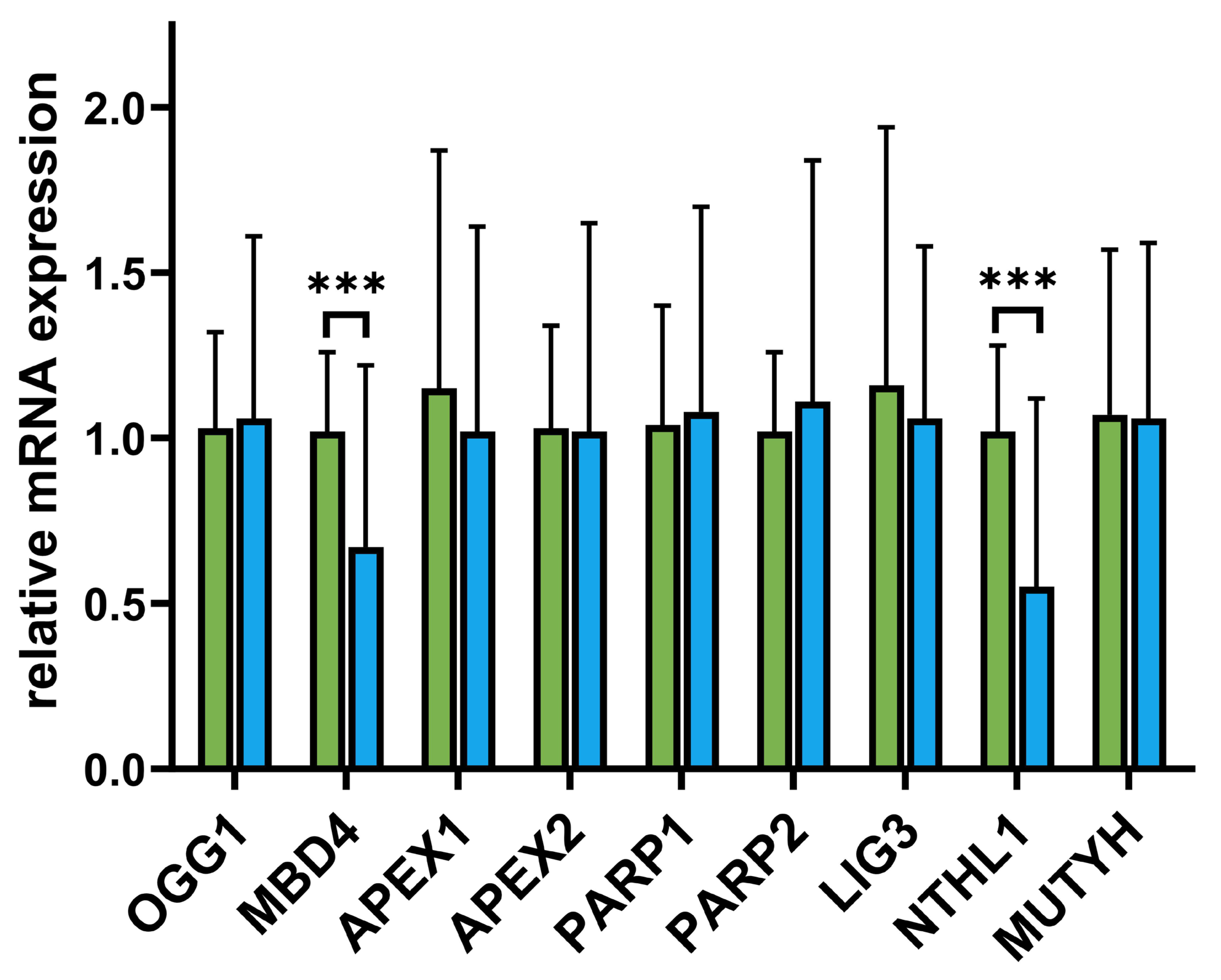

3.5. Associations Between DNA Repair Efficiency (DReff) and MS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ford, H. Clinical Presentation and Diagnosis of Multiple Sclerosis. Clin. Med. Lond. Engl. 2020, 20, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple Sclerosis: Clinical Aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Clarelli, F.; Pignolet, B.; Mascia, E.; Sorosina, M.; Misra, K.; Ferrè, L.; Bucciarelli, F.; Manouchehrinia, A.; Moiola, L.; et al. Vitamin D Affects the Risk of Disease Activity in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2025, 96, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Scazzone, C.; Agnello, L.; Bivona, G.; Lo Sasso, B.; Ciaccio, M. Vitamin D and Genetic Susceptibility to Multiple Sclerosis. Biochem. Genet. 2021, 59, 1–30. [Google Scholar] [CrossRef]
- Schreiner, T.-G.; Genes, T.-M. Obesity and Multiple Sclerosis-A Multifaceted Association. J. Clin. Med. 2021, 10, 2689. [Google Scholar] [CrossRef]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 Lymphocytes Promote Blood-Brain Barrier Disruption and Central Nervous System Inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4+ T Cells in Multiple Sclerosis. Cell 2018, 175, 85–100. [Google Scholar] [CrossRef]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Höftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative Damage in Multiple Sclerosis Lesions. Brain J. Neurol. 2011, 134, 1914–1924. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Salgado-Cámara, P.; García-Martín, E.; Agúndez, J.A.G. Oxidative Stress Markers in Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 6289. [Google Scholar] [CrossRef] [PubMed]
- Gonsette, R.E. Neurodegeneration in Multiple Sclerosis: The Role of Oxidative Stress and Excitotoxicity. J. Neurol. Sci. 2008, 274, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Tadesse, S.; Kidane, D. Significance of Base Excision Repair to Human Health. Int. Rev. Cell Mol. Biol. 2021, 364, 163–193. [Google Scholar] [CrossRef]
- Briggs, F.B.S.; Goldstein, B.A.; McCauley, J.L.; Zuvich, R.L.; De Jager, P.L.; Rioux, J.D.; Ivinson, A.J.; Compston, A.; Hafler, D.A.; Hauser, S.L.; et al. Variation Within DNA Repair Pathway Genes and Risk of Multiple Sclerosis. Am. J. Epidemiol. 2010, 172, 217–224. [Google Scholar] [CrossRef]
- Collins, A.R. The Comet Assay for DNA Damage and Repair: Principles, Applications, and Limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Filipek, B.; Poplawski, T. Elevated Level of DNA Damage and Impaired Repair of DNA Oxidative Damage in Multiple Sclerosis Patients (P7-6.010). Neurology 2024, 102, 7185. [Google Scholar] [CrossRef]
- Galita, G.; Brzezińska, O.; Gulbas, I.; Sarnik, J.; Poplawska, M.; Makowska, J.; Poplawski, T. Increased Sensitivity of PBMCs Isolated from Patients with Rheumatoid Arthritis to DNA Damaging Agents Is Connected with Inefficient DNA Repair. J. Clin. Med. 2020, 9, 988. [Google Scholar] [CrossRef]
- Galita, G.; Sarnik, J.; Zajac, G.; Brzezinska, O.; Budlewski, T.; Poplawska, M.; Przybyłowska-Sygut, K.; Joanna, M.S.; Poplawski, T. The Association between Inefficient Repair of Oxidative DNA Lesions and Common Polymorphisms of the Key Base Excision Repair Genes as Well as Their Expression Levels in Patients with Rheumatoid Arthritis. Arch. Med. Sci. 2023. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Kozera, B.; Rapacz, M. Reference Genes in Real-Time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to Do Successful Gene Expression Analysis Using Real-Time PCR. Methods San. Diego Calif. 2010, 50, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Azqueta, A.; Langie, S.A.S.; Slyskova, J.; Collins, A.R. Measurement of DNA Base and Nucleotide Excision Repair Activities in Mammalian Cells and Tissues Using the Comet Assay—A Methodological Overview. DNA Repair 2013, 12, 1007–1010. [Google Scholar] [CrossRef]
- Ladeira, C.; Møller, P.; Giovannelli, L.; Gajski, G.; Haveric, A.; Bankoglu, E.E.; Azqueta, A.; Gerić, M.; Stopper, H.; Cabêda, J.; et al. The Comet Assay as a Tool in Human Biomonitoring Studies of Environmental and Occupational Exposure to Chemicals—A Systematic Scoping Review. Toxics 2024, 12, 270. [Google Scholar] [CrossRef] [PubMed]
- Duez, P.; Dehon, G.; Kumps, A.; Dubois, J. Statistics of the Comet Assay: A Key to Discriminate between Genotoxic Effects. Mutagenesis 2003, 18, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Field, N.; Cohen, T.; Struelens, M.J.; Palm, D.; Cookson, B.; Glynn, J.R.; Gallo, V.; Ramsay, M.; Sonnenberg, P.; Maccannell, D.; et al. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): An Extension of the STROBE Statement. Lancet Infect. Dis. 2014, 14, 341–352. [Google Scholar] [CrossRef]
- Azqueta, A.; Langie, S.A.S.; Boutet-Robinet, E.; Duthie, S.; Ladeira, C.; Møller, P.; Collins, A.R.; Godschalk, R.W.L. Working Group 5 of the hCOMET project (CA15132) DNA Repair as a Human Biomonitoring Tool: Comet Assay Approaches. Mutat. Res. Rev. Mutat. Res. 2019, 781, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Nandy, A.; Basu, A.; Ghosh, A. Robust Inference for Skewed Data in Health Sciences. J. Appl. Stat. 2022, 49, 2093–2123. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alape-Girón, A.; Flores-Díaz, M.; Echeverri-McCandless, A.; Parajeles-Vindas, A. Oxidative Stress Involvement in the Molecular Pathogenesis and Progression of Multiple Sclerosis: A Literature Review. Rev. Neurosci. 2024, 35, 355–371. [Google Scholar] [CrossRef]
- Wang, P.-F.; Jiang, F.; Zeng, Q.-M.; Yin, W.-F.; Hu, Y.-Z.; Li, Q.; Hu, Z.-L. Mitochondrial and Metabolic Dysfunction of Peripheral Immune Cells in Multiple Sclerosis. J. Neuroinflamm. 2024, 21, 28. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH Oxidase Expression in Active Multiple Sclerosis Lesions in Relation to Oxidative Tissue Damage and Mitochondrial Injury. Brain J. Neurol. 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lei, Y.; Rezk, A.; Espinoza, D.A.; Wang, J.; Feng, H.; Zhang, B.; Barcelos, I.P.; Zhang, H.; Yu, J.; et al. Oxidative Phosphorylation Regulates B Cell Effector Cytokines and Promotes Inflammation in Multiple Sclerosis. Sci. Immunol. 2024, 9, eadk0865. [Google Scholar] [CrossRef] [PubMed]
- Vladimirova, O.; O’Connor, J.; Cahill, A.; Alder, H.; Butunoi, C.; Kalman, B. Oxidative Damage to DNA in Plaques of MS Brains. Mult. Scler. Houndmills Basingstoke Engl. 1998, 4, 413–418. [Google Scholar] [CrossRef]
- Manasa, N.; Naik, K.B.; Singh, M.; Moiz, A.A.; Kudala, V.R.; John, N.K. Patient’s Knowledge and Perception of Endodontics Treatment: An Observational Study. J. Pharm. Bioallied Sci. 2023, 15, S571–S574. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T. Peroxidation of Lipoproteins in Multiple Sclerosis. J. Neurol. Sci. 2011, 311, 92–97. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative Stress in Multiple Sclerosis: Central and Peripheral Mode of Action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ Consumption by PARP1 in Response to DNA Damage Triggers Metabolic Shift Critical for Damaged Cell Survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Tanakaya, K.; Kumamoto, K.; Tada, Y.; Eguchi, H.; Ishibashi, K.; Idani, H.; Tachikawa, T.; Akagi, K.; Okazaki, Y.; Ishida, H. A Germline MBD4 Mutation Was Identified in a Patient with Colorectal Oligopolyposis and Early-onset Cancer: A Case Report. Oncol. Rep. 2019, 42, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Quintana, V.G.; Sweasy, J.B. NTHL1 in Genomic Integrity, Aging and Cancer. DNA Repair 2020, 93, 102920. [Google Scholar] [CrossRef]
- di Penta, A.; Moreno, B.; Reix, S.; Fernandez-Diez, B.; Villanueva, M.; Errea, O.; Escala, N.; Vandenbroeck, K.; Comella, J.X.; Villoslada, P. Oxidative Stress and Proinflammatory Cytokines Contribute to Demyelination and Axonal Damage in a Cerebellar Culture Model of Neuroinflammation. PLoS ONE 2013, 8, e54722. [Google Scholar] [CrossRef]
- González-Díaz, C.A.; Suárez-Souto, M.A.; Pérez-Soto, E.; Gómez-López, M.; Munguía-Cervantes, J.E.; Pérez-Vielma, N.M.; Sánchez-Monroy, V. The Human 8-oxoG DNA Glycosylase 1 (OGG1) Ser326Cys Polymorphism in Infertile Men. Biomedicines 2024, 12, 2286. [Google Scholar] [CrossRef]
- Valinluck, V.; Tsai, H.-H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative Damage to Methyl-CpG Sequences Inhibits the Binding of the Methyl-CpG Binding Domain (MBD) of Methyl-CpG Binding Protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Gong, Y.; Dai, J.; Wu, X.; Gu, J. Genetic Variations in Base Excision Repair Pathway and Risk of Bladder Cancer: A Case-Control Study in the United States. Mol. Carcinog. 2015, 54, 50–57. [Google Scholar] [CrossRef]
- Stratigopoulou, M.; van Dam, T.P.; Guikema, J.E.J. Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences. Front. Immunol. 2020, 11, 1084. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, P.; Gęgotek, A.; Žarković, N.; Skrzydlewska, E. Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 723. [Google Scholar] [CrossRef]
- de Melo, J.T.A.; de Souza Timoteo, A.R.; Lajus, T.B.P.; Brandão, J.A.; de Souza-Pinto, N.C.; Menck, C.F.M.; Campalans, A.; Radicella, J.P.; Vessoni, A.T.; Muotri, A.R.; et al. XPC Deficiency Is Related to APE1 and OGG1 Expression and Function. Mutat. Res. 2016, 784, 25–33. [Google Scholar] [CrossRef]
- Smith, C.M.; Gupta, G.P. Polymerase θ Inhibition Steps on the cGAS Pedal. J. Clin. Investig. 2023, 133, e170660. [Google Scholar] [CrossRef]
- Escrig-Larena, J.I.; Delgado-Pulido, S.; Mittelbrunn, M. Mitochondria during T Cell Aging. Semin. Immunol. 2023, 69, 101808. [Google Scholar] [CrossRef]
- Wang, K.K.W.; Yang, Z.; Yue, J.K.; Zhang, Z.; Winkler, E.A.; Puccio, A.M.; Diaz-Arrastia, R.; Lingsma, H.F.; Yuh, E.L.; Mukherjee, P.; et al. Plasma Anti-Glial Fibrillary Acidic Protein Autoantibody Levels during the Acute and Chronic Phases of Traumatic Brain Injury: A Transforming Research and Clinical Knowledge in Traumatic Brain Injury Pilot Study. J. Neurotrauma 2016, 33, 1270–1277. [Google Scholar] [CrossRef]
- Ellen, O.; Ye, S.; Nheu, D.; Dass, M.; Pagnin, M.; Ozturk, E.; Theotokis, P.; Grigoriadis, N.; Petratos, S. The Heterogeneous Multiple Sclerosis Lesion: How Can We Assess and Modify a Degenerating Lesion? Int. J. Mol. Sci. 2023, 24, 11112. [Google Scholar] [CrossRef]
- Shaw, P.; Kumar, N.; Sahun, M.; Smits, E.; Bogaerts, A.; Privat-Maldonado, A. Modulating the Antioxidant Response for Better Oxidative Stress-Inducing Therapies: How to Take Advantage of Two Sides of the Same Medal? Biomedicines 2022, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Perrone, V.; Veronesi, C.; Giacomini, E.; Citraro, R.; Dell’Orco, S.; Lena, F.; Paciello, A.; Resta, A.M.; Nica, M.; Ritrovato, D.; et al. The Epidemiology, Treatment Patterns and Economic Burden of Different Phenotypes of Multiple Sclerosis in Italy: Relapsing-Remitting Multiple Sclerosis and Secondary Progressive Multiple Sclerosis. Clin. Epidemiol. 2022, 14, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Katchur, S.R.; Jiang, Y.; Briand, J.; Schaber, M.; Kreatsoulas, C.; Schwartz, B.; Thrall, S.; Davis, A.M.; Duvall, S.; et al. Small Molecule-Mediated Allosteric Activation of the Base Excision Repair Enzyme 8-Oxoguanine DNA Glycosylase and Its Impact on Mitochondrial Function. Sci. Rep. 2022, 12, 14685. [Google Scholar] [CrossRef] [PubMed]
- Renaudin, X.; Campalans, A. Modulation of OGG1 Enzymatic Activities by Small Molecules, Promising Tools and Current Challenges. DNA Repair 2025, 149, 103827. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | MS Patients (n = 70) |
|---|---|
| Age (mean ± SD) | 42. 36 (±12.08) |
| Female | 42.59 (±11.57) |
| Male | 41.56 (±14.06) |
| MS type | |
| Relapsing–Remitting (RRMS) | 63 |
| Primary Progressive (PPMS) Secondary Progressive (SPMS) | 6 1 |
| EDSS score (mean ± SD) | 3.62 (±1.62) |
| Duration of the disease (mean ± SD) | 9.9 (±7.1) |
| The course of the disease at the time of blood collection | |
| Relapse | 23 |
| Remission | 47 |
| Treatment | |
| Cladribine | 1 |
| Dimethyl fumarate | 4 |
| Glatiramer acetate | 3 |
| Natalizumab | 28 |
| Ocrelizumab | 21 |
| Ofatumumab | 3 |
| No DMT treatment | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipek, B.; Macieja, A.; Binda, A.; Szelenberger, R.; Gorniak, L.; Miller, E.; Swiderek-Matysiak, M.; Stasiolek, M.; Majsterek, I.; Poplawski, T. Oxidative DNA Damage and Repair Dynamics in Multiple Sclerosis: Insights from Comet Assay Kinetics, Base Excision Repair Gene Expression, and Genotype Analysis. Biomolecules 2025, 15, 756. https://doi.org/10.3390/biom15060756
Filipek B, Macieja A, Binda A, Szelenberger R, Gorniak L, Miller E, Swiderek-Matysiak M, Stasiolek M, Majsterek I, Poplawski T. Oxidative DNA Damage and Repair Dynamics in Multiple Sclerosis: Insights from Comet Assay Kinetics, Base Excision Repair Gene Expression, and Genotype Analysis. Biomolecules. 2025; 15(6):756. https://doi.org/10.3390/biom15060756
Chicago/Turabian StyleFilipek, Beata, Anna Macieja, Aleksandra Binda, Rafal Szelenberger, Leslaw Gorniak, Elzbieta Miller, Mariola Swiderek-Matysiak, Mariusz Stasiolek, Ireneusz Majsterek, and Tomasz Poplawski. 2025. "Oxidative DNA Damage and Repair Dynamics in Multiple Sclerosis: Insights from Comet Assay Kinetics, Base Excision Repair Gene Expression, and Genotype Analysis" Biomolecules 15, no. 6: 756. https://doi.org/10.3390/biom15060756
APA StyleFilipek, B., Macieja, A., Binda, A., Szelenberger, R., Gorniak, L., Miller, E., Swiderek-Matysiak, M., Stasiolek, M., Majsterek, I., & Poplawski, T. (2025). Oxidative DNA Damage and Repair Dynamics in Multiple Sclerosis: Insights from Comet Assay Kinetics, Base Excision Repair Gene Expression, and Genotype Analysis. Biomolecules, 15(6), 756. https://doi.org/10.3390/biom15060756