
LPS2336, a New TREK-1 Channel Activator Identified by High Throughput Screening

 , , , ,
, , , ,  , , , , and
, , , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Models
2.2. Chemical Synthesis
2.3. Cell Lines
2.4. Thallium Assay
2.5. Patch Clamp
2.6. Behavioral Tests
2.6.1. Hargreaves Test
2.6.2. Open Field
2.6.3. Horizontal Bar Test
2.6.4. Rotarod
2.7. Body Temperature Monitoring
2.8. Data Analysis
3. Results
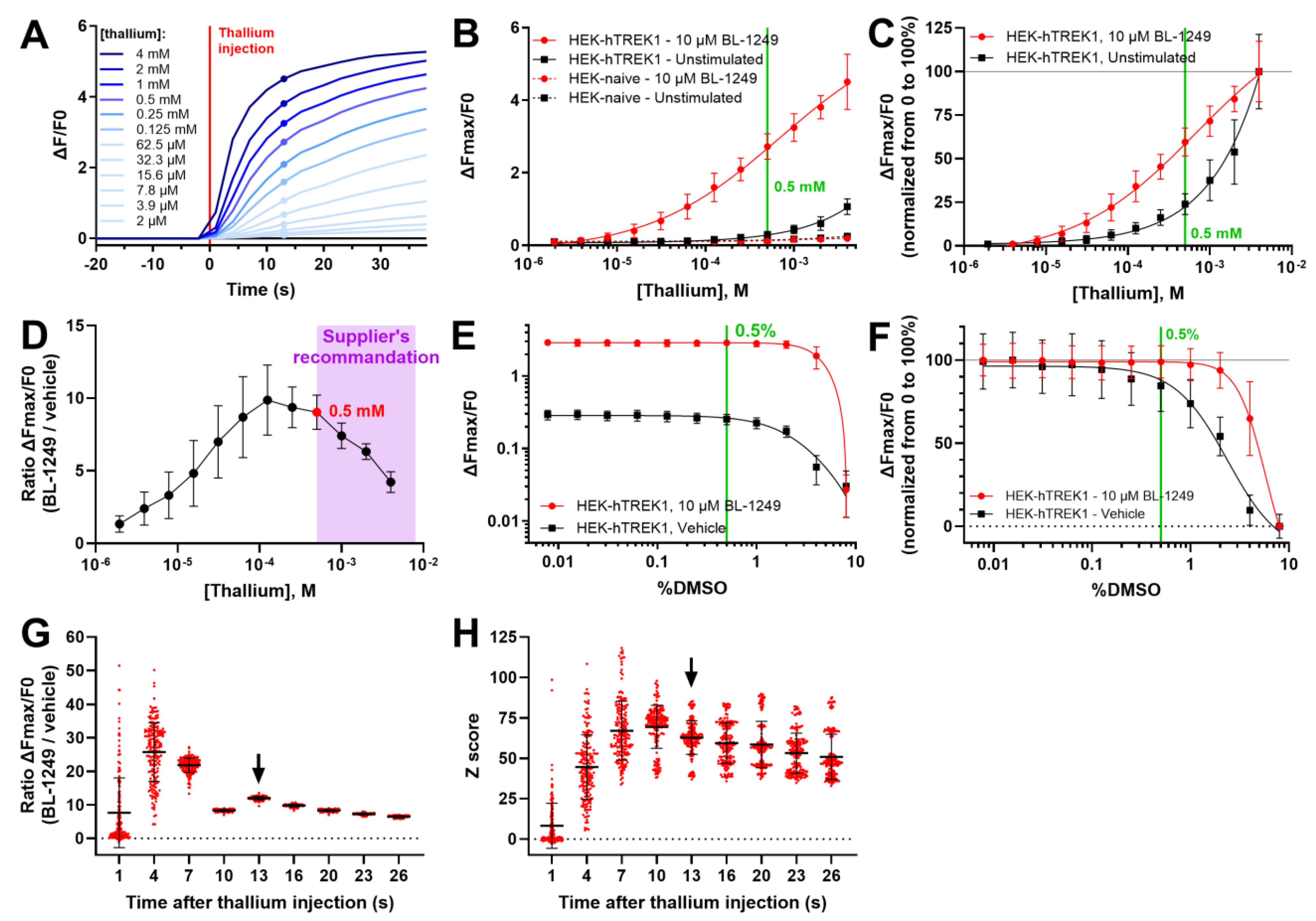
3.1. Optimization of the Thallium Assay
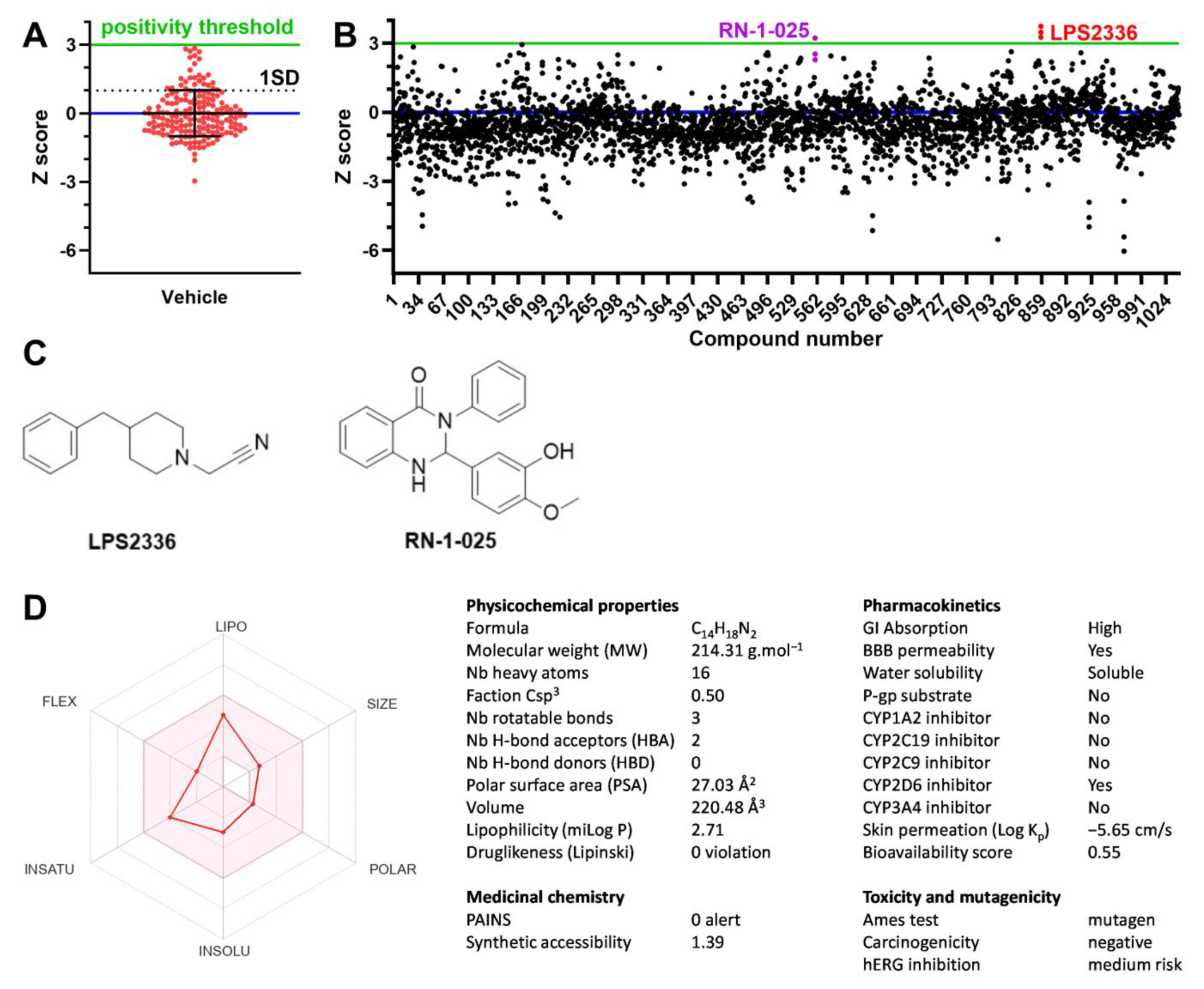
3.2. High Throughput Screening for TREK-1 Activators
3.3. Confirmation of TREK-1 Activation by LPS2336
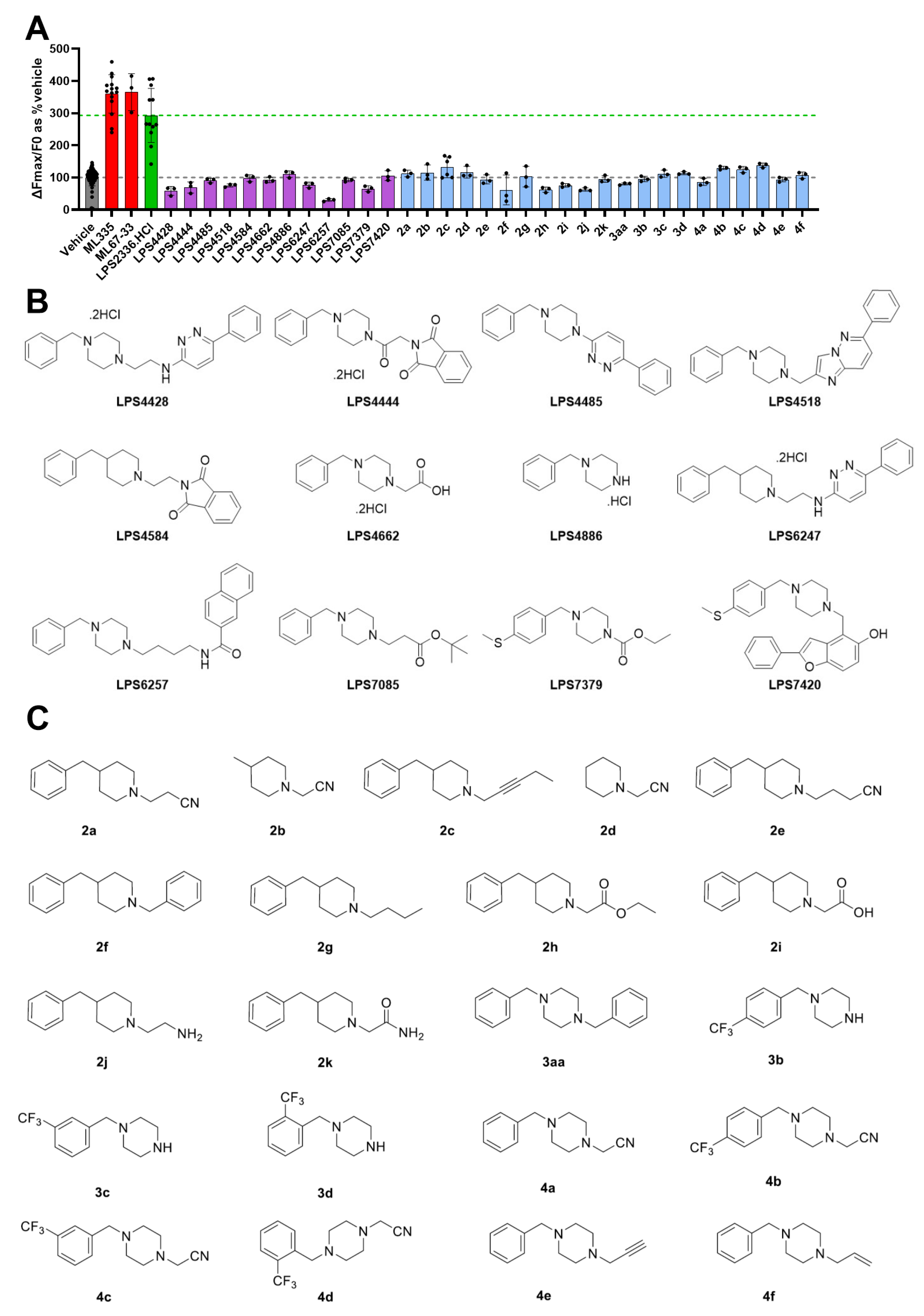
3.4. Synthesis and Evaluation of LPS2336 Analogs
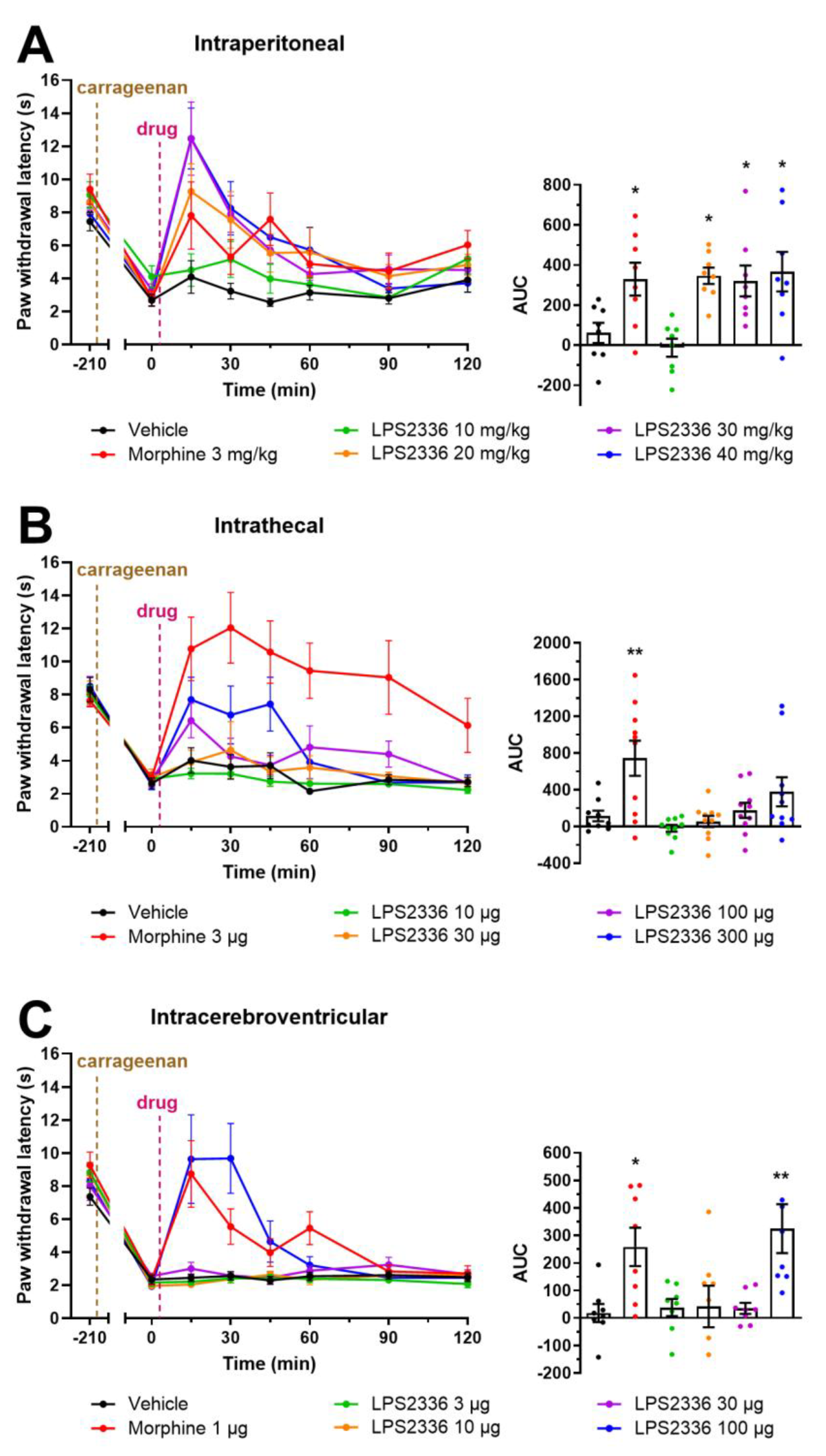
3.5. Effect of LPS2336 on Pain Thresholds
3.6. Evaluation of the Adverse Effects Induced by LPS2336
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAS | Chemical Abstract Service |
| DCM | DiChloroMethane |
| DIPEA | DiIsoPropylEthylAmine |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | DiMethylSulfOxide |
| EC50 | Half maximal Effective Concentration |
| ESI | ElectroSpray Ionization |
| HEK | Human Embryonic Kidney |
| HEPES | (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) |
| HPLC | High Performance Liquid Chromatography |
| HRMS | High Resolution Mass Spectrometry |
| HTS | High Throughput Screening |
| K2P | Two-pore-domain K+ channel |
| KO | Knock-Out |
| NMR | Nuclear Magnetic Resonance |
| ORF | Open Reading Frame |
| TASK | TWIK-Related Acid-Sensitive Potassium Channels |
| THF | TetraHydroFuran |
| TREK | TWIK-related K+ channel |
| TWIK | Tandem pore domains in Weak rectifying K+ channel |
| WT | Wild-Type |
References
- Fink, M.; Duprat, F.; Lesage, F.; Reyes, R.; Romey, G.; Heurteaux, C.; Lazdunski, M. Cloning, Functional Expression and Brain Localization of a Novel Unconventional Outward Rectifier K+ Channel. EMBO J. 1996, 15, 6854–6862. [Google Scholar] [CrossRef] [PubMed]
- Meadows, H.J.; Benham, C.D.; Cairns, W.; Gloger, I.; Jennings, C.; Medhurst, A.D.; Murdock, P.; Chapman, C.G. Cloning, Localisation and Functional Expression of the Human Orthologue of the TREK-1 Potassium Channel. Pflugers Arch. 2000, 439, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Maingret, F.; Lauritzen, I.; Patel, A.J.; Heurteaux, C.; Reyes, R.; Lesage, F.; Lazdunski, M.; Honoré, E. TREK-1 Is a Heat-Activated Background K+ Channel. EMBO J. 2000, 19, 2483–2491. [Google Scholar] [CrossRef]
- Hervieu, G.J.; Cluderay, J.E.; Gray, C.W.; Green, P.J.; Ranson, J.L.; Randall, A.D.; Meadows, H.J. Distribution and Expression of TREK-1, a Two-Pore-Domain Potassium Channel, in the Adult Rat CNS. Neuroscience 2001, 103, 899–919. [Google Scholar] [CrossRef]
- Talley, E.M.; Solorzano, G.; Lei, Q.; Kim, D.; Bayliss, D.A. Cns Distribution of Members of the Two-Pore-Domain (KCNK) Potassium Channel Family. J. Neurosci. 2001, 21, 7491–7505. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Rennie, G.; Chapman, C.G.; Meadows, H.; Duckworth, M.D.; Kelsell, R.E.; Gloger, I.I.; Pangalos, M.N. Distribution Analysis of Human Two Pore Domain Potassium Channels in Tissues of the Central Nervous System and Periphery. Brain Res. Mol. Brain Res. 2001, 86, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Alloui, A.; Zimmermann, K.; Mamet, J.; Duprat, F.; Noël, J.; Chemin, J.; Guy, N.; Blondeau, N.; Voilley, N.; Rubat-Coudert, C.; et al. TREK-1, a K+ Channel Involved in Polymodal Pain Perception. EMBO J. 2006, 25, 2368–2376. [Google Scholar] [CrossRef]
- Ray, P.; Torck, A.; Quigley, L.; Wangzhou, A.; Neiman, M.; Rao, C.; Lam, T.; Kim, J.-Y.; Kim, T.H.; Zhang, M.Q.; et al. Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: An RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research. Pain 2018, 159, 1325–1345. [Google Scholar] [CrossRef]
- Noël, J.; Zimmermann, K.; Busserolles, J.; Deval, E.; Alloui, A.; Diochot, S.; Guy, N.; Borsotto, M.; Reeh, P.; Eschalier, A.; et al. The Mechano-Activated K+ Channels TRAAK and TREK-1 Control Both Warm and Cold Perception. EMBO J. 2009, 28, 1308–1318. [Google Scholar] [CrossRef]
- García, G.; Martínez-Rojas, V.A.; Murbartián, J. TREK-1 Potassium Channels Participate in Acute and Long-Lasting Nociceptive Hypersensitivity Induced by Formalin in Rats. Behav. Brain Res. 2021, 413, 113446. [Google Scholar] [CrossRef]
- Ávalos Prado, P.; Landra-Willm, A.; Verkest, C.; Ribera, A.; Chassot, A.-A.; Baron, A.; Sandoz, G. TREK Channel Activation Suppresses Migraine Pain Phenotype. iScience 2021, 24, 102961. [Google Scholar] [CrossRef] [PubMed]
- Heurteaux, C.; Lucas, G.; Guy, N.; El Yacoubi, M.; Thümmler, S.; Peng, X.-D.; Noble, F.; Blondeau, N.; Widmann, C.; Borsotto, M.; et al. Deletion of the Background Potassium Channel TREK-1 Results in a Depression-Resistant Phenotype. Nat. Neurosci. 2006, 9, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Heurteaux, C.; Guy, N.; Laigle, C.; Blondeau, N.; Duprat, F.; Mazzuca, M.; Lang-Lazdunski, L.; Widmann, C.; Zanzouri, M.; Romey, G.; et al. TREK-1, a K+ Channel Involved in Neuroprotection and General Anesthesia. EMBO J. 2004, 23, 2684–2695. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tian, Y.; Huang, Q.; Wan, Y.; Xu, L.; Wang, W.; Pan, D.; Zhu, S.; Xie, M. Deficiency of TREK-1 Potassium Channel Exacerbates Blood-Brain Barrier Damage and Neuroinflammation after Intracerebral Hemorrhage in Mice. J. Neuroinflamm. 2019, 16, 96. [Google Scholar] [CrossRef]
- Chen, T.; Qian, X.; Zhu, J.; Yang, L.-K.; Wang, Y.-H. Controlled Decompression Attenuates Compressive Injury Following Traumatic Brain Injury via TREK-1-Mediated Inhibition of Necroptosis and Neuroinflammation. Oxid. Med. Cell Longev. 2021, 2021, 4280951. [Google Scholar] [CrossRef]
- Unudurthi, S.D.; Wu, X.; Qian, L.; Amari, F.; Onal, B.; Li, N.; Makara, M.A.; Smith, S.A.; Snyder, J.; Fedorov, V.V.; et al. Two-Pore K+ Channel TREK-1 Regulates Sinoatrial Node Membrane Excitability. J. Am. Heart Assoc. 2016, 5, e002865. [Google Scholar] [CrossRef]
- Mondéjar-Parreño, G. Unraveling the Role of K2P Channels in Atrial Fibrillation. Front. Biosci. 2022, 14, 31. [Google Scholar] [CrossRef]
- Schwingshackl, A.; Teng, B.; Makena, P.; Ghosh, M.; Sinclair, S.E.; Luellen, C.; Balasz, L.; Rovnaghi, C.; Bryan, R.M.; Lloyd, E.E.; et al. Deficiency of the Two-Pore-Domain Potassium Channel TREK-1 Promotes Hyperoxia-Induced Lung Injury. Crit. Care Med. 2014, 42, e692–e701. [Google Scholar] [CrossRef]
- Monaghan, K.; Baker, S.A.; Dwyer, L.; Hatton, W.C.; Sik Park, K.; Sanders, K.M.; Koh, S.D. The Stretch-Dependent Potassium Channel TREK-1 and Its Function in Murine Myometrium. J. Physiol. 2011, 589, 1221–1233. [Google Scholar] [CrossRef]
- Pineda, R.H.; Hypolite, J.; Lee, S.; Carrasco, A.; Iguchi, N.; Meacham, R.B.; Malykhina, A.P. Altered Detrusor Contractility and Voiding Patterns in Mice Lacking the Mechanosensitive TREK-1 Channel. BMC Urol. 2019, 19, 40. [Google Scholar] [CrossRef]
- Voloshyna, I.; Besana, A.; Castillo, M.; Matos, T.; Weinstein, I.B.; Mansukhani, M.; Robinson, R.B.; Cordon-Cardo, C.; Feinmark, S.J. TREK-1 Is a Novel Molecular Target in Prostate Cancer. Cancer Res. 2008, 68, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Innamaa, A.; Jackson, L.; Asher, V.; van Schalkwyk, G.; Warren, A.; Keightley, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and Effects of Modulation of the K2P Potassium Channels TREK-1 (KCNK2) and TREK-2 (KCNK10) in the Normal Human Ovary and Epithelial Ovarian Cancer. Clin. Transl. Oncol. 2013, 15, 910–918. [Google Scholar] [CrossRef]
- Maingret, F.; Patel, A.J.; Lesage, F.; Lazdunski, M.; Honoré, E. Mechano- or Acid Stimulation, Two Interactive Modes of Activation of the TREK-1 Potassium Channel. J. Biol. Chem. 1999, 274, 26691–26696. [Google Scholar] [CrossRef] [PubMed]
- Devilliers, M.; Busserolles, J.; Lolignier, S.; Deval, E.; Pereira, V.; Alloui, A.; Christin, M.; Mazet, B.; Delmas, P.; Noel, J.; et al. Activation of TREK-1 by Morphine Results in Analgesia without Adverse Side Effects. Nat. Commun. 2013, 4, 2941. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.; Bennis, K.; Vivier, D.; Pereira, V.; Chatelain, C.F.; Chapuy, E.; Deokar, H.; Busserolles, J.; Lesage, F.; Eschalier, A.; et al. Synthesis and Structure-Activity Relationship Study of Substituted Caffeate Esters as Antinociceptive Agents Modulating the TREK-1 Channel. Eur. J. Med. Chem. 2014, 75, 391–402. [Google Scholar] [CrossRef]
- Busserolles, J.; Ben Soussia, I.; Pouchol, L.; Marie, N.; Meleine, M.; Devilliers, M.; Judon, C.; Schopp, J.; Clémenceau, L.; Poupon, L.; et al. TREK1 Channel Activation as a New Analgesic Strategy Devoid of Opioid Adverse Effects. Br. J. Pharmacol. 2020, 177, 4782–4795. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Huang, L.; Fu, J.; Han, C.; Fang, J.; Liao, P.; Chen, Z.; Mo, Y.; Sun, P.; Liao, D.; et al. A TREK Channel Family Activator with Well-Defined Structure-Activation Relationship for Pain and Neurogenic Inflammation. J. Med. Chem. 2020, 63, 3665–3677. [Google Scholar] [CrossRef]
- Delanne-Cuménal, M.; Lamoine, S.; Meleine, M.; Aissouni, Y.; Prival, L.; Fereyrolles, M.; Barbier, J.; Cercy, C.; Boudieu, L.; Schopp, J.; et al. The TREK-1 Potassium Channel Is Involved in Both the Analgesic and Anti-Proliferative Effects of Riluzole in Bone Cancer Pain. Biomed. Pharmacother. 2024, 176, 116887. [Google Scholar] [CrossRef]
- Vivier, D.; Soussia, I.B.; Rodrigues, N.; Lolignier, S.; Devilliers, M.; Chatelain, F.C.; Prival, L.; Chapuy, E.; Bourdier, G.; Bennis, K.; et al. Development of the First Two-Pore Domain Potassium Channel TWIK-Related K+ Channel 1-Selective Agonist Possessing in Vivo Antinociceptive Activity. J. Med. Chem. 2017, 60, 1076–1088. [Google Scholar] [CrossRef]
- Weaver, C.D.; Harden, D.; Dworetzky, S.I.; Robertson, B.; Knox, R.J. A Thallium-Sensitive, Fluorescence-Based Assay for Detecting and Characterizing Potassium Channel Modulators in Mammalian Cells. J. Biomol. Screen. 2004, 9, 671–677. [Google Scholar] [CrossRef]
- Dadi, P.K.; Vierra, N.C.; Days, E.; Dickerson, M.T.; Vinson, P.N.; Weaver, C.D.; Jacobson, D.A. Selective Small Molecule Activators of TREK-2 Channels Stimulate Dorsal Root Ganglion c-Fiber Nociceptor Two-Pore-Domain Potassium Channel Currents and Limit Calcium Influx. ACS Chem. Neurosci. 2017, 8, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.D.; Veale, E.L.; McCoull, D.; Tickle, D.C.; Large, J.M.; Ococks, E.; Gothard, G.; Kettleborough, C.; Mathie, A.; Jerman, J. Terbinafine Is a Novel and Selective Activator of the Two-Pore Domain Potassium Channel TASK3. Biochem. Biophys. Res. Commun. 2017, 493, 444–450. [Google Scholar] [CrossRef]
- Wright, P.D.; McCoull, D.; Walsh, Y.; Large, J.M.; Hadrys, B.W.; Gaurilcikaite, E.; Byrom, L.; Veale, E.L.; Jerman, J.; Mathie, A. Pranlukast Is a Novel Small Molecule Activator of the Two-Pore Domain Potassium Channel TREK2. Biochem. Biophys. Res. Commun. 2019, 520, 35–40. [Google Scholar] [CrossRef]
- De Vries, W.N.; Binns, L.T.; Fancher, K.S.; Dean, J.; Moore, R.; Kemler, R.; Knowles, B.B. Expression of Cre Recombinase in Mouse Oocytes: A Means to Study Maternal Effect Genes. Genesis 2000, 26, 110–112. [Google Scholar] [CrossRef]
- Bagriantsev, S.N.; Ang, K.-H.; Gallardo-Godoy, A.; Clark, K.A.; Arkin, M.R.; Renslo, A.R.; Minor, D.L. A High-Throughput Functional Screen Identifies Small Molecule Regulators of Temperature- and Mechano-Sensitive K2P Channels. ACS Chem. Biol. 2013, 8, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Lolicato, M.; Arrigoni, C.; Mori, T.; Sekioka, Y.; Bryant, C.; Clark, K.A.; Minor, D.L. K2P2.1(TREK-1):Activator Complexes Reveal a Cryptic Selectivity Filter Binding Site. Nature 2017, 547, 364–368. [Google Scholar] [CrossRef]
- Goktug, A.N.; Chai, S.C.; Chen, T. Data Analysis Approaches in High Throughput Screening; IntechOpen: London, UK, 2013; ISBN 978-953-51-0906-8. [Google Scholar]
- Pope, L.; Arrigoni, C.; Lou, H.; Bryant, C.; Gallardo-Godoy, A.; Renslo, A.R.; Minor, D.L. Protein and Chemical Determinants of BL-1249 Action and Selectivity for K2P Channels. ACS Chem. Neurosci. 2018, 9, 3153–3165. [Google Scholar] [CrossRef]
- Patel, A.J.; Honoré, E.; Maingret, F.; Lesage, F.; Fink, M.; Duprat, F.; Lazdunski, M. A Mammalian Two Pore Domain Mechano-Gated S-like K+ Channel. EMBO J. 1998, 17, 4283–4290. [Google Scholar] [CrossRef]
- Takahira, M.; Sakurai, M.; Sakurada, N.; Sugiyama, K. Fenamates and Diltiazem Modulate Lipid-Sensitive Mechano-Gated 2P Domain K(+) Channels. Pflugers Arch. 2005, 451, 474–478. [Google Scholar] [CrossRef]
- Duprat, F.; Lesage, F.; Patel, A.J.; Fink, M.; Romey, G.; Lazdunski, M. The Neuroprotective Agent Riluzole Activates the Two P Domain K+ Channels TREK-1 and TRAAK. Mol. Pharmacol. 2000, 57, 906–912. [Google Scholar] [CrossRef]
- Loucif, A.J.C.; Saintot, P.; Liu, J.; Antonio, B.M.; Zellmer, S.G.; Yoger, K.; Veale, E.L.; Wilbrey, A.; Omoto, K.; Cao, L.; et al. GI-530159, a Novel, Selective, Mechanosensitive Two-pore-domain Potassium (K2P) Channel Opener, Reduces Rat Dorsal Root Ganglion Neuron Excitability. Br. J. Pharmacol. 2018, 175, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Kennard, L.E.; Chumbley, J.R.; Ranatunga, K.M.; Armstrong, S.J.; Veale, E.L.; Mathie, A. Inhibition of the Human Two-Pore Domain Potassium Channel, TREK-1, by Fluoxetine and Its Metabolite Norfluoxetine. Br. J. Pharmacol. 2005, 144, 821–829. [Google Scholar] [CrossRef]
- Contreras, J.-M.; Parrot, I.; Sippl, W.; Rival, Y.M.; Wermuth, C.G. Design, Synthesis, and Structure−Activity Relationships of a Series of 3-[2-(1-Benzylpiperidin-4-Yl)Ethylamino]Pyridazine Derivatives as Acetylcholinesterase Inhibitors. J. Med. Chem. 2001, 44, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Noel, R.; Gupta, N.; Pons, V.; Goudet, A.; Garcia-Castillo, M.D.; Michau, A.; Martinez, J.; Buisson, D.-A.; Johannes, L.; Gillet, D.; et al. N-Methyldihydroquinazolinone Derivatives of Retro-2 with Enhanced Efficacy against Shiga Toxin. J. Med. Chem. 2013, 56, 3404–3413. [Google Scholar] [CrossRef]
- Poupon, L.; Lamoine, S.; Pereira, V.; Barriere, D.A.; Lolignier, S.; Giraudet, F.; Aissouni, Y.; Meleine, M.; Prival, L.; Richard, D.; et al. Targeting the TREK-1 Potassium Channel via Riluzole to Eliminate the Neuropathic and Depressive-like Effects of Oxaliplatin. Neuropharmacology 2018, 140, 43–61. [Google Scholar] [CrossRef]
- Raphemot, R.; Weaver, C.D.; Denton, J.S. High-Throughput Screening for Small-Molecule Modulators of Inward Rectifier Potassium Channels. J. Vis. Exp. 2013, 71, 4209. [Google Scholar] [CrossRef]
- Buijs, T.J.; McNaughton, P.A. The Role of Cold-Sensitive Ion Channels in Peripheral Thermosensation. Front. Cell Neurosci. 2020, 14, 262. [Google Scholar] [CrossRef]
- Wechselberger, M.; Wright, C.L.; Bishop, G.A.; Boulant, J.A. Ionic Channels and Conductance-Based Models for Hypothalamic Neuronal Thermosensitivity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R518–R529. [Google Scholar] [CrossRef] [PubMed]
- Pisani, D.F.; Beranger, G.E.; Corinus, A.; Giroud, M.; Ghandour, R.A.; Altirriba, J.; Chambard, J.-C.; Mazure, N.M.; Bendahhou, S.; Duranton, C.; et al. The K+ Channel TASK1 Modulates β-Adrenergic Response in Brown Adipose Tissue through the Mineralocorticoid Receptor Pathway. FASEB J. 2016, 30, 909–922. [Google Scholar] [CrossRef]
- Amata, E.; Dichiara, M.; Arena, E.; Pittalà, V.; Pistarà, V.; Cardile, V.; Graziano, A.C.E.; Fraix, A.; Marrazzo, A.; Sortino, S.; et al. Novel Sigma Receptor Ligand–Nitric Oxide Photodonors: Molecular Hybrids for Double-Targeted Antiproliferative Effect. J. Med. Chem. 2017, 60, 9531–9544. [Google Scholar] [CrossRef]
- Ruchelman, A.L.; Houghton, P.J.; Zhou, N.; Liu, A.; Liu, L.F.; LaVoie, E.J. 5-(2-Aminoethyl)Dibenzo[c,h][1,6]Naphthyridin-6-Ones: Variation of N-Alkyl Substituents Modulates Sensitivity to Efflux Transporters Associated with Multidrug Resistance. J. Med. Chem. 2005, 48, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Takata, Y. Anodic Cyanation of Tertiary Aliphatic and Heterocyclic Amines. J. Org. Chem. 1977, 42, 2973–2977. [Google Scholar] [CrossRef]
- McGonagle, F.I.; MacMillan, D.S.; Murray, J.; Sneddon, H.F.; Jamieson, C.; Watson, A.J.B. Development of a Solvent Selection Guide for Aldehyde-Based Direct Reductive Amination Processes. Green Chem. 2013, 15, 1159–1165. [Google Scholar] [CrossRef]
- Cai, J.; Wathey, B. A Novel Traceless Solid Phase Tertiary Amine Synthesis Based on Merrifield Resin. Tetrahedron Letters 2001, 42, 1383–1385. [Google Scholar] [CrossRef]
- Özturan Özer, E.; Tan, O.U.; Ozadali, K.; Küçükkılınç, T.; Balkan, A.; Uçar, G. Synthesis, Molecular Modeling and Evaluation of Novel N’-2-(4-Benzylpiperidin-/Piperazin-1-Yl)Acylhydrazone Derivatives as Dual Inhibitors for Cholinesterases and Aβ Aggregation. Bioorg. Med. Chem. Lett. 2013, 23, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.H.; Mahajan, U.S.; Bhalerao, D.S.; Akamanchi, K.G. Novel and Facile Transformation of N,N-Disubstituted Glycylamides into Corresponding Cyanamides by Using Pentavalent Iodine Reagents in Combination with Tetraethylammonium Bromide. Synlett 2007, 18, 2815–2818. [Google Scholar] [CrossRef]
- Putt, K.S.; Chen, G.W.; Pearson, J.M.; Sandhorst, J.S.; Hoagland, M.S.; Kwon, J.-T.; Hwang, S.-K.; Jin, H.; Churchwell, M.I.; Cho, M.-H.; et al. Small-Molecule Activation of Procaspase-3 to Caspase-3 as a Personalized Anticancer Strategy. Nat. Chem. Biol. 2006, 2, 543–550. [Google Scholar] [CrossRef]
- Ferla, S.; Manganaro, R.; Benato, S.; Paulissen, J.; Neyts, J.; Jochmans, D.; Brancale, A.; Bassetto, M. Rational Modifications, Synthesis and Biological Evaluation of New Potential Antivirals for RSV Designed to Target the M2-1 Protein. Bioorg. Med. Chem. 2020, 28, 115401. [Google Scholar] [CrossRef]
- Liu, G.; Lynch, J.K.; Freeman, J.; Liu, B.; Xin, Z.; Zhao, H.; Serby, M.D.; Kym, P.R.; Suhar, T.S.; Smith, H.T.; et al. Discovery of Potent, Selective, Orally Bioavailable Stearoyl-CoA Desaturase 1 Inhibitors. J. Med. Chem. 2007, 50, 3086–3100. [Google Scholar] [CrossRef]
- Saxena, M.; Agarwal, S.K.; Patnaik, G.K.; Saxena, A.K. Synthesis, Biological Evaluation, and Quantitative Structure-Activity Relationship Analysis of [.Beta.-(Aroylamino)Ethyl]Piperazines and -Piperidines and [2-[(Arylamino)Carbonyl]Ethyl]Piperazines, -Piperidines, -Pyrazinopyridoindoles, and -Pyrazinoisoquinolines. A New Class of Potent H1 Antagonists. J. Med. Chem. 1990, 33, 2970–2976. [Google Scholar] [CrossRef]
- Jia, Z.; Wen, H.; Huang, S.; Luo, Y.; Gao, J.; Wang, R.; Wan, K.; Xue, W. “Click” Assembly of Novel Dual Inhibitors of AChE and MAO-B from Pyridoxine Derivatives for the Treatment of Alzheimer’s Disease. Heterocycl. Commun. 2022, 28, 18–25. [Google Scholar] [CrossRef]
- Murty, M.S.R.; Jyothirmai, B.; Radha Krishna, P.; Yadav, J.S. Zinc Mediated Alkylation of Cyclic Secondary Amines. Synth. Commun. 2003, 33, 2483–2486. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyer, R.; Bony, R.; Maugis, M.; Schopp, J.; Leroux, M.; Michelin, C.; Gonthier, L.; Grzeskiewicz, Q.; Jouannet, A.; Aissouni, Y.; et al. LPS2336, a New TREK-1 Channel Activator Identified by High Throughput Screening. Biomolecules 2025, 15, 740. https://doi.org/10.3390/biom15050740
Boyer R, Bony R, Maugis M, Schopp J, Leroux M, Michelin C, Gonthier L, Grzeskiewicz Q, Jouannet A, Aissouni Y, et al. LPS2336, a New TREK-1 Channel Activator Identified by High Throughput Screening. Biomolecules. 2025; 15(5):740. https://doi.org/10.3390/biom15050740
Chicago/Turabian StyleBoyer, Romane, Romane Bony, Maxence Maugis, Julien Schopp, Marion Leroux, Clément Michelin, Laurie Gonthier, Quentin Grzeskiewicz, Alexandre Jouannet, Youssef Aissouni, and et al. 2025. "LPS2336, a New TREK-1 Channel Activator Identified by High Throughput Screening" Biomolecules 15, no. 5: 740. https://doi.org/10.3390/biom15050740
APA StyleBoyer, R., Bony, R., Maugis, M., Schopp, J., Leroux, M., Michelin, C., Gonthier, L., Grzeskiewicz, Q., Jouannet, A., Aissouni, Y., Didier, B., Gulea, M., Girard, N., Cintrat, J.-C., Dumeige, A., Busserolles, J., Ducki, S., & Lolignier, S. (2025). LPS2336, a New TREK-1 Channel Activator Identified by High Throughput Screening. Biomolecules, 15(5), 740. https://doi.org/10.3390/biom15050740