Abstract
Multiple myeloma (MM) remains an incurable hematologic malignancy due to the inevitable development of drug resistance, particularly in relapsed or refractory cases. Post-translational modifications (PTMs), including phosphorylation, ubiquitination, acetylation, and glycosylation, play pivotal roles in regulating protein function, stability, and interactions, thereby influencing MM pathogenesis and therapeutic resistance. This review comprehensively explores the mechanisms by which dysregulated PTMs contribute to drug resistance in MM, focusing on their impact on key signaling pathways, metabolic reprogramming, and the tumor microenvironment. We highlight how PTMs modulate drug uptake, alter drug targets, and regulate cell survival signals, ultimately promoting resistance to PIs, IMiDs, and other therapeutic agents. Furthermore, we discuss emerging therapeutic strategies targeting PTM-related pathways, which offer promising avenues for overcoming resistance to treatment. By integrating preclinical and clinical insights, this review underscores the potential of PTM-targeted therapies to enhance treatment efficacy and improve patient outcomes in MM.
1. Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by the clonal expansion of plasma cells within the bone marrow, leading to the excessive production of monoclonal immunoglobulins [1]. This abnormal proliferation of plasma cells results in a cascade of clinical manifestations, including hypercalcemia, renal impairment, anemia, and osteolytic bone lesions, commonly referred to as the CRAB criteria (calcium elevation, renal failure, anemia, and bone lesions) [2]. Despite significant advancements in MM therapy, the disease remains incurable due to the emergence of therapeutic resistance, particularly in patients with relapsed or refractory MM (RRMM) [3]. Consequently, understanding the molecular mechanisms of RRMM and developing novel therapeutic strategies to overcome resistance is critical to improving patient outcomes.
Traditional treatment options for MM include chemotherapy, proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), and autologous hematopoietic stem cell transplantation (ASCT). Alkylating agents like melphalan and cyclophosphamide disrupt DNA replication, leading to tumor cell death [4,5]. PIs such as bortezomib and carfilzomib prevent protein degradation in MM cells, inducing apoptosis [6,7,8]. IMiDs, like lenalidomide and pomalidomide, boost immune-mediated destruction of myeloma cells while modulating tumor microenvironments [9]; ASCT is employed for eligible patients (particularly younger individuals with adequate organ function) by harvesting the patient’s own hematopoietic stem cells, administering high-dose melphalan to eliminate residual myeloma cells, and then reinfusing the stem cells to restore bone marrow function [10]. These approaches have extended patient survival, but most patients eventually develop resistance or relapse after prolonged use.
Recent advances in MM treatment have introduced innovative therapies such as monoclonal antibodies (mAbs), chimeric antigen receptor T-cell (CAR-T) therapy, and bispecific T-cell engagers (BiTEs). CAR-T therapy, targeting B-cell maturation antigen (BCMA), has demonstrated remarkable efficacy in RRMM patients [11,12]. Monoclonal antibodies like daratumumab, targeting CD38, have significantly improved outcomes when combined with traditional therapies [13]. Meanwhile, BiTEs are showing promise by bridging T-cells to myeloma cells, enhancing immune-mediated destruction [14]. Although these treatments represent groundbreaking advancements, challenges like accessibility and resistance remain, highlighting the need for further research to refine these therapies and enhance patient outcomes.
Post-translational modifications (PTMs) are crucial regulators of cellular processes, playing key roles in protein function, stability, localization, and interactions. In the context of MM, the dysregulation of PTMs has been implicated in both disease pathogenesis and therapeutic resistance [15,16]. PTMs, such as phosphorylation, acetylation, and ubiquitination, are involved in the regulation of tumor suppressor proteins and oncogenes, contributing to tumor progression, metastasis, and drug resistance. Thus, understanding the interplay between PTMs and therapeutic resistance offers a promising avenue for the development of novel combination therapies to improve patient outcomes.
Roles of histone modifications on MM drug resistance have been extensively reviewed elsewhere [17,18]. Here, we mainly focus on recent advances in the study of PTMs and their contribution to therapeutic resistance in MM, with an emphasis on identifying novel therapeutic targets and strategies to improve patient outcomes.
2. Major PTMs Implicated in MM Pathogenesis and Progression
Proteins, biomacromolecules that are synthesized by ribosomes and subsequently processed by the endoplasmic reticulum (ER) and Golgi apparatus, play essential roles in cellular functions. Throughout their lifecycle, proteins undergo various PTMs, which typically involve the covalent attachment of small molecular groups to specific amino acid residues under enzymatic catalysis [19,20]. Common modifications implicated in MM include phosphorylation, ubiquitination, methylation, acetylation, SUMOylation (modification by small ubiquitin-related modifier), and glycosylation, often occurring at amino acid residues such as lysine, serine, threonine, arginine, and tyrosine [2]. Studies have shown that PTMs are functionally diverse and closely related to the onset and progression of numerous diseases, including MM, neurological disorders, immune diseases, and cardiovascular diseases [21,22,23].
Generally, PTMs are installed on amino acid residues by enzymes that could be termed “writers”, and can be removed by another group of enzymes termed “erasers” [24]. Such dynamic modifications exert a multitude of functions, as mentioned above. In this part, we recapitulate PTMs implicated in the pathogenesis and progression of MM (Figure 1).
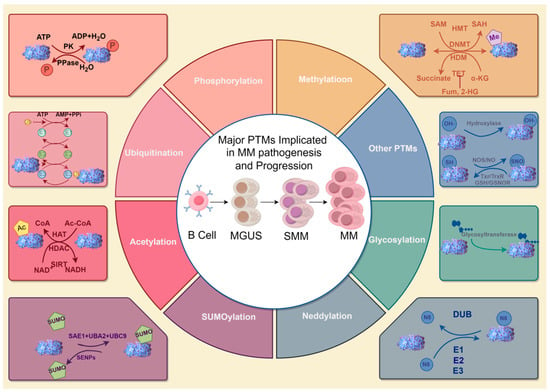
Figure 1.
A summary of major PTMs implicated in MM pathogenesis and progression. The center illustrates the clinical progression stages of MM (MGUS → SMM → MM), defined by diagnostic criteria including monoclonal protein levels, bone marrow plasma cell infiltration, and CRAB symptoms. The surrounding circular diagrams depict key PTMs (e.g., phosphorylation, SUMOylation) that drive molecular pathogenesis across disease stages. While no specific PTM is exclusively linked to a single clinical stage, dysregulated PTMs collectively contribute to malignant transformation and drug resistance during progression. Abbreviations: MGUS, Monoclonal Gammopathy of Undetermined Significance; SMM, Smoldering MM; MM, MM; ATP, Adenosine Triphosphate; ADP, Adenosine Diphosphate; PK, Pyruvate Kinase; PPase, Pyrophosphatase; SAM, S-Adenosylmethionine; HMT, Histone Methyltransferase; SAH, S-Adenosylhomocysteine; DNMT, DNA Methyltransferase; MD, Methyltransferase Domain; HDM, Histone Demethylase; TET, Ten-Eleven Translocation methylcytosine dioxygenase; α-KG, Alpha-Ketoglutarate; Fum, Fumarate; 2-HG, 2-Hydroxyglutarate; AMP, Adenosine Monophosphate; PPI, Inorganic Pyrophosphate; NOS, Nitric Oxide Synthase; NO, Nitric Oxide; SNO, S-Nitrosothiol; GSH, Glutathione; GSNOR, Glutathione-Dependent Formaldehyde Dehydrogenase; CoA, Coenzyme A; Ac-CoA, Acetyl-Coenzyme A; HAT, Histone Acetyltransferase; ADA, Adenosine Deaminase; NADH, Nicotinamide Adenine Dinucleotide (Reduced Form); SAE1, SUMO Activating Enzyme Subunit 1; UBA2, Ubiquitin-like Modifier Activating Enzyme 2; UBC9, Ubiquitin Conjugating Enzyme 9; SENPs, Sentrin/SUMO-specific Proteases; DUB, Deubiquitinating Enzyme; E1, Ubiquitin-Activating Enzyme; E2, Ubiquitin-Conjugating Enzyme; E3, Ubiquitin Ligase. The figure was created with Figdraw. Hu, S. (2025) https://www.figdraw.com/ (accessed on 19 March 2025).
2.1. Phosphorylation
Phosphorylation involves the covalent attachment of a phosphate group to an amino acid residue under the catalysis of protein kinases [25,26]. Protein phosphatases are responsible for removing the phosphate group from proteins. It is one of the most abundant and common PTMs in eukaryotes and plays a crucial role in intracellular signal transduction, regulating various biological processes such as the cell cycle, differentiation, transformation, development, and peptide hormone responses [27]. Phosphorylation plays a crucial role in the pathogenesis and progression of MM. It is involved in the regulation of various signaling pathways that drive the proliferation, survival, and metastasis of MM cells. For instance, the phosphorylation of signal transducer and activator of transcription 3 (STAT3) is a key event in MM progression. STAT3 activation, often mediated by phosphorylation at Tyr705, promotes cell proliferation, survival, and immune evasion in MM cells [28,29].
Moreover, phosphorylation events are implicated in the regulation of metabolic reprogramming in MM. For example, the phosphorylation of AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) influences the metabolic state of MM cells, contributing to their survival and progression [30,31]. Phosphorylation of histone proteins and other epigenetic regulators also plays a role in the transcriptional dysregulation observed in MM [32,33]. Phosphorylation-mediated signaling pathways and metabolic reprogramming are central to the pathogenesis and progression of MM, highlighting the importance of targeting these pathways for therapeutic intervention.
2.2. Acetylation
Acetylation is a process involving the attachment of an acetyl group (-COCH3) to a protein molecule, which includes two types: N-terminal acetylation (Nt-acetylation) and internal acetylation. N-terminal acetylation refers to the covalent modification on the α-amino group of the first amino acid residue at the N-terminus of the protein, while internal acetylation occurs on the ε-amino group of lysine residues within proteins [34]. This acetylation modification is mainly catalyzed by two types of enzymes: histone acetyltransferases (HATs) are responsible for adding acetyl groups, while histone deacetylases (HDACs) are responsible for their removal [35]. Non-histone protein acetylation plays a significant role in the pathogenesis and progression of MM. For instance, the acetylation of APE1 (apurinic/apyrimidinic endonuclease 1) at lysine residues K6 and K7 has been shown to regulate MDR1 (multidrug resistance protein 1) expression, thereby influencing drug efflux and resistance to alkylating agents like melphalan [36]. Additionally, the ADA2B subunit of the SAGA complex regulates MYC(c-Myc) expression through acetylation, which is crucial for maintaining oncogenic programs in MM [37]. KDM6A regulates immune recognition genes in MM through H3K27 acetylation, highlighting its role in immune evasion [38,39]. Furthermore, metabolic regulation through non-histone protein acetylation is also a key factor. It was found that ENO1, a MYC target gene, promotes MM progression by enhancing mitophagy through YWHAZ acetylation, contributing to metabolic regulation [40,41,42]. These findings highlight the importance of non-histone protein acetylation in driving MM progression.
2.3. Ubiquitination
Ubiquitination is a widespread protein modification in eukaryotes that involves the attachment of ubiquitin, a highly conserved protein composed of 76 amino acids, to target proteins. This modification is catalyzed by the E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases. Erasers of ubiquitination include a broad range of deubiquitinating enzymes, namely ubiquitin-specific proteases (USPs) and ubiquitin C-terminal hydrolases (UCHs) [43,44]. Ubiquitination serves as a dynamic regulatory switch, controlling protein stability, subcellular localization, activity, and protein-protein interactions [45,46,47]. In the context of MM, dysregulated ubiquitination not only drives oncogenic signaling and disease progression but also profoundly impacts therapeutic resistance.
In MM, the ubiquitin-proteasome system (UPS) is critical for maintaining protein homeostasis. MM cells rely heavily on the UPS to degrade excess monoclonal immunoglobulins and misfolded proteins generated by their high proliferative rate [16]. However, dysfunction of the UPS can lead to proteotoxic stress, which paradoxically contributes to disease progression by activating compensatory survival pathways (e.g., NF-κB and HSF1) [16,48]. For example, the E3 ubiquitin ligase CRL4CRBN is hijacked by IMiDs (lenalidomide/pomalidomide) to degrade IKZF1/3, key transcription factors sustaining MM cell survival [48]. Conversely, mutations or downregulation of CRBN disrupt IMiD-induced degradation, representing a primary mechanism of IMiD resistance [49].
Beyond its role in protein degradation, ubiquitination dynamically modulates signaling pathways that drive MM pathogenesis. For instance, TRAF6-mediated K63-linked ubiquitination of AKT enhances its activation, promoting PI3K/AKT/mTOR signaling and cell survival [50]. Additionally, deubiquitinating enzymes (DUBs) such as USP7 and USP14 counteract ubiquitination, stabilizing oncoproteins (e.g., Mcl-1, NEK2) and contributing to resistance against proteasome inhibitors like bortezomib [51,52]. These DUBs also protect MM cells from ER stress-induced apoptosis by deubiquitinating key sensors like IRE1α, disrupting the UPR pathway [53].
Overall, ubiquitination acts as a double-edged sword in MM: while it maintains protein quality control and regulates physiological signaling, its dysregulation underpins both disease progression and therapeutic evasion. Targeting aberrant ubiquitination pathways (e.g., E3 ligases, DUBs) has emerged as a promising strategy to overcome resistance and restore treatment sensitivity.
2.4. Methylation
Methylation is the process of introducing methyl groups into proteins, which can occur on various amino acids such as arginine, lysine, and histidine. Methylation is involved in regulating protein-protein interactions, thereby exerting a significant impact on a series of crucial cellular events, such as transcriptional stress response, aging, protein repair, and neuronal differentiation [54]. Nonhistone protein methylation plays a crucial role in the development and progression of MM by modulating the function of transcription factors, signaling molecules, and other regulatory proteins. For example, the histone methyltransferase MMSET (also known as NSD2) is frequently overexpressed in MM due to the t(4;14) chromosomal translocation. MMSET catalyzes the methylation of histone H3 lysine 36 (H3K36me2), leading to global changes in histone methylation patterns and chromatin structure [55,56]. This aberrant methylation can dysregulate genes involved in cell cycle control, apoptosis, and DNA repair [57]. Promoter methylation of the ABCG2 gene, which encodes an ATP-binding cassette transporter, enhances drug efflux capacity by upregulating its expression, contributing to chemoresistance against agents such as topotecan and doxorubicin [58,59].
Another important example is the methylation of the suppressor of cytokine signaling 1 (SOCS1), a key regulator of cytokine signaling and immune response. In MM, SOCS1 methylation is often associated with increased resistance to apoptosis and enhanced cell survival. Studies have shown that demethylation agents, such as 5-azacytidine, can reverse SOCS1 methylation, leading to its reactivation and increased sensitivity of MM cells to apoptosis [60]. Additionally, methylation of nonhistone proteins can affect the interaction between myeloma cells and the bone marrow microenvironment, which is crucial for disease progression [61].
2.5. SUMOylation
SUMOylation is the process of covalently attaching small ubiquitin-like modifier (SUMO) proteins to target proteins as a post-translational modification. This process is mediated by specific enzymes known as SUMOylation writers and erasers [62,63]. Writers include SUMO E1-activating enzyme (SAE1/SAE2), SUMO E2-conjugating enzyme (UBC9), and various SUMO E3 ligases (such as PIAS proteins), which facilitate the attachment of SUMO to target proteins. On the other hand, erasers are SUMO-specific proteases (SENPs), which remove SUMO from modified proteins, thus reversing the modification [64]. Abnormalities in SUMOylation can affect the sensitivity and drug resistance of MM cells through various mechanisms. For instance, SUMOylation of β-catenin is associated with poor prognosis in MM. Inhibiting SUMOylation downregulates β-catenin levels by promoting its ubiquitin-proteasomal degradation, thereby suppressing the Wnt/β-catenin signaling pathway [65]. This suggests that SUMOylation of β-catenin contributes to the aberrant proliferation of MM cells.
Additionally, the SUMOylation pathway is significantly enhanced in MM patients compared to normal plasma cells. High expression of SUMOylation-related enzymes, such as UBE2I and PIAS1, is associated with poor survival outcomes. This indicates that dysregulated SUMOylation is a critical factor in MM progression and may serve as a potential therapeutic target [62]. Moreover, SUMOylation can regulate protein stability in MM cells. For example, SUMOylation of Rep78, a regulatory protein, affects its stability and function, suggesting that SUMOylation may impact viral replication and MM cell survival [66].
2.6. Neddylation
Neddylation is a post-translational modification that involves the covalent attachment of the ubiquitin-like protein NEDD8 to substrate proteins, particularly cullins, which are components of Cullin-RING Ligases (CRLs) [67,68,69]. CRLs are the largest family of E3 ubiquitin ligases and play a critical role in regulating protein degradation through the UPS. Aberrant regulation of this pathway has been implicated in the pathogenesis of MM [68,69]. Dysregulation of the neddylation pathway can lead to imbalances in protein homeostasis, which is essential for maintaining cellular functions. This disruption is particularly relevant in MM, where the UPS is already compromised due to the high protein synthesis load of malignant plasma cells [70]. Neddylation is also involved in the activation of various oncogenic pathways in MM. For example, the neddylation of cullins is essential for the function of CRLs, which regulate the degradation of key proteins involved in cell cycle progression, apoptosis, and DNA repair. Overactivation of these pathways can contribute to tumor growth and progression [69,70]. The bone marrow microenvironment in MM is highly immunosuppressive, and neddylation may play a role in modulating this environment. Studies have shown that inhibition of neddylation can enhance the expression of NK cell-activating ligands (e.g., MICA and MICB) on MM cells, making them more susceptible to immune-mediated killing [71].
2.7. Glycosylation
Glycosylation, the enzymatic process of attaching sugar moieties (glycans) to proteins or lipids, is a critical post-translational modification that influences protein folding, stability, and function. This process occurs in two primary forms: N-glycosylation, where glycans are attached to the nitrogen atom of asparagine side chains [72,73], and O-glycosylation, where glycans are linked to the oxygen atom of serine or threonine residues [74,75]. Both N-glycosylation and O-glycosylation play significant roles in the pathogenesis of MM, affecting cell signaling, immune evasion, and disease progression. Studies have shown that monoclonal immunoglobulins produced by MM cells often exhibit altered glycosylation patterns compared to normal immunoglobulins. For instance, a decrease in galactosylation and sialylation, along with an increase in fucosylation, is commonly observed in MM patients [73,76]. These changes are present across all International Staging System (ISS) stages, including low-risk ISS I, indicating that glycosylation alterations are an early event in MM development. Abnormal glycosylation of immunoglobulins has also been linked to bone loss in MM. Specifically, reduced galactosylation and sialylation of IgG in MM patients with bone disease suggest that glycosylation status can influence the interaction between MM cells and the bone microenvironment [77]. This finding highlights the potential role of glycosylation in promoting osteoclast differentiation and bone resorption.
Glycosylation changes in MM are not static and can evolve with disease progression. For example, difucosylation of diantennary glycans decreases as MM progresses, indicating that glycosylation profiles may serve as biomarkers for disease stage and progression [73]. Additionally, glycosylation alterations in the immunoglobulin variable region (Fab) have been identified as potential risk factors for disease progression in plasma cell disorders [72]. Genome-wide association studies (GWAS) have identified loci associated with IgG glycosylation that also show pleiotropy with autoimmune diseases and hematological cancers, including MM [78]. These genetic insights suggest that glycosylation pathways are intricately linked to the molecular mechanisms underlying MM pathogenesis.
2.8. Other PTMs
Hydroxylation and nitrosylation are emerging as important PTMs involved in the pathogenesis and progression of MM. Hydroxylation, catalyzed by enzymes such as prolyl hydroxylases, plays a role in stabilizing hypoxia-inducible factor-1 alpha (HIF-1α), which promotes tumor adaptation to hypoxic conditions [79,80]. In MM, the methylation of EGLN3, a prolyl hydroxylase, is associated with poor prognosis and advanced disease stages, suggesting that hydroxylation may contribute to disease progression [79]. Nitrosylation, specifically S-nitrosylation, has also been implicated in MM. This modification can inhibit the activity of key signaling pathways such as STAT3 and NF-κB, which are often hyperactivated in MM cells [81]. It was shown that S-nitrosothiols, such as S-nitroso-N-acetylcysteine (SNAC), can inhibit MM cell proliferation and survival by targeting these pathways [81]. This suggests that nitrosylation may be a potential therapeutic target for overcoming drug resistance and improving patient outcomes.
Collectively, various PTMs are critical for regulating cellular processes and maintaining cell function, playing a particularly significant role in the pathogenesis, progression and drug resistance of MM (Table 1). Understanding these modifications is essential for identifying new therapeutic targets and developing more effective treatments for MM.

Table 1.
Comprehensive Summary of PTM Types, Key Proteins, and Mechanisms in MM Pathogenesis and Drug Resistance.
Table 1.
Comprehensive Summary of PTM Types, Key Proteins, and Mechanisms in MM Pathogenesis and Drug Resistance.
| PTM Type | Key Proteins/Molecules | Mechanism in MM | References |
|---|---|---|---|
| Phosphorylation | |||
| STAT3 (Tyr705) | Phosphorylated STAT3 activates NF-κB/STAT3 signaling, promoting proliferation, survival, and immune evasion. | [28,29] | |
| AMPK/mTOR | Phosphorylation regulates metabolic reprogramming; hyperactivation supports MM survival under stress. | [30,82] | |
| MAFb | GSK3-mediated phosphorylation stabilizes MAFb, disrupting proteasome inhibitor targets and inducing resistance. | [83] | |
| Akt/GSK-3β | Akt phosphorylates and inactivates GSK-3β, stabilizing c-Myc to drive drug resistance. | [84] | |
| MKK4/7-JNK | GCK-induced phosphorylation activates RAS-mutant MAPK signaling, promoting adaptive resistance. | [29] | |
| Acetylation | |||
| ADA2B (SAGA complex) | Acetylates histones to regulate c-Myc expression, sustaining oncogenic programs. | [37] | |
| KDM6A | Modulates H3K27 acetylation to suppress immune recognition genes, enabling immune evasion. | [38,39] | |
| YWHAZ | Acetylation by ENO1 enhances mitophagy, supporting metabolic adaptation in MM progression. | [40] | |
| APE1 (K6/K7) | Acetylation enhances DNA repair via BER and upregulates MDR1, promoting melphalan resistance. | [36] | |
| Ubiquitination | |||
| CRL4CRBN | IMiDs recruit CRBN to ubiquitinate IKZF1/3 for proteasomal degradation; mutations confer resistance. | [48] | |
| IKZF1/IKZF3 | Degradation by IMiDs suppresses MM survival; loss of ubiquitination leads to IMiD resistance. | [85,86] | |
| HMGB1 | MALAT-1-induced ubiquitination promotes autophagy and inhibits apoptosis, driving drug resistance. | [87] | |
| BRCC36 | Cleaves K63-ubiquitin chains on CRBN, stabilizing it to enhance IMiD sensitivity. | [49] | |
| NEK2/USP7/TRIP13 | USP7 deubiquitinates NEK2, stabilizing it to promote chromosomal instability and PI resistance. | [51,88] | |
| Methylation | |||
| MMSET (NSD2) | Catalyzes H3K36me2, dysregulating tumor suppressor genes and promoting MM progression. | [55,56] | |
| SOCS1 | Methylation silences SOCS1, enhancing cytokine signaling and resistance to apoptosis. | [60] | |
| EGLN3 | Methylation of EGLN3 (prolyl hydroxylase) correlates with hypoxia adaptation and poor prognosis. | [79] | |
| SUMOylation | |||
| β-catenin | SUMOylation stabilizes β-catenin, activating Wnt signaling to drive proliferation and drug resistance. | [65] | |
| IκBα | SENP2 deficiency increases SUMOylation of IκBα, activating NF-κB and bortezomib resistance. | [89] | |
| IRF4/c-Myc | SUMOylation stabilizes IRF4/c-Myc; inhibition by TAK-981 restores lenalidomide sensitivity. | [90] | |
| Neddylation | |||
| CRLs (Cullin-RING ligases) | Neddylation activates CRLs; inhibition by MLN4924 stabilizes pro-apoptotic proteins (e.g., NOXA). | [69,91] | |
| REDD1 | Neddylation blockade stabilizes REDD1, inhibiting PI3K/AKT/mTOR and overcoming resistance. | [91] | |
| Glycosylation | |||
| α4β1/α4β7 integrins | Sialylation enhances adhesion to bone marrow stroma, promoting CAM-DR and drug sanctuary. | [92,93,94] | |
| CD38/PSGL-1 | Sialylation masks CD38 epitopes (daratumumab resistance); PSGL-1 binds Siglec-7 to suppress NK activity. | [76,93] | |
| IgG (Fab region) | Altered glycosylation in the Fab region correlates with disease progression and bone loss. | [72,78] | |
| Hydroxylation | |||
| HIF-1α | Hydroxylation stabilizes HIF-1α under hypoxia, promoting tumor adaptation and survival. | [79] | |
| Nitrosylation | |||
| STAT3/NF-κB | S-nitrosylation inhibits STAT3/NF-κB activity; SNAC reverses hyperactivation to restore drug sensitivity. | [81] | |
| Deubiquitination | |||
| USP14/UCHL5 | Deubiquitinate misfolded proteins to reduce proteotoxic stress, conferring PI resistance. | [53] | |
| Deacetylation | |||
| HDAC1 | Deacetylates histones and non-histones (e.g., HPV), enhancing DNA repair and resistance to DNA damage. | [95] | |
3. PTM Targeting Therapies in MM
Targeting PTMs of proteins has emerged as a promising therapeutic strategy in the treatment of MM. Several classes of drugs that modulate PTMs are currently in clinical use or under investigation, offering new avenues for more effective MM management.
3.1. PIs
PIs are a cornerstone of MM therapy, targeting the UPS, a critical PTM pathway responsible for protein degradation. By inhibiting the proteasome, these drugs induce the accumulation of ubiquitinated proteins, leading to ER stress, activation of the unfolded protein response (UPR), and ultimately apoptosis in MM cells [45,96,97]. The first-in-class proteasome inhibitor, bortezomib, has significantly improved outcomes in both newly diagnosed and relapsed/refractory MM. It targets the 26S proteasome, leading to the accumulation of pro-apoptotic proteins such as NOXA and BIM, and is often used in combination with dexamethasone and other agents. Its subcutaneous administration has reduced the risk of peripheral neuropathy. Carfilzomib, the second-generation proteasome inhibitor, offers improved specificity for the proteasome’s chymotrypsin-like activity and reduced neurotoxicity compared to bortezomib [98,99,100,101,102]. It is approved for RRMM and is often used in combination with dexamethasone and lenalidomide. Ixazomib, the first oral proteasome inhibitor, provides convenience and improved patient adherence [103]. It is approved for RRMM in combination with lenalidomide and dexamethasone, offering a well-tolerated option for maintenance therapy. Together, these agents have revolutionized MM treatment, significantly improving overall survival (OS) and progression-free survival (PFS) by disrupting protein homeostasis, a critical vulnerability in MM cells.
3.2. IMiDs
While not direct PTM-targeting drugs, IMiDs modulate PTMs by recruiting specific substrates to the cereblon E3 ubiquitin ligase complex, leading to their ubiquitination and degradation [104]. This mechanism disrupts MM cell survival pathways and enhances immune-mediated tumor cell killing. Lenalidomide, approved for both newly diagnosed and relapsed/refractory MM, induces the degradation of IKZF1 and IKZF3, transcription factors critical for MM cell survival [85,86]. It is often used in combination with dexamethasone and PIs and is a mainstay of maintenance therapy. Pomalidomide, a more potent IMiD, is approved for RRMM and has shown efficacy in patients refractory to lenalidomide and bortezomib, making it a valuable option for heavily pretreated patients [105,106]. IMiDs have transformed MM treatment, with lenalidomide-based regimens significantly improving OS and PFS. Their ability to modulate PTMs through the cereblon pathway has made them a cornerstone of MM therapy, offering durable responses and improved outcomes across all stages of the disease.
3.3. Histone Deacetylase (HDAC) Inhibitors
HDAC inhibitors target epigenetic modifications by inhibiting the deacetylation of histones and non-histone proteins, leading to the reactivation of tumor suppressor genes, disruption of MM cell proliferation, and induction of apoptosis [107]. Panobinostat is the first and currently the only FDA-approved HDAC inhibitor for MM, though its approval status may have evolved in recent years [108]. Approved for RRMM in combination with bortezomib and dexamethasone, panobinostat has shown efficacy in patients who have become refractory to PIs and IMiDs, highlighting its role in overcoming drug resistance. It enhances the activity of PIs by further disrupting protein homeostasis, making it a valuable addition to combination therapies [109,110].
4. Role of PTMs in Drug Resistance Mechanisms in MM
PTMs play a pivotal role in the pathogenesis and progression of MM, particularly in the development of drug resistance. These modifications, including phosphorylation, methylation, acetylation, and ubiquitination, among others, regulate key cellular processes such as protein function, stability, and interactions. In the context of drug resistance, PTMs can alter drug uptake, efflux, target activity, and cell survival pathways, ultimately enabling MM cells to evade therapeutic interventions (Figure 2). Understanding the mechanisms by which PTMs contribute to drug resistance is crucial for developing targeted strategies to overcome treatment challenges in MM.
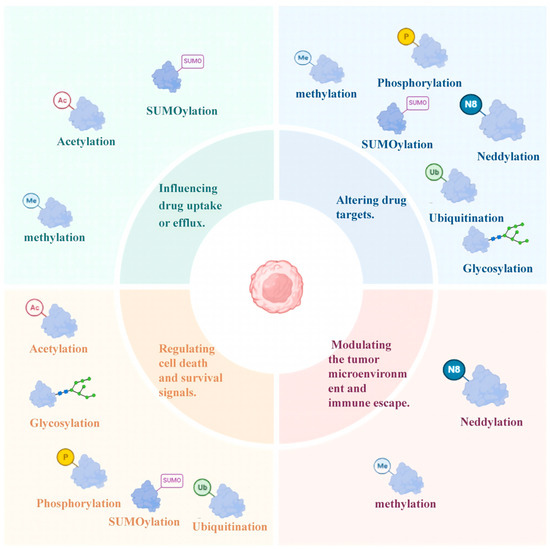
Figure 2.
Four Major Mechanisms of PTM-Mediated Drug Resistance in MM. This figure summarizes the key PTMs involved in the drug resistance of MM, organized into four functional categories (distinguished by colored background blocks) and their corresponding PTMs (annotated with colored icons and abbreviations). The figure was created in Biorender. Hu, S. (2025) https://BioRender.com (accessed on 19 March 2025).
4.1. Influencing Drug Uptake or Efflux
4.1.1. SUMOylation
A study conducted by Heynen et al. highlights the correlation between an overactive SUMO signaling pathway and resistance to carfilzomib, a proteasome inhibitor utilized in MM therapy. SUMOylation, a type of PTM, can modulate the function or expression of drug transporter proteins, thereby potentially altering their capacity to manage drug uptake and efflux (Figure 3A). This modification might decrease the intracellular concentration of therapeutic agents, consequently diminishing their efficacy [16,111]. Gaining insights into how SUMOylation influences the activity of drug transporter proteins is crucial for devising strategies aimed at enhancing drug accumulation within drug-resistant myeloma cells.
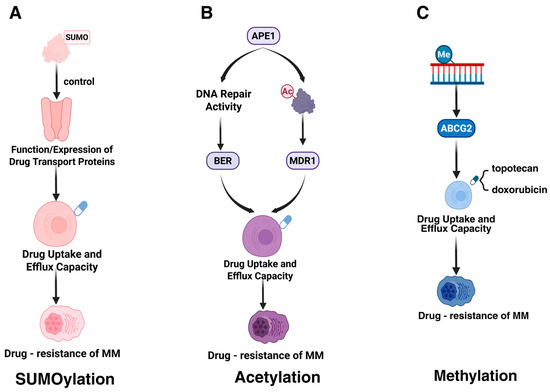
Figure 3.
PTMs affecting drug uptake or efflux. This figure summarizes the impact of three PTMs on drug uptake or efflux capacity and their association with drug resistance in MM. (A) SUMOylation regulates the function or expression of drug transport proteins, affecting drug uptake and efflux, leading to MM drug resistance. (B) Acetylation modification of the APE1 protein may enhance DNA repair activity through the base excision repair (BER) pathway, thereby altering drug accumulation within cells and promoting MM drug resistance. (C) Methylation modification of the ABCG2 protein is associated with the efflux capacity of drugs such as topotecan and doxorubicin, further exacerbating MM drug resistance. The figure was created in Biorender. Hu, S. (2025) https://BioRender.com (accessed on 19 March 2025).
4.1.2. Acetylation
The acetylation modification of APE1 (specifically at lysine residues K6 and K7) enhances cellular resistance to melphalan by regulating the expression of multidrug resistance protein 1 (MDR1), thereby promoting drug efflux (Figure 3B) [36]. Additionally, the DNA repair activity of APE1 mediates resistance through the base excision repair (BER) pathway, which repairs melphalan-induced DNA damage [36].
4.1.3. Methylation
The methylation modification of the ABCG2 promoter enhances drug efflux capacity by regulating the expression level of the ABCG2 gene, thereby influencing drug resistance in MM [58,59]. High expression of ABCG2, functioning as an ATP-binding cassette (ABC) transporter, mediates the active efflux of chemotherapeutic agents (e.g., topotecan, doxorubicin), reducing intracellular drug concentrations (Figure 3C). Additionally, exposure to chemotherapeutic drugs can further upregulate ABCG2 expression, aggravating drug resistance [112].
4.2. Altering Drug Targets
4.2.1. Phosphorylation
Qiang et al. discovered that the MAFb protein is overexpressed in MM cells as a result of the t(14;20) translocation or Igλ insertion, with its protein levels being regulated by PTMs (Figure 4A). GSK3 is capable of phosphorylating the MAFb protein, thereby controlling its degradation. PIs can impede the degradation of the MAFb protein, leading to its accumulation in both the nucleus and cytoplasm [83]. This accumulation can disrupt drug targets and contribute to cellular resistance against PIs. GCK promotes the proliferation and survival of RAS-mutant MM cells by inducing phosphorylation-mediated activation of the MKK4/7-JNK signaling pathway. Inhibiting GCK blocks this phosphorylation process, leading to the degradation of key transcription factors (e.g., IKZF1/3, c-MYC), thereby overcoming resistance to PIs (e.g., bortezomib) and IMiDs [29]. Akt-mediated phosphorylation of GSK-3β (p-GSK-3β) suppresses its kinase activity, impairing its ability to degrade c-Myc. This results in c-Myc protein stabilization and accumulation, promoting MM cell survival and drug resistance [84].
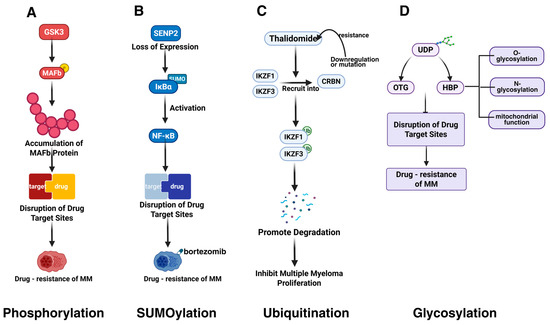
Figure 4.
PTMs altering drug targets in MM. This figure illustrates four key PTMs that disrupt drug target sites and contribute to drug resistance in MM. (A) GSK3-mediated phosphorylation stabilizes MAPb protein, destabilizing drug targets and promoting resistance. (B) Loss of SENP2 triggers IκBα SUMOylation, activating NF-κB signaling and enhancing resistance. (C) CRBN downregulation or mutations cause IMiD resistance. (D) Upregulated mitochondrial biogenesis markers (PGC1α, SIRT1), enhanced antioxidant capacity, and maintained mitochondrial integrity reduce apoptosis and promote resistance. The figure was created in Biorender. Hu, S. (2025) https://BioRender.com (accessed on 19 March 2025).
4.2.2. SUMOylation
In MM, the overexpression of the SUMO E2 conjugating enzyme Ubc9, the SUMO E3 ligase PIAS1, and the SUMOylation-inducing factor, the tumor suppressor ARF, is associated with poor prognosis [113]. Ubc9, a central protein in SUMOylation, may modulate drug resistance by altering the properties of drug targets [63]. Another study found that deficiency in SENP2 expression leads to increased SUMOylation of IκBα and subsequent NF-κB activation, which can induce bortezomib resistance (Figure 4B) [89]. This resistance may be mediated by changes in the activity or expression levels of drug targets.
Van Andel et al. found that the deletion of the tumor suppressor CYLD, overexpression of the transcriptional co-activator BCL9, and SUMOylation of β-catenin can abnormally activate the Wnt signaling pathway, potentially altering drug target activity or expression levels [114]. Alterations of SUMOylation also affect the resistance of MM to lenalidomide. The lenalidomide-resistant MMR10R cell line shows higher levels of SUMO E1 (SAE2) and overall SUMOylation compared to the lenalidomide-sensitive MM1S cell line [90]. Inhibition of SUMOylation with the SUMO E1 inhibitor TAK-981 significantly increases myeloma cell sensitivity to lenalidomide. SUMOylation may also modulate drug sensitivity by regulating the IRF4-Myc pathway, with lenalidomide resistance associated with overexpression of IRF4 and c-Myc [90]. TAK-981 can reduce IRF4 and c-Myc expression, affecting drug targets and resistance by decreasing DOT1L and histone H3 lysine 79 methylation, reducing IRF4 gene transcription, and enhancing IRF4 protein degradation.
4.2.3. Ubiquitination
BRCC36, an interacting protein of CRBN (a substrate receptor of cullin 4-RING E3 ligase (CRL4)), is considered a primary target of the immunomodulatory drug thalidomide. Thalidomide and its structural analogs lenalidomide and pomalidomide promote the ubiquitination-mediated degradation of two essential transcription factors, IKZF1 (Ikaros) and IKZF3 (Aiolos), by directly recruiting them to CRBN, thereby inhibiting the proliferation of myeloma cells. BRCC36 protects CRBN from degradation by cleaving K63-linked polyubiquitin chains (Figure 4C) [49]. The compound SHIN1 up-regulates BRCC36 by binding to SHMT2, increasing CRBN levels and affecting lenalidomide sensitivity in MM cells.
Katia et al. found that blocking the transient receptor potential vanilloid receptor 1 (TRPV1) affects MM drug resistance [115]. The TRPV1 antagonist AMG9810 inhibits MM cell viability and synergizes with bortezomib. AMG9810 inhibits CXCR4 expression and activity, overcoming matrix-mediated drug resistance and regulating the ubiquitin signaling pathway, down-regulating 38 proteins related to ubiquitination during bortezomib/AMG9810 co-treatment and abolishing bortezomib-induced ubiquitination of cytoplasmic and mitochondrial proteins, thus altering drug targets and resistance [115]. IMiD agents suppress MM cell growth by CRBN-dependent ubiquitination and degradation of IKZF1/3. However, reduced CRBN expression or mutations impair this process, leading to IMiD resistance [30]. In contrast to IMiDs, GCK inhibitors overcome IMiD resistance by inducing ubiquitination and degradation of key transcription factors IKZF1/3 through a CRBN-independent mechanism [29].
4.2.4. Deubiquitination
The reversal of ubiquitination by deubiquitinating enzymes (DUBs) is also implicated in MM drug resistance. High expression of DUBs such as Usp9x, Usp24, and Usp7 is associated with poor prognosis and may affect drug efficacy by stabilizing key pro-survival proteins like Mcl-1 [52]. The deubiquitinase USP7 interacts with NEK2 (a key regulator of chromosomal instability) and inhibits its ubiquitination, thereby stabilizing NEK2 protein levels. Overexpression of both USP7 and NEK2 in MM cells correlates with chemotherapy resistance and poor prognosis [51]. Notably, TRIP13 enhances USP7-mediated deubiquitination of NEK2 by forming a complex with USP7, which further stabilizes NEK2 and promotes resistance to PIs [88]. Additionally, USP7 stabilizes tumor suppressors like PTEN and p53, while TRIP13 modulates their subcellular localization, collectively contributing to altered drug responses [88]. CRBN downregulation or mutations prevent IMiDs from effectively recruiting IKZF1/3 to the E3 ubiquitin ligase complex, thereby blocking their degradation. This failure to degrade IKZF1/3 results in an inability to suppress c-Myc and interferon regulatory factor-4 (IRF4), ultimately driving IMiD resistance [84].
4.2.5. Glycosylation
Aberrant glycosylation also affects MM drug resistance. Bortezomib-resistant U266-R cells exhibit higher levels of glycosylated uridine diphosphate (UDP) derivatives and increased activity of the hexosamine biosynthesis pathway (HBP) compared to bortezomib-sensitive U266-S cells (Figure 4D) [115]. HBP regulates protein O- and N-glycosylation and mitochondrial function. O-linked N-acetylglucosamine transferase (OGT), involved in glycosylation, is more highly expressed in U266-R, while O-GlcNAcase (OGA), involved in deglycosylation, is less expressed. After bortezomib treatment, U266-R maintains higher concentrations of glycosylated substrates, unlike U266-S, which may alter drug targets and resistance.
4.2.6. Neddylation
The inhibition of the NEDD8-activating enzyme (NAE) blocks the activity of CRL (Cullin-RING ubiquitin ligase), leading to the stabilization and upregulation of the REDD1 protein. MLN4924, a NAE inhibitor, disrupts CRL-mediated ubiquitination by blocking neddylation, resulting in the accumulation of CRL substrates (e.g., p27, CDT1, NRF2) and REDD1 stabilization. REDD1 inhibits the PI3K/AKT/mTOR signaling pathway, thereby counteracting growth factor-mediated survival signals (e.g., IL-6, IGF-1) [91]. Importantly, MLN4924 demonstrates efficacy in bortezomib-resistant MM cells (e.g., ANBL-6-VSR), suggesting that its mechanism is independent of proteasome inhibition [116].
4.3. Regulating Cell Death and Survival Signals
The UPR is a cellular adaptive mechanism that activates in response to ER stress, enabling cells to mitigate proteotoxicity and maintain survival under drug-induced pressure. Moderate UPR activation helps alleviate ER stress caused by excessive protein synthesis, such as the production of monoclonal immunoglobulins in MM cells. However, sustained or overwhelming UPR activation can trigger cell death through JNK- or ER stress-dependent apoptotic pathways. MM cells initially rely on UPR to manage proteotoxic stress, making them sensitive to proteasome inhibitors (PIs). Over time, drug resistance develops due to reduced expression of key UPR regulators (e.g., XBP1), highlighting the dual role of UPR in MM therapy [117].
4.3.1. Phosphorylation
Phosphorylation modifications exert a core role in drug resistance in MM by regulating multiple key signaling pathways (Figure 5A). AMG9810 enhances the sensitivity of MM cells to drugs and overcomes resistance by upregulating C/EBP homologous protein (CHOP) to induce ER stress, triggering the UPR [115]. Concurrently, it reduces the expression of anti-apoptotic proteins BCL-2 and MCL-1 in a dose-dependent manner, inhibits phosphorylation of the mTOR downstream target pS6, and activates executor caspase-3 [118]. On the other hand, GCK (MAP4K2) drives cell proliferation and adaptive resistance by phosphorylating and activating the MKK4/7-JNK pathway, sustaining RAS mutation-dependent MAPK signaling hyperactivation. This phosphorylation-dependent MAPK overactivation not only promotes survival but may also enhance chemotherapy resistance by regulating downstream transcription factors such as c-JUN [29].
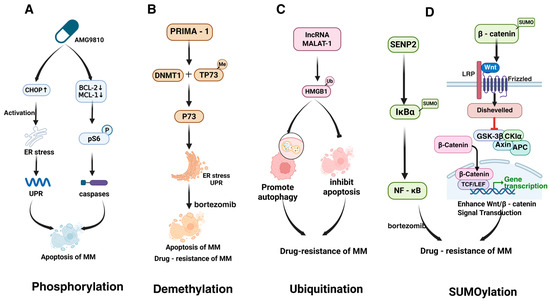
Figure 5.
PTMs regulate cell death and survival in MM. This figure highlights key PTMs that regulate cell death and survival signals in MM. (A) Phosphorylation activates CHOP1 and ER stress pathways, inducing apoptosis. (B) Demethylation by PRIMA-1 depletes DNMT1 and restores sensitivity in bortezomib-resistant cells. (C) Ubiquitination of HMGB1, induced by lncRNA MALAT-1, promotes autophagy and inhibits apoptosis. (D) SUMOylation of IκBα and β-catenin activates NF-κB and Wnt/β-catenin signaling, reducing apoptosis and promoting drug resistance. The figure was created in Biorender. Hu, S. (2025) https://BioRender.com/ (accessed on 19 March 2025).
The phosphorylation status of the Akt/mTOR signaling pathway exhibits dual regulatory properties in MM drug resistance [119]. INPP4B enhances the efficacy of bortezomib by suppressing phosphorylation of Akt at Ser473, thereby attenuating PI3K/Akt/mTOR pathway activity, whereas loss of INPP4B leads to Akt hyperactivation and drug resistance [120,121,122]. Simultaneously, Akt-mediated phosphorylation inactivates EZH2, reducing H3K27me3 levels and derepressing pro-survival genes. This results in sustained expression of anti-apoptotic genes (e.g., BCL-2 family members), promoting cell adhesion-mediated drug resistance (CAM-DR) [61,123,124]. The crosstalk between epigenetic regulation and phosphorylation modifications underscores the complexity of signal integration within the tumor microenvironment.
Phosphorylation-driven activation of the NF-κB and STAT3 pathways further expands the network dimensions of resistance mechanisms. Docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) activate the NF-κB pathway by increasing phosphorylation of its subunit p65 at Ser536, enhancing anti-apoptotic capacity and reducing sensitivity to bortezomib [125]. However, the timing of pre-treatment with DHA/EPA enhances toxicity, while co-administration causes antagonism—highlighting the critical role of temporal regulation. Additionally, JAK kinase promotes phosphorylation of STAT3 at Tyr705, upregulating anti-apoptotic targets such as BCL-XL and MCL-1, thereby constructing multilayered survival barriers for MM cells. These phosphorylation events collectively shape a dynamic signaling network in which hyperactivation of pro-survival pathways (e.g., Akt/mTOR, NF-κB, STAT3) synergizes with epigenetic regulation (e.g., EZH2 inactivation) to drive drug resistance [81,126].
4.3.2. Demethylation
Small molecule PRIMA-1 was shown to influence drug resistance in MM through demethylation of p73. PRIMA-1 induces ER stress and the UPR by depleting DNA methyltransferase 1 (DNMT1), leading to TP73 demethylation and enhanced UPR [127]. Notably, PRIMA-1 exhibits greater toxicity in p53-deficient cells, suggesting that p73 may act as an alternative tumor suppressor. Knockdown of p73 attenuates PRIMA-1-induced apoptosis and ER stress marker upregulation, while p73 overexpression enhances UPR marker expression and growth inhibition (Figure 5B). PRIMA-1 also synergizes with bortezomib and partially restores sensitivity in bortezomib-resistant cells, potentially by amplifying UPR and modulating apoptotic pathways to overcome drug resistance [127].
4.3.3. Ubiquitination
Zhuang et al. reported that ubiquitin-activating enzyme inhibitors can reverse MM resistance to PIs by inducing the UPR [127]. MM cells rely on UPR to alleviate ER stress caused by excessive M protein production, making them initially sensitive to PIs [117]. However, drug resistance development is associated with reduced expression of key UPR regulators, such as XBP1. Multiple PTMs of HMGB1 were also shown to promote MM drug resistance. HMGB1, which participates in DNA damage repair and autophagy, is highly expressed in MM and negatively correlates with patient survival (Figure 5C). For instance, lncRNA MALAT-1 induces HMGB1 ubiquitination at the post-translational level, increasing its expression, promoting autophagy, inhibiting apoptosis, and ultimately affecting drug sensitivity [87].
4.3.4. Deubiquitination
USP14 and UCHL5 are highly expressed in MM and contribute to drug resistance by deubiquitinating misfolded or unfolded proteins, thereby reducing intracellular stress levels. Inhibition of these DUBs reduces MM cell viability, suppresses proliferation, and restores drug sensitivity in resistant cells [53]. Additionally, certain DUBs stabilize anti-apoptotic proteins like Mcl-1, modulating apoptotic pathways and influencing MM cell sensitivity to drugs [52].
4.3.5. Glycosylation
Mitochondria-related changes have been implicated in MM drug resistance. For example, mitochondrial biogenesis markers PGC1α and SIRT1 are significantly upregulated in bortezomib-resistant U266-R cells compared to sensitive U266-S cells [88]. Notably, aberrant O-GlcNAcylation of mitochondrial proteins (e.g., VDAC1 and Complex I subunits) enhances oxidative phosphorylation efficiency, supporting energy demands in resistant cells [128]. U266-R cells also exhibit increased mitochondrial mass, elevated expression of the antioxidant enzyme GSTK1, and higher glutathione (GSH) concentrations. Furthermore, enhanced hexosamine biosynthesis pathway (HBP) activity in resistant cells promotes protein O-GlcNAcylation, which stabilizes anti-apoptotic BCL-2 family proteins and sustains mitochondrial membrane potential [129]. Unlike U266-S cells, which undergo mitochondrial depolarization and downregulation of cytochrome b and ATP synthase following bortezomib treatment, U266-R cells maintain mitochondrial integrity, suggesting that mitochondrial adaptations may reduce apoptosis and contribute to drug resistance [88].
4.3.6. SUMOylation
Xie et al. demonstrated that downregulation of the SUMO-specific protease SENP2 in MM activates NF-κB by regulating the SUMOylation of IκBα, leading to bortezomib resistance [130]. Another study further highlighted that SUMOylation of β-catenin enhances Wnt/β-catenin signaling, promoting MM cell proliferation, reducing apoptosis, and impairing drug sensitivity (Figure 5D). Loss of CYLD stabilizes β-catenin and enhances β-catenin/TCF-mediated transcription, while BCL9 overexpression amplifies Wnt signaling, tumor cell proliferation, migration, and vascular endothelial growth factor (VEGF) expression, all of which may contribute to drug resistance [114].
4.3.7. Deacetylation
Deacetylation (mediated by HDAC1) enhances the stability of HP1γ and strengthens its interaction with MDC1, promoting homologous recombination repair (HR) and improving cellular tolerance to DNA damage [95]. Concurrently, deacetylation induces HP1γ nuclear condensation through liquid-liquid phase separation (LLPS), which increases chromatin accessibility and upregulates the expression of drug resistance-related genes (e.g., CD40, FOS, JUN). These mechanisms collectively reduce sensitivity to PIs by enhancing DNA repair efficiency and activating survival pathways [95]
4.4. Modulating the Tumor Microenvironment and Immune Escape
4.4.1. Sialylation
The drug resistance of MM is closely associated with sialyltransferase (STs)-mediated tumor cell-microenvironment interactions and immune escape (Figure 6). Studies demonstrate that STs enhance the binding capacity of MM cells to adhesion molecules (e.g., VCAM1, MADCAM1, and E-selectin) on bone marrow endothelial cells by catalyzing sialylation modifications of integrins α4β1/α4β7 and selectin ligands (e.g., SLea/x), thereby driving tumor cell homing to the bone marrow microenvironment (BMM) [92,93,94]. This homing process enables MM cells to acquire the protective barrier of BMM, which reduces exposure to chemotherapeutic agents (e.g., bortezomib) through physical isolation while activating cell adhesion-mediated drug resistance (CAM-DR). Notably, in a human MM mouse model, stromal cells in BMM activate the STAT3 signaling pathway via direct contact and secretion of factors such as IL-6, forming a positive feedback loop that persistently upregulates key enzymes like ST3GAL6 and ST6GAL1. This further promotes sialylation of membrane proteins such as CD147, enhancing tumor cell proliferation, metastasis, and microenvironmental adaptation [131].
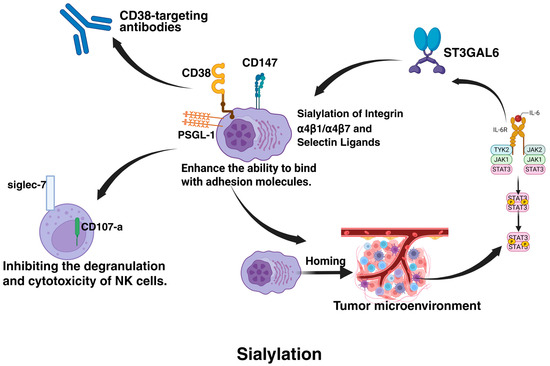
Figure 6.
Sialylation-mediated regulation of the tumor microenvironment and immune escape in MM. This figure illustrates how sialylation promotes MM progression by enhancing tumor homing to the bone marrow microenvironment via integrin and selectin ligand modifications, reducing chemotherapeutic exposure and activating cell adhesion-mediated drug resistance (CAM-DR). Stromal cell-derived IL-6 activates STAT3, upregulating sialyltransferases (ST3GAL6/ST6GAL1) in a positive feedback loop, further driving sialylation (e.g., CD147) to enhance tumor proliferation. Sialylated PSGL-1 binds Siglec-7 on NK cells, suppressing cytotoxicity, while sialylation masks CD38 epitopes, impairing antibody recognition. The figure was created in Biorender. Hu, S. (2025) https://BioRender.com (accessed on 19 March 2025).
At the immunomodulatory level, sialylation modifications establish a dual inhibitory barrier. On the one hand, sialylated PSGL-1 binds to the inhibitory receptor Siglec-7 on natural killer (NK) cells, significantly suppressing NK cell degranulation (reduced CD107a expression) and cytotoxicity (diminished IFN-γ release) [76]. On the other hand, sialylation-induced epitope masking of CD38 directly impairs the recognition efficiency of CD38-targeted antibodies like daratumumab, leading to resistance in immunotherapy. This immunosuppressive microenvironment can be reprogrammed through ST inhibitors [93]. The pan-sialyltransferase inhibitor 3F-Neu5Ac not only reduces sialylation levels of α4 integrins, weakening their ligand-binding affinity but also remodels immune cell composition—promoting infiltration of CD8+ T cells and NK cells while reducing regulatory T cell populations—thereby synergistically enhancing anti-tumor immune responses [131,132].
From a therapeutic perspective, 3F-Neu5Ac operates through two critical mechanisms: it inhibits ST3GAL6-catalyzed generation of E-selectin ligands, blocking MM cell homing to BMM and restricting tumor cell entry into drug sanctuaries; simultaneously, it disrupts glycosylation-dependent maturation of integrin α4 chains, directly impairing tumor-stromal cell adhesion-dependent survival signals [133]. In animal models, this dual action significantly enhances bortezomib sensitivity and prolongs survival [133]. However, it is noteworthy that this inhibitor fails to reverse IL-6-mediated stromal cell-induced resistance in vitro, suggesting that fully overcoming drug resistance requires combinatorial targeting of microenvironmental signaling pathways [131]. These findings provide a theoretical foundation for developing sialylation-targeted combination therapies, emphasizing the necessity to concurrently address intrinsic tumor cell resistance mechanisms and microenvironment-mediated extrinsic protective effects.
4.4.2. Methylation
PRMT1 mediates methylation modifications that downregulate the expression of major histocompatibility complex class II (MHC II) genes, impairing the function of antigen-presenting cells and subsequently suppressing the expression of T cell activation markers CD25/CD69 [134]. This process contributes to the formation of an immunosuppressive microenvironment. Authors showed that inhibiting PRMT1 can restore MHC II-mediated antigen presentation efficiency, promote T cell activation, and provide a potential therapeutic target for reversing immune escape [134].
4.4.3. Neddylation
Neddylation modifications regulate the activity of CRLs, the stability of the IMiD target CRBN, and the transcriptional network (IRF4/IKZF3/MEIS2), thereby influencing the immune escape capabilities and drug responses of MM cells and contributing to drug resistance. Inhibition of neddylation enhances immune recognition and restores IMiD sensitivity [71]. Additionally, TGFβ promotes immune escape and drug resistance in MM by suppressing the expression of natural killer (NK) cell activation receptors (e.g., NKG2D) and their effector functions (e.g., degranulation). Neddylation inhibition can partially reverse TGFβ-mediated suppression of NK cells, restoring NKG2D expression and perforin polarization, thereby enhancing NK cell-mediated killing of MM cells [71,116]. Furthermore, neddylation inhibition enhances the antibody-dependent cellular cytotoxicity (ADCC) effects mediated by daratumumab (anti-CD38) and elotuzumab (anti-SLAMF7) by upregulating Rac1/RhoA GTPase and F-actin levels in NK cells, improving immune synapse function, and overcoming resistance to monoclonal antibody therapy in MM [116].
5. Overcoming Drug Resistance in MM via Targeting PTMs
The critical role of PTMs in MM drug resistance highlights the potential for targeting these modifications to overcome resistance. Given the reversible nature of many PTMs and the availability of specific inhibitors, several strategies can be employed to modulate aberrant PTMs and restore drug sensitivity. This section recapitulates various approaches, including FDA-approved drugs, preclinical inhibitors, gene editing technologies, and novel therapeutic modalities like PROTACs and LYSOTACs, to target PTMs and overcome drug resistance in MM (Table 2).
Table 2.
Strategies to Overcome Drug Resistance in MM by Targeting PTMs.
Table 2.
Strategies to Overcome Drug Resistance in MM by Targeting PTMs.
| Strategy Category | Drug/Technology Name | Targeted PTM | Target/Pathway | Mechanism of Action | Ref |
|---|---|---|---|---|---|
| FDA-Approved Drugs | |||||
| Tyrosine Kinase Inhibitors (TKI) | Dasatinib | Phosphorylation | Src, BCR-ABL | Inhibits Src and BCR-ABL kinase activity, blocks PI3K/AKT and MAPK phosphorylation signaling, reverses resistance to PIs (PIs, e.g., bortezomib) and IMiDs (IMiDs, e.g., lenalidomide). | [135,136,137,138] |
| Tyrosine Kinase Inhibitors (TKI) | Imatinib | Phosphorylation | BCR-ABL | Binds specifically to the ATP-binding site of the BCR-ABL fusion protein, inhibits STAT5 and RAS/MAPK phosphorylation, and suppresses MM (MM) cell proliferation. | [135,136,137] |
| mTOR Inhibitor | Everolimus | Phosphorylation | mTORC1 (not mTORC2) | Inhibits mTORC1 phosphorylation activity, downregulates 4EBP1 and S6K1 signaling, blocks protein synthesis, and enhances sensitivity to PIs. | [135,136,137] |
| HDAC Inhibitor | Panobinostat | Acetylation, SUMOylation (indirect) | HDAC1/2/3/6 | Inhibits HDAC6-mediated deacetylation of SUMOylation enzymes (e.g., SENP1), increases histone (H3K9/K14) and non-histone (e.g., HSP90) acetylation, and activates pro-apoptotic genes (BIM, NOXA). | [139,140,141,142,143] |
| HDAC Inhibitor | Vorinostat | Acetylation | HDAC1/2/3 (Class I) | Selectively inhibits Class I HDACs, enhances histone H3/H4 acetylation, promotes RelA acetylation to block its nuclear translocation, and reverses NF-κB-mediated drug resistance. | [141,142,143] |
| Preclinical Inhibitors | |||||
| SUMO E1 Inhibitor | TAK-981 | SUMOylation | SUMO E1 enzyme (SAE1/SAE2) | Covalently inhibits SUMO activation, reduces SUMO modification of IRF4 and c-Myc, and enhances CRBN-dependent degradation induced by lenalidomide. | [90,144,145,146] |
| Neddylation Inhibitor | MLN4924 | Ubiquitination (via Neddylation) | NEDD8-activating enzyme (NAE) | Inhibits neddylation of CRL complexes, leading to accumulation of pro-apoptotic proteins (NOXA, BIM) and stabilization of IκBα to suppress NF-κB signaling. | [69,147] |
| Glycosylation Inhibitor | OGT inhibitors (e.g., OSMI-1) | Glycosylation (O-GlcNAcylation) | O-GlcNAc transferase (OGT) | Reduces O-GlcNAc modification of β-catenin (Ser112) and c-Myc, inhibits Wnt/β-catenin and MYC signaling, and reverses bortezomib resistance. | [129,148] |
| Gene Editing Technologies | |||||
| CRISPR/Cas9 | Ubc9/USP7 knockout | SUMOylation, Ubiquitination | Ubc9 (SUMO E2), USP7 | Knockout of Ubc9 blocks SUMO-PML modification and inhibits DNA repair; knockout of USP7 stabilizes p53 and PTEN protein levels, inducing apoptosis. | [149,150,151,152,153,154,155] |
| siRNA/shRNA | SAE2/USP14 targeting | SUMOylation, Ubiquitination | SAE2, USP14 | siRNA-mediated SAE2 silencing inhibits SUMO activation; shRNA-mediated USP14 silencing enhances ubiquitinated protein degradation and downregulates IRF4. | [156,157,158,159,160,161] |
| ASO | MDM2/USP7 targeting | Ubiquitination | MDM2, USP7 | Antisense oligonucleotides (ASOs) suppress MDM2 or USP7 expression, stabilize ubiquitination levels of p53 or PTEN, and promote apoptosis. | [162,163] |
| Novel Therapeutic Modalities | |||||
| PROTACs | IRF4/c-Myc degraders | Ubiquitination | IRF4, c-Myc | Bifunctional molecules recruit CRBN or VHL to induce ubiquitination and degradation of IRF4 or c-Myc, directly eliminating drug resistance-associated transcription factors. | [164,165,166,167,168] |
| LYSOTACs | Wnt pathway-targeted degraders | Lysosomal degradation | LRP6, β-catenin | Target LRP6 or β-catenin for lysosomal degradation, inhibits Wnt signaling, and restore bortezomib sensitivity. | [169,170,171,172,173,174,175,176] |
| Combination Therapies | |||||
| SUMO inhibitor + PI | TAK-981 + Bortezomib | SUMOylation + Ubiquitination | SUMO pathway + Proteasome | TAK-981 induces misfolded protein accumulation, synergizing with bortezomib to activate the UPR (UPR) and ER stress, triggering apoptosis. | [70,111] |
| HDAC inhibitor + IMiD | Panobinostat + Lenalidomide | Acetylation + Ubiquitination | HDACs + CRBN | Panobinostat upregulates CRBN expression, enhancing lenalidomide-induced ubiquitination and degradation of IKZF1/3, thereby suppressing the IRF4-MYC axis. | [40,84,177] |
5.1. FDA-Approved Drugs Targeting PTMs
FDA-approved tyrosine kinase inhibitors (TKIs) and mTOR inhibitors, such as dasatinib, imatinib, everolimus, and temsirolimus, target phosphorylation-dependent signaling pathways [135,136]. These drugs inhibit kinases like Src, BCR-ABL, and mTOR, which are often hyperactivated in drug-resistant MM. By modulating phosphorylation, these agents can restore sensitivity to PIs (PIs) and IMiDs (IMiDs) [137]. For example, dasatinib has shown potential in preclinical studies to overcome resistance by targeting phosphorylation-dependent survival pathways [136,138].
HDAC inhibitors, such as panobinostat, modulate acetylation levels of histones and non-histone proteins, influencing gene expression and protein function [139,140]. HDAC inhibitors have shown promise in overcoming resistance to PIs and IMiDs by restoring the expression of pro-apoptotic genes and enhancing the acetylation of drug targets. While no FDA-approved drugs specifically target SUMOylation, certain agents like HDAC inhibitors (e.g., panobinostat and vorinostat) can indirectly modulate SUMOylation pathways [141,142,143]. Panobinostat is approved for MM, inhibits HDACs, and has been shown to impact SUMOylation, potentially restoring sensitivity to PIs and IMiDs [143]. The repurposing of FDA-approved drugs to target PTMs offers a promising strategy for overcoming drug resistance in MM. Future research should focus on identifying novel PTM targets, developing more specific inhibitors, and exploring the potential of combination therapies.
5.2. Preclinical Inhibitors Targeting PTMs
Novel SUMOylation and ubiquitination inhibitors show promise in overcoming drug resistance in MM. SUMOylation inhibitors, such as TAK-981, a SUMO E1 inhibitor, have demonstrated potential in preclinical studies to increase sensitivity to lenalidomide by reducing the expression of key resistance regulators like IRF4 and c-Myc [90,178]. Similarly, neddylation-dependent ubiquitination inhibitors like MLN4924 (a NEDD8-activating enzyme inhibitor) have shown promise in preclinical models by blocking the activity of cullin-RING ligases (CRLs), essential for ubiquitination [69,147]. By inhibiting CRL activity, MLN4924 can induce the accumulation of pro-apoptotic proteins and enhance the efficacy of PIs. Additionally, targeting specific E3 ubiquitin ligases or deubiquitinases (DUBs) may provide more targeted approaches for overcoming resistance [45,104,179]. Glycosylation inhibitors also offer potential strategies to address drug resistance in MM [128]. Inhibitors of glycosylation enzymes, such as O-GlcNAc transferase (OGT) inhibitors, have shown potential in preclinical studies to restore drug sensitivity by modulating glycosylation levels [129,148]. Such small molecules are promising agents for overcoming drug resistance in MM.
5.3. Gene Editing Technologies
5.3.1. CRISPR/Cas9
The CRISPR/Cas9 system has emerged as a powerful tool for precisely editing genes involved in PTMs, offering a direct approach to modulate key pathways MM [149]. By targeting enzymes central to SUMOylation, ubiquitination, or glycosylation, CRISPR/Cas9 can be used to either activate or inactivate specific PTMs, thereby modulating drug resistance pathways [150,151,152,153]. For instance, knocking out the SUMO E2 conjugating enzyme Ubc9 or the deubiquitinating enzyme USP7 has been shown to restore sensitivity to therapeutic agents by disrupting resistance mechanisms [154,155]. Additionally, CRISPR/Cas9 can be used to study the functional consequences of specific PTMs in MM, providing valuable insights into potential therapeutic targets and enabling the development of more effective treatments.
5.3.2. Oligonucleotide-Based Interventions
Oligonucleotide-based strategies, such as RNA interference (RNAi) technologies, provide a powerful and versatile approach to selectively silence genes involved in PTMs (PTMs) [156,157,158]. RNAi tools, including small interfering RNA (siRNA) and short hairpin RNA (shRNA), can be designed to target and reduce the expression of key enzymes in PTM pathways, such as SUMOylation (e.g., SAE2) or deubiquitinating enzymes (e.g., USP14) [159,160,161]. By modulating these enzymes, RNAi can decrease specific PTMs and enhance drug sensitivity in cancer cells. The reversible and tunable nature of RNAi-based approaches allows researchers to precisely explore the roles of PTMs in drug resistance and identify potential therapeutic targets. This flexibility makes RNAi a valuable tool not only for preclinical studies but also for potential clinical applications in MM. Beyond RNAi, other oligonucleotide-based interventions, such as antisense oligonucleotides (ASOs), are also being explored for their ability to modulate gene expression and PTM-related pathways, further expanding the therapeutic potential of this approach [162,163].
5.4. Novel Therapeutic Modalities
5.4.1. Proteolysis-Targeting Chimeras (PROTACs)
PROTACs are bifunctional molecules that recruit E3 ubiquitin ligases to target proteins for degradation [164,165]. By leveraging the ubiquitination pathway, PROTACs can selectively degrade proteins involved in drug resistance, such as IRF4 or c-Myc [166]. For example, a PROTAC targeting IRF4 could overcome lenalidomide resistance by reducing the levels of this transcription factor [167]. This approach offers a promising strategy for targeting undruggable proteins and overcoming resistance in MM. Recent studies have demonstrated the potential of PROTACs to enhance the efficacy of existing therapies by degrading key resistance proteins, thereby restoring sensitivity to treatments like PIs and IMiDs [165,168].
5.4.2. Lysosome-Targeting Chimeras (LYSOTACs)
LYSOTACs represent a novel class of therapeutics that target proteins for lysosomal degradation [169,170]. Unlike PROTACs, which rely on the UPS, LYSOTACs utilize the lysosomal pathway to degrade target proteins. This approach could be particularly useful for targeting membrane-bound or extracellular proteins involved in drug resistance [169,170,171]. For instance, a LYSOTAC targeting components of the Wnt signaling pathway could reduce β-catenin levels and restore sensitivity to PIs [172,173,174]. By leveraging the lysosomal pathway, LYSOTACs offer a complementary strategy to PROTACs, expanding the range of targetable proteins and providing new opportunities to overcome resistance mechanisms in MM [175,176].
5.5. Combination Therapies
Combining PTM-targeting agents with existing therapies may provide a synergistic effect in overcoming drug resistance. For example, combining a SUMOylation inhibitor with a PI could enhance the accumulation of misfolded proteins and induce apoptosis in resistant MM cells [70,111]. Similarly, combining an HDAC inhibitor with an IMiD could restore the expression of pro-apoptotic genes and enhance the degradation of drug targets [84,177]. Rational combination therapies that target multiple PTMs simultaneously may offer a more effective strategy for overcoming resistance in MM. By addressing multiple resistance pathways, these combinations could provide a more comprehensive and durable therapeutic effect.
5.6. Personalized Therapy
The current treatment landscape for multiple myeloma remains largely rooted in a “one-size-fits-all” approach despite overwhelming evidence of the disease’s molecular and clinical heterogeneity. This paradigm fails to address critical variations in PTM-driven resistance mechanisms across patients, often resulting in suboptimal outcomes. As we enter the era of precision medicine, targeting individual patients’ unique PTM signatures—from phosphorylation patterns to glycosylation profiles—represents a transformative opportunity to overcome therapeutic resistance.
5.6.1. Personalized CAR-T Therapy
BCMA, a critical target of CAR-T therapy, exhibits membrane surface expression regulated by both glycosylation and proteolytic cleavage. Research indicates that N-glycosylation modifications of BCMA significantly impact CAR-T cell recognition efficiency toward its epitopes, with specific glycosylation patterns potentially serving as biomarkers to predict CAR-T efficacy [180,181]. Notably, ADAM10 and γ-secretase-mediated proteolytic cleavage of BCMA releases soluble BCMA (sBCMA), resulting in antigen loss and immune evasion [3,182]. Clinical data demonstrate that dynamic sBCMA levels correlate strongly with tumor burden and prognosis (hazard ratio [HR] = 2.1, p < 0.001) [182]. Combining γ-secretase inhibitors (e.g., LY3039478) reduces sBCMA production, enhancing CAR-T cell targeting efficiency for membrane-bound BCMA by 40% [182].
5.6.2. Targeted Interventions in Epigenetic Modifiers and Personalized Therapy
The t(4;14) translocation upregulates the MMSET (WHSC1) gene, leading to aberrant histone methylation modifications and conferring resistance to proteasome inhibitors [183]. Preclinical studies demonstrate that small-molecule inhibitors targeting MMSET can enhance sensitivity to bortezomib [184,185]. HDAC inhibitors, such as panobinostat, when combined with proteasome inhibitors, significantly prolonged PFS in relapsed/refractory patients in the phase III PANORAMA-1 trial (median PFS 12 vs. 8.1 months, p = 0.01) [182,186,187,188]. While challenges remain in standardizing PTM assessment and validating predictive algorithms, the field is moving decisively toward a future where MM treatment is tailored not just to cytogenetic risk groups but to each patient’s molecular vulnerability landscape.
6. The Challenges in Targeting PTMs for Therapy
6.1. Complexity and Dynamic Nature of PTMs
PTMs, including phosphorylation, acetylation, ubiquitination, glycosylation, and palmitoylation, are essential for regulating protein function, stability, and localization. In MM, abnormal PTM patterns can drive disease progression and affect treatment responses. However, the complexity of these modifications arises from their context-dependent roles, where different cell types, proteins, and disease states can influence how PTMs behave [15,189]. Importantly, crosstalk between different PTMs adds another layer of complexity. Lysine residues emerge as particularly contested sites, serving as substrates for acetylation, ubiquitination, SUMOylation, methylation, and other modifications [190,191]. This molecular “crowding” creates several layers of biological complexity: (1) Direct competition between modifications—for instance, acetylation and ubiquitination often vie for the same lysine, with acetylation typically stabilizing proteins while ubiquitination marks them for degradation; (2) Sequential dependencies where one modification enables another—phosphorylation frequently creates docking sites for E3 ubiquitin ligases, coupling signaling to protein turnover; (3) Bistable switches where mutually exclusive modifications create digital regulatory nodes [189]. The dynamic nature makes it challenging to target a specific PTM without inadvertently disrupting other critical cellular functions. For example, targeting the enzymes responsible for phosphorylation or acetylation might alter multiple downstream pathways, complicating the therapeutic strategy. This requires developing therapies that balance efficacy while minimizing unintended effects in non-cancerous tissues. Recent advances in structural biology and covalent inhibitor design are beginning to meet this challenge, as demonstrated by the development of lysine-targeting warheads that can distinguish between acetylation and ubiquitination sites based on local conformational features.
6.2. Heterogeneity and Plasticity of MM Cells
The heterogeneity and plasticity of cancer cells present significant barriers to PTM-targeted therapies in MM [16,192,193,194]. Different patients may exhibit distinct PTM patterns, even within the same subtype of MM, leading to varied responses to treatment. Furthermore, cancer cells can adapt by changing their PTM profiles in response to therapy, leading to treatment resistance [111,195,196,197]. This variability requires personalized therapeutic approaches that consider each patient’s unique PTM landscape, as a one-size-fits-all treatment is unlikely to be effective across the diverse patient population seen in MM.
6.3. Technological Limitations
The clinical translation of PTM-targeted therapies is hindered by the limitations of current technologies used to detect and analyze PTMs. Although advancements in proteomics have enhanced our ability to map PTM landscapes, these methods are not yet fully optimized for routine clinical use [198,199]. In MM, identifying PTM biomarkers that correlate with disease progression or therapeutic response is crucial to advancing these therapies. Additionally, rigorous clinical trials are required to ensure the safety and efficacy of PTM-targeted treatments, given the complexity of PTMs in cancer biology and their involvement in broader regulatory networks.
While PTMs offer promising therapeutic targets in MM, addressing the complexity, heterogeneity, and technological limitations associated with these modifications is essential for translating PTM-targeted therapies into clinical practice. Future research should focus on developing more precise and personalized approaches to overcome these challenges and improve outcomes for patients with MM.
7. Concluding Remarks
MM remains a challenging hematologic malignancy, primarily due to the inevitable development of therapeutic resistance. PTMs, including phosphorylation, acetylation, ubiquitination, and glycosylation, play pivotal roles in regulating protein function, stability, and cellular interactions. Dysregulation of these modifications contributes significantly to MM pathogenesis, tumor progression, and the emergence of drug resistance, particularly in relapsed or refractory cases. Understanding the molecular mechanisms underlying PTM-mediated resistance is essential for developing effective therapeutic strategies.
This review highlights the critical roles of PTMs in driving drug resistance in MM. Dysregulated PTMs—including phosphorylation, acetylation, ubiquitination, and glycosylation—alter key signaling pathways (e.g., NF-κB, STAT3, Wnt/β-catenin), metabolic reprogramming, and interactions with the tumor microenvironment, enabling MM cells to evade therapies such as PIs and IMiDs. Notably, PTM-mediated mechanisms mainly include modulation of drug uptake/efflux, altering drug targets, regulating cell death and survival, and immune evasion. Emerging strategies, such as HDAC inhibitors, SUMOylation blockers, and ubiquitin-proteasome modulators, demonstrate preclinical success in reversing resistance. However, challenges persist due to PTM crosstalk, tumor heterogeneity, and adaptive responses in the bone marrow niche.
To translate these insights into clinical breakthroughs, future efforts should prioritize (1) Mechanistic studies to decode context-dependent PTM interactions (e.g., SUMOylation-ubiquitination competition) using single-cell proteomics; (2) Biomarker development via high-throughput PTM profiling to stratify patients for targeted therapies; (3) Innovative therapeutics, including isoform-specific PTM inhibitors (e.g., HDAC6-selective agents) and combination regimens (e.g., neddylation inhibitors + anti-CD38 antibodies); and (4) Microenvironmental targeting to disrupt PTM-driven stromal protection (e.g., CRISPR-edited CAR-T cells against glycosylated BCMA). Collaborative interdisciplinary research—integrating epigenomics, structural biology, and AI-driven drug design—will be essential to overcome resistance and achieve durable remissions in MM.
In conclusion, this review underscores PTMs as central players in MM drug resistance and advocates for their therapeutic targeting. By integrating mechanistic insights with innovative technologies, we envision a paradigm shift toward precision medicine in MM, where PTM modulation could transform relapsed/refractory cases into manageable chronic conditions. Future research must bridge the gap between molecular understanding and clinical application to realize this potential.
Author Contributions
Conceptualization, Y.M.; writing—original draft preparation, S.H., J.X., W.C., H.J. and X.W.; writing—review and editing, Y.M. and S.H.; supervision, Y.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Natural Science Foundation of Xinjiang Uygur Autonomous Region [grant number 2024D01E17], National Natural Science Foundation of China [grant number 82460714], Xinjiang Uygur Autonomous Region Tianchi Talent Introduction Plan (“TianChi Excellent Award for Young Doctoral Talents”), State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia Fund [grant number SKL-HIDCA-2023-JY6], Xinjiang Medical University National Young Talent Cultivation Program [grant number XYD2024GR05], and the Undergraduate Innovation Training Program of Xinjiang Medical University (Project Number: X202410760050).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We gratefully acknowledge Na Bu (Zhejiang University Women’s Hospital) for her valuable suggestions and critical feedback during the revision of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ABC | ATP Binding Cassette |
| ASOs | Antisense Oligonucleotides |
| BER | Base Excision Repair |
| BiTEs | Bispecific T-Cell Engagers |
| BCMA | B Cell Maturation Antigen |
| BMM | Bone Marrow Microenvironment |
| CAM DR | Cell Adhesion-Mediated Drug Resistance |
| CAR-T | Chimeric Antigen Receptor T-Cell |
| CHOP | C EBP Homologous Protein |
| CIN | Chromosomal Instability |
| CRBN | Cereblon |
| CRLs | Cullin-RING Ligases |
| DHA | Docosahexaenoic Acid |
| DUBs | Deubiquitinating Enzymes |
| EPA | Eicosapentaenoic Acid |
| ER | Endoplasmic Reticulum |
| GWAS | Genome-Wide Association Studies |
| HATs | Histone Acetyltransferases |
| HBP | Hexosamine Biosynthesis Pathway |
| HDACs | Histone Deacetylases |
| HIF-1α | Hypoxia Inducible Factor 1 Alpha |
| IMiDs | Immunomodulatory Drugs |
| IRF4 | Interferon Regulatory Factor 4 |
| ISS | International Staging System |
| LLPS | Liquid-Liquid Phase Separation |
| LYSOTACs | Lysosome-Targeting Chimeras |
| mAbs | Monoclonal Antibodies |
| MDR1 | Multidrug Resistance Protein 1 |
| MM | Multiple Myeloma |
| mTOR | Mammalian Target of Rapamycin |
| NAE | NEDD8-Activating Enzyme |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| OGT | O-GlcNAc Transferase |
| OS | Overall Survival |
| PFS | Progression-Free Survival |
| PIs | Proteasome Inhibitors |
| PROTACs | Proteolysis-Targeting Chimeras |
| PTMs | Post Translational Modifications |
| RRMM | Relapsed Refractory Multiple Myeloma |
| SENPs | SUMO-Specific Proteases |
| SNAC | S-Nitroso-N-Acetylcysteine |
| SOCS1 | Suppressor of Cytokine Signaling 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| STs | Sialyltransferases |
| TKIs | Tyrosine Kinase Inhibitors |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| UCHs | Ubiquitin C-Terminal Hydrolases |
| UPR | Unfolded Protein Response |
| UPS | Ubiquitin-Proteasome System |
| USPs | Ubiquitin-Specific Proteases |
| Wnt | Wingless Integrated Signaling Pathway |
References
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Brigle, K.; Rogers, B. Pathobiology and Diagnosis of Multiple Myeloma. Semin. Oncol. Nurs. 2017, 33, 225–236. [Google Scholar] [CrossRef]
- Rodriguez-Otero, P.; Ailawadhi, S.; Arnulf, B.; Patel, K.; Cavo, M.; Nooka, A.K.; Manier, S.; Callander, N.; Costa, L.J.; Vij, R.; et al. Ide-cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2023, 388, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.A.; Mouhieddine, T.H.; Ortiz, R.J.; Richter, J. Revisiting the role of alkylating agents in multiple myeloma: Up-to-date evidence and future perspectives. Crit. Rev. Oncol. Hematol. 2023, 187, 104040. [Google Scholar] [CrossRef] [PubMed]
- Esma, F.; Salvini, M.; Troia, R.; Boccadoro, M.; Larocca, A.; Pautasso, C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert. Opin. Pharmacother. 2017, 18, 1127–1136. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Scott, K.; Hayden, P.J.; Will, A.; Wheatley, K.; Coyne, I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst. Rev. 2016, 4, Cd010816. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Raza, S.; Safyan, R.A.; Lentzsch, S. Immunomodulatory Drugs (IMiDs) in Multiple Myeloma. Curr. Cancer Drug Targets 2017, 17, 846–857. [Google Scholar] [CrossRef]
- Ricciuti, G.; Falcone, A.; Cascavilla, N.; Martinelli, G.; Cerchione, C. Autologous stem cell transplantation in multiple myeloma. Panminerva Med. 2020, 62, 220–224. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Viola, D.; Dona, A.; Caserta, E.; Troadec, E.; Besi, F.; McDonald, T.; Ghoda, L.; Gunes, E.G.; Sanchez, J.F.; Khalife, J.; et al. Daratumumab induces mechanisms of immune activation through CD38+ NK cell targeting. Leukemia 2021, 35, 189–200. [Google Scholar] [CrossRef]
- Cai, H.; Kakiuchi-Kiyota, S.; Hendricks, R.; Zhong, S.; Liu, L.; Adedeji, A.O.; Chan, P.; Schutten, M.M.; Kamath, A.V.; Ovacik, M.A. Nonclinical Pharmacokinetics, Pharmacodynamics, and Translational Model of RO7297089, A Novel Anti-BCMA/CD16A Bispecific Tetravalent Antibody for the Treatment of Multiple Myeloma. Aaps J. 2022, 24, 100. [Google Scholar] [CrossRef]
- Dunphy, K.; Dowling, P.; Bazou, D.; O’Gorman, P. Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers. Cancers 2021, 13, 1930. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Schick, M.; Keller, U.; Krönke, J. Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy. Cancers 2020, 12, 3764. [Google Scholar] [CrossRef]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Hammarén, H.M.; Savitski, M.M.; Baek, S.H. Control of protein stability by post-translational modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef]
- Pan, S.; Chen, R. Pathological implication of protein post-translational modifications in cancer. Mol. Asp. Med. 2022, 86, 101097. [Google Scholar] [CrossRef]
- Powley, I.R.; Hughes, M.A.; Cain, K.; MacFarlane, M. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing procaspase-8 maturation and cycling within the complex. Oncogene 2016, 35, 5629–5640. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef]
- Dikic, I.; Schulman, B.A. An expanded lexicon for the ubiquitin code. Nat. Rev. Mol. Cell Biol. 2023, 24, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T. Protein phosphatase 1--targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [CrossRef]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef]
- Pawson, T.; Scott, J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997, 278, 2075–2080. [Google Scholar] [CrossRef]
- Li, L.; Hu, X.; Nkwocha, J.; Sharma, K.; Kmieciak, M.; Mann, H.; Zhou, L.; Grant, S. Non-canonical role for the ataxia-telangiectasia-Rad3 pathway in STAT3 activation in human multiple myeloma cells. Cell Oncol 2023, 46, 1369–1380. [Google Scholar] [CrossRef]
- Li, S.; Fu, J.; Yang, J.; Ma, H.; Bhutani, D.; Mapara, M.Y.; Marcireau, C.; Lentzsch, S. Targeting the GCK pathway: A novel and selective therapeutic strategy against RAS-mutated multiple myeloma. Blood 2021, 137, 1754–1764. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Li, Q.; Gao, S.; Liu, S.; Ma, J.; Xie, Y.; Wang, J.; Cao, Z.; Liu, Z. Inositol Polyphosphate 4-Phosphatase Type II Is a Tumor Suppressor in Multiple Myeloma. Front. Oncol. 2021, 11, 785297. [Google Scholar] [CrossRef]
- Murphy, C.S.; DeMambro, V.E.; Fadel, S.; Fairfield, H.; Garter, C.A.; Rodriguez, P.; Qiang, Y.W.; Vary, C.P.H.; Reagan, M.R. Inhibition of Acyl-CoA Synthetase Long Chain Isozymes Decreases Multiple Myeloma Cell Proliferation and Causes Mitochondrial Dysfunction. bioRxiv 2024. [Google Scholar] [CrossRef]
- Jia, Y.; Yu, X.; Liu, R.; Shi, L.; Jin, H.; Yang, D.; Zhang, X.; Shen, Y.; Feng, Y.; Zhang, P.; et al. PRMT1 methylation of WTAP promotes multiple myeloma tumorigenesis by activating oxidative phosphorylation via m6A modification of NDUFS6. Cell Death Dis. 2023, 14, 512. [Google Scholar] [CrossRef]
- Yao, Y.; Park, W.D.; Morelli, E.; Samur, M.K.; Kwiatkowski, N.P.; Shirasaki, R.; Xu, Y.; Chakraborty, C.; Nabet, B.; Ches, M.; et al. Aberrant CDK7 Activity Drives the Cell Cycle and Transcriptional Dysregulation to Support Multiple Myeloma Growth: An Attractive Molecular Vulnerability. Blood 2021, 138, 2687. [Google Scholar] [CrossRef]
- Varland, S.; Osberg, C.; Arnesen, T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 2015, 15, 2385–2401. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, L.; Li, M.; Du, J.; Zhou, L.; Yang, S.; Zeng, L.; Li, Z.; Wang, G.; Wang, D. Functional analysis of the involvement of apurinic/apyrimidinic endonuclease 1 in the resistance to melphalan in multiple myeloma. BMC Cancer 2014, 14, 11. [Google Scholar] [CrossRef]
- Chen, Y.C.; Bhaskara, G.B.; Lu, Y.; Lin, K.; Dent, S.Y.R. The SAGA acetyltransferase module is required for the maintenance of MAF and MYC oncogenic gene expression programs in multiple myeloma. Genes. Dev. 2024, 38, 738–754. [Google Scholar] [CrossRef] [PubMed]
- Dupéré-Richer, D.; Riva, A.; Barwick, B.G.; Maji, S.; Casellas Román, H.; Li, J.; De, U.; Sobh, A.; Quickstad, G.; Piper, C.; et al. KDM6A regulates immune response genes in multiple myeloma. Blood 2024, 144, 1508–1520. [Google Scholar] [CrossRef]
- Ezponda, T.; Dupéré-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef]
- Gao, X.; Feng, Q.; Zhang, Q.; Zhang, Y.; Hu, C.; Zhang, L.; Zhang, H.; Wang, G.; Hu, K.; Ma, M.; et al. Targeting enolase 1 reverses bortezomib resistance in multiple myeloma through YWHAZ/Parkin axis. J. Biomed. Sci. 2025, 32, 9. [Google Scholar] [CrossRef]
- Gu, Z.; Xia, J.; Xu, H.; Frech, I.; Tricot, G.; Zhan, F. NEK2 Promotes Aerobic Glycolysis in Multiple Myeloma Through Regulating Splicing of Pyruvate Kinase. J. Hematol. Oncol. 2017, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.Y.; Stewart, A.K.; Sooknanan, R.R.; Henderson, G.; Hawley, T.S.; Reimold, A.M.; Glimcher, L.H.; Baumann, H.; Malek, L.T.; Hawley, R.G. Identification of c-myc promoter-binding protein and X-box binding protein 1 as interleukin-6 target genes in human multiple myeloma cells. Int. J. Oncol. 1999, 15, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Deng, Z.; Ding, P.; Qiang, W.; Lu, Y.; Gao, S.; Hu, Y.; Yang, Y.; Du, J.; Gu, C. A novel protein encoded by circHNRNPU promotes multiple myeloma progression by regulating the bone marrow microenvironment and alternative splicing. J. Exp. Clin. Cancer Res. 2022, 41, 85. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, X.L.; Wang, H.F.; Guo, T.; Yao, J.; Jiang, Z.S.; Pei, Q. The prognostic significance of ubiquitination-related genes in multiple myeloma by bioinformatics analysis. BMC Med. Genom. 2024, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Maimaitiyiming, Y.; Yang, T.; Wang, Q.Q.; Feng, Y.; Chen, Z.; Björklund, M.; Wang, F.; Hu, C.; Hsu, C.H.; Naranmandura, H. Heat Treatment Promotes Ubiquitin-Mediated Proteolysis of SARS-CoV-2 RNA Polymerase and Decreases Viral Load. Research 2022, 2022, 9802969. [Google Scholar] [CrossRef]
- Maimaitiyiming, Y.; Wang, Q.Q.; Yang, C.; Ogra, Y.; Lou, Y.; Smith, C.A.; Hussain, L.; Shao, Y.M.; Lin, J.; Liu, J.; et al. Hyperthermia Selectively Destabilizes Oncogenic Fusion Proteins. Blood Cancer Discov. 2021, 2, 388–401. [Google Scholar] [CrossRef]
- Gao, S.; Geng, C.; Song, T.; Lin, X.; Liu, J.; Cai, Z.; Cang, Y. Activation of c-Abl Kinase Potentiates the Anti-myeloma Drug Lenalidomide by Promoting DDA1 Protein Recruitment to the CRL4 Ubiquitin Ligase. J. Biol. Chem. 2017, 292, 3683–3691. [Google Scholar] [CrossRef]
- Wang, B.; Li, M.; Cao, D.; Sun, Q.; Yu, W.; Ma, J.; Ren, H.; Xu, G.; Zhou, L. Lys-63-specific deubiquitinase BRCC36 enhances the sensitivity of multiple myeloma cells to lenalidomide by inhibiting lysosomal degradation of cereblon. Cell Mol. Life Sci. 2024, 81, 349. [Google Scholar] [CrossRef]
- Jia, H.; Liu, C.; Ge, F.; Xiao, C.; Lu, C.; Wang, T.; He, Q. Identification of ubiquitinated proteins from human multiple myeloma U266 cells by proteomics. Biomed. Environ. Sci. 2011, 24, 422–430. [Google Scholar] [CrossRef]
- Li, C.; Xia, J.; Franqui-Machin, R.; Chen, F.; He, Y.; Ashby, T.C.; Teng, F.; Xu, H.; Liu, D.; Gai, D.; et al. TRIP13 modulates protein deubiquitination and accelerates tumor development and progression of B cell malignancies. J. Clin. Investig. 2021, 131, e146893. [Google Scholar] [CrossRef] [PubMed]
- Lub, S.; Maes, K.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Van Valckenborgh, E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 2016, 7, 6521–6537. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ozcan, U. Unfolded protein response signaling and metabolic diseases. J. Biol. Chem. 2014, 289, 1203–1211. [Google Scholar] [CrossRef]
- Kim, H.J.; Ha, S.; Lee, H.Y.; Lee, K.J. ROSics: Chemistry and proteomics of cysteine modifications in redox biology. Mass. Spectrom. Rev. 2015, 34, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Berardi, A.; Kaestner, C.L.; Ghitti, M.; Quilici, G.; Cocomazzi, P.; Li, J.; Ballabio, F.; Zucchelli, C.; Knapp, S.; Licht, J.D.; et al. The C-terminal PHDVC5HCH tandem domain of NSD2 is a combinatorial reader of unmodified H3K4 and tri-methylated H3K27 that regulates transcription of cell adhesion genes in multiple myeloma. Nucleic Acids Res. 2025, 53, gkae1121. [Google Scholar] [CrossRef] [PubMed]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef]
- de Krijger, I.; van der Torre, J.; Peuscher, M.H.; Eder, M.; Jacobs, J.J.L. H3K36 dimethylation by MMSET promotes classical non-homologous end-joining at unprotected telomeres. Oncogene 2020, 39, 4814–4827. [Google Scholar] [CrossRef]
- Wang, L.; Lin, N.; Li, Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol. Rep. 2019, 41, 1678–1690. [Google Scholar] [CrossRef]
- Niebudek, K.; Balcerczak, E.; Mirowski, M.; Pietrzak, J.; Zawadzka, I.; Żebrowska-Nawrocka, M. The contribution of ABCG2 G34A and C421A polymorphisms to multiple myeloma susceptibility. Onco Targets Ther. 2019, 12, 1655–1660. [Google Scholar] [CrossRef]
- Wang, M.M.; Zhu, Q.; Ren, Z.H.; Zou, L.F.; Dou, H.J.; Hu, J.P. Arsenic trioxide induces socs-1 gene demethylation in myeloma cell lines. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2008, 16, 1064–1068. [Google Scholar]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D., Jr.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qian, J.; Yang, Y.; Gu, C. Novel insights into the impact of the SUMOylation pathway in hematological malignancies (Review). Int. J. Oncol. 2021, 59, 73. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, L.; Jiang, D.; Wei, W.; Nasir, M.F.; Khan, M.S.; Yousafi, Q.; Liu, X.; Fu, X.; Li, X.; et al. Sumoylation as an Emerging Target in Therapeutics against Cancer. Curr. Pharm. Des. 2020, 26, 4764–4776. [Google Scholar] [CrossRef]
- Huang, H.J.; Zhou, L.L.; Fu, W.J.; Zhang, C.Y.; Jiang, H.; Du, J.; Hou, J. β-catenin SUMOylation is involved in the dysregulated proliferation of myeloma cells. Am. J. Cancer Res. 2015, 5, 309–320. [Google Scholar] [PubMed]
- Weger, S.; Hammer, E.; Heilbronn, R. SUMO-1 modification regulates the protein stability of the large regulatory protein Rep78 of adeno associated virus type 2 (AAV-2). Virology 2004, 330, 284–294. [Google Scholar] [CrossRef]
- Wu, S.; Yu, L. Targeting cullin-RING ligases for cancer treatment: Rationales, advances and therapeutic implications. Cytotechnology 2016, 68, 1–8. [Google Scholar] [CrossRef]
- Ying, J.; Zhang, M.; Qiu, X.; Lu, Y. Targeting the neddylation pathway in cells as a potential therapeutic approach for diseases. Cancer Chemother. Pharmacol. 2018, 81, 797–808. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr. Pharm. Des. 2013, 19, 3215–3225. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Dechowdhury, R. Therapeutically targeting the SUMOylation, Ubiquitination and Proteasome pathways as a novel anticancer strategy. Target. Oncol. 2010, 5, 281–289. [Google Scholar] [CrossRef]
- Petillo, S.; Capuano, C.; Molfetta, R.; Fionda, C.; Mekhloufi, A.; Pighi, C.; Antonangeli, F.; Zingoni, A.; Soriani, A.; Petrucci, M.T.; et al. Immunomodulatory effect of NEDD8-activating enzyme inhibition in Multiple Myeloma: Upregulation of NKG2D ligands and sensitization to Natural Killer cell recognition. Cell Death Dis. 2021, 12, 836. [Google Scholar] [CrossRef] [PubMed]
- Langerhorst, P.; Baerenfaenger, M.; Kulkarni, P.; Nadal, S.; Wijnands, C.; Post, M.A.; Noori, S.; vanDuijn, M.M.; Joosten, I.; Dejoie, T.; et al. N-linked glycosylation of the M-protein variable region: Glycoproteogenomics reveals a new layer of personalized complexity in multiple myeloma. Clin. Chem. Lab. Med. 2024, 62, 1626–1635. [Google Scholar] [CrossRef]
- Zhang, Z.; Westhrin, M.; Bondt, A.; Wuhrer, M.; Standal, T.; Holst, S. Serum protein N-glycosylation changes in multiple myeloma. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 960–970. [Google Scholar] [CrossRef]
- Renfrow, M.B.; Mackay, C.L.; Chalmers, M.J.; Julian, B.A.; Mestecky, J.; Kilian, M.; Poulsen, K.; Emmett, M.R.; Marshall, A.G.; Novak, J. Analysis of O-glycan heterogeneity in IgA1 myeloma proteins by Fourier transform ion cyclotron resonance mass spectrometry: Implications for IgA nephropathy. Anal. Bioanal. Chem. 2007, 389, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Smith, A.D.; Poulsen, K.; Kilian, M.; Julian, B.A.; Mestecky, J.; Novak, J.; Renfrow, M.B. Naturally occurring structural isomers in serum IgA1 o-glycosylation. J. Proteome Res. 2012, 11, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Mittermayr, S.; Lê, G.N.; Clarke, C.; Millán Martín, S.; Larkin, A.M.; O’Gorman, P.; Bones, J. Polyclonal Immunoglobulin G N-Glycosylation in the Pathogenesis of Plasma Cell Disorders. J. Proteome Res. 2017, 16, 748–762. [Google Scholar] [CrossRef]
- Westhrin, M.; Kovcic, V.; Zhang, Z.; Moen, S.H.; Nedal, T.M.V.; Bondt, A.; Holst, S.; Misund, K.; Buene, G.; Sundan, A.; et al. Monoclonal immunoglobulins promote bone loss in multiple myeloma. Blood 2020, 136, 2656–2666. [Google Scholar] [CrossRef]
- Lauc, G.; Huffman, J.E.; Pučić, M.; Zgaga, L.; Adamczyk, B.; Mužinić, A.; Novokmet, M.; Polašek, O.; Gornik, O.; Krištić, J.; et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 2013, 9, e1003225. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Dasoula, A.; Shah, R.; Syed, N.; Papoudou-Bai, A.; Coley, H.M.; Dranitsaris, G.; Bourantas, K.L.; Stebbing, J.; Crook, T. The prolyl-hydroxylase EGLN3 and not EGLN1 is inactivated by methylation in plasma cell neoplasia. Eur. J. Haematol. 2010, 84, 47–51. [Google Scholar] [CrossRef]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef]
- Kim, J.; Choi, S.; Saxena, N.; Singh, A.K.; Singh, I.; Won, J.S. Regulation of STAT3 and NF-κB activations by S-nitrosylation in multiple myeloma. Free Radic. Biol. Med. 2017, 106, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.S.; Fairfield, H.; DeMambro, V.E.; Fadel, S.; Gartner, C.A.; Karam, M.; Potts, C.; Rodriguez, P.; Qiang, Y.W.; Hamidi, H.; et al. Inhibition of acyl-CoA synthetase long-chain isozymes decreases multiple myeloma cell proliferation and causes mitochondrial dysfunction. Mol. Oncol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.W.; Ye, S.; Huang, Y.; Chen, Y.; Van Rhee, F.; Epstein, J.; Walker, B.A.; Morgan, G.J.; Davies, F.E. MAFb protein confers intrinsic resistance to proteasome inhibitors in multiple myeloma. BMC Cancer 2018, 18, 724. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Imai, Y.; Kaito, Y.; Murayama, T.; Sato, K.; Ishida, T.; Yamamoto, J.; Ito, T.; Futami, M.; Ri, M.; et al. Small-molecule HDAC and Akt inhibitors suppress tumor growth and enhance immunotherapy in multiple myeloma. J. Exp. Clin. Cancer Res. 2021, 40, 110. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Krönke, J.; Hurst, S.N.; Ebert, B.L. Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 2014, 3, e941742. [Google Scholar] [CrossRef]
- Gao, D.; Lv, A.E.; Li, H.P.; Han, D.H.; Zhang, Y.P. LncRNA MALAT-1 Elevates HMGB1 to Promote Autophagy Resulting in Inhibition of Tumor Cell Apoptosis in Multiple Myeloma. J. Cell Biochem. 2017, 118, 3341–3348. [Google Scholar] [CrossRef]
- Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Wissam Saab, M.; Tavazzi, B.; et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules 2020, 10, 696. [Google Scholar] [CrossRef]
- Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. [Google Scholar] [CrossRef]
- Du, L.; Liu, W.; Pichiorri, F.; Rosen, S.T. SUMOylation inhibition enhances multiple myeloma sensitivity to lenalidomide. Cancer Gene Ther. 2023, 30, 567–574. [Google Scholar] [CrossRef]
- Gu, Y.; Kaufman, J.L.; Bernal, L.; Torre, C.; Matulis, S.M.; Harvey, R.D.; Chen, J.; Sun, S.Y.; Boise, L.H.; Lonial, S. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 2014, 123, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.; Sarkar, S.; Natoni, A.; Stark, J.C.; Riley, N.M.; Bertozzi, C.R.; Carlsten, M.; O’Dwyer, M.E. Targeting hypersialylation in multiple myeloma represents a novel approach to enhance NK cell-mediated tumor responses. Blood Adv. 2022, 6, 3352–3366. [Google Scholar] [CrossRef] [PubMed]
- Natoni, A.; Macauley, M.S.; O’Dwyer, M.E. Targeting Selectins and Their Ligands in Cancer. Front. Oncol. 2016, 6, 93. [Google Scholar] [CrossRef]
- Natoni, A.; Smith, T.A.G.; Keane, N.; McEllistrim, C.; Connolly, C.; Jha, A.; Andrulis, M.; Ellert, E.; Raab, M.S.; Glavey, S.V.; et al. E-selectin ligands recognised by HECA452 induce drug resistance in myeloma, which is overcome by the E-selectin antagonist, GMI-1271. Leukemia 2017, 31, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, S.; Xie, Y.; Jiang, H.; Guo, J.; Wang, Y.; Peng, Z.; Hu, M.; Wang, M.; Wang, J.; et al. Deacetylation induced nuclear condensation of HP1γ promotes multiple myeloma drug resistance. Nat. Commun. 2023, 14, 1290. [Google Scholar] [CrossRef]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Narayanan, S.; Cai, C.Y.; Assaraf, Y.G.; Guo, H.Q.; Cui, Q.; Wei, L.; Huang, J.J.; Ashby, C.R., Jr.; Chen, Z.S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updat. 2020, 48, 100663. [Google Scholar] [CrossRef]
- Pigneux, A.; Mahon, F.X.; Moreau-Gaudry, F.; Uhalde, M.; de Verneuil, H.; Lacombe, F.; Reiffers, J.; Milpied, N.; Praloran, V.; Belloc, F. Proteasome inhibition specifically sensitizes leukemic cells to anthracyclin-induced apoptosis through the accumulation of Bim and Bax pro-apoptotic proteins. Cancer Biol. Ther. 2007, 6, 603–611. [Google Scholar] [CrossRef][Green Version]
- Gomez-Bougie, P.; Wuillème-Toumi, S.; Ménoret, E.; Trichet, V.; Robillard, N.; Philippe, M.; Bataille, R.; Amiot, M. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res. 2007, 67, 5418–5424. [Google Scholar] [CrossRef]
- Saj, F.; Nisha, Y.; Ganesan, P.; Kayal, S.; Kar, R.; Halanaik, D.; Dubashi, B. Efficacy and safety of pomalidomide, bortezomib, and dexamethasone combination chemotherapy for newly diagnosed multiple myeloma: POMACE Phase II Study. Blood Cancer J. 2023, 13, 45. [Google Scholar] [CrossRef]
- Jannuzzi, A.T.; Arslan, S.; Yilmaz, A.M.; Sari, G.; Beklen, H.; Méndez, L.; Fedorova, M.; Arga, K.Y.; Karademir Yilmaz, B.; Alpertunga, B. Higher proteotoxic stress rather than mitochondrial damage is involved in higher neurotoxicity of bortezomib compared to carfilzomib. Redox Biol. 2020, 32, 101502. [Google Scholar] [CrossRef] [PubMed]
- Karademir, B.; Sari, G.; Jannuzzi, A.T.; Musunuri, S.; Wicher, G.; Grune, T.; Mi, J.; Hacioglu-Bay, H.; Forsberg-Nilsson, K.; Bergquist, J.; et al. Proteomic approach for understanding milder neurotoxicity of Carfilzomib against Bortezomib. Sci. Rep. 2018, 8, 16318. [Google Scholar] [CrossRef] [PubMed]
- Haq, M.; Pellegrini, M.V.; Thumallapally, N. Ixazomib. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2025. [Google Scholar]
- Barankiewicz, J.; Salomon-Perzyński, A.; Misiewicz-Krzemińska, I.; Lech-Marańda, E. CRL4(CRBN) E3 Ligase Complex as a Therapeutic Target in Multiple Myeloma. Cancers 2022, 14, 4492. [Google Scholar] [CrossRef]
- Mark, T.M.; Coleman, M.; Niesvizky, R. Preclinical and clinical results with pomalidomide in the treatment of relapsed/refractory multiple myeloma. Leuk. Res. 2014, 38, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Schiller, G.J.; Samaras, C.; Sebag, M.; Berdeja, J.; Ganguly, S.; Matous, J.; Song, K.; Seet, C.S.; Talamo, G.; et al. Pomalidomide, dexamethasone, and daratumumab in relapsed refractory multiple myeloma after lenalidomide treatment. Leukemia 2020, 34, 3286–3297. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]
- Sivaraj, D.; Green, M.M.; Gasparetto, C. Panobinostat for the management of multiple myeloma. Future Oncol. 2017, 13, 477–488. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Laubach, J.P.; Richter, J.; Stricker, S.; Spencer, A.; Richardson, P.G.; Chari, A. Panobinostat From Bench to Bedside: Rethinking the Treatment Paradigm for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 752–765. [Google Scholar] [CrossRef]
- Richardson, P.G.; Harvey, R.D.; Laubach, J.P.; Moreau, P.; Lonial, S.; San-Miguel, J.F. Panobinostat for the treatment of relapsed or relapsed/refractory multiple myeloma: Pharmacology and clinical outcomes. Expert. Rev. Clin. Pharmacol. 2016, 9, 35–48. [Google Scholar] [CrossRef]
- Heynen, G.; Baumgartner, F.; Heider, M.; Patra, U.; Holz, M.; Braune, J.; Kaiser, M.; Schäffer, I.; Bamopoulos, S.A.; Ramberger, E.; et al. SUMOylation inhibition overcomes proteasome inhibitor resistance in multiple myeloma. Blood Adv. 2023, 7, 469–481. [Google Scholar] [CrossRef]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Fang, Y.; Wu, X.; Xu, T.; Hu, T.; Xu, Y.; Ma, P.; Wang, Q.; Shu, Y. The emerging roles of SUMOylation in the tumor microenvironment and therapeutic implications. Exp. Hematol. Oncol. 2023, 12, 58. [Google Scholar] [CrossRef]
- van Andel, H.; Kocemba, K.A.; Spaargaren, M.; Pals, S.T. Aberrant Wnt signaling in multiple myeloma: Molecular mechanisms and targeting options. Leukemia 2019, 33, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Rosenberg, E.; Dimenshtein-Voevoda, V.; Sirovsky, Y.; Vladimirsky, J.; Magen, H.; Ostrovsky, O.; Shimoni, A.; Bromberg, Z.; Weiss, L.; et al. Blocking of Transient Receptor Potential Vanilloid 1 (TRPV1) promotes terminal mitophagy in multiple myeloma, disturbing calcium homeostasis and targeting ubiquitin pathway and bortezomib-induced unfolded protein response. J. Hematol. Oncol. 2020, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Petillo, S.; Sproviero, E.; Loconte, L.; Cuollo, L.; Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Cerboni, C.; Petrucci, M.T.; et al. NEDD8-activating enzyme inhibition potentiates the anti-myeloma activity of natural killer cells. Cell Death Dis. 2023, 14, 438. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 91. [Google Scholar] [CrossRef]
- Li, S.; Bode, A.M.; Zhu, F.; Liu, K.; Zhang, J.; Kim, M.O.; Reddy, K.; Zykova, T.; Ma, W.Y.; Carper, A.L.; et al. TRPV1-antagonist AMG9810 promotes mouse skin tumorigenesis through EGFR/Akt signaling. Carcinogenesis 2011, 32, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.; Kumar, S. PI3K/AKT/mTOR pathway in multiple myeloma: From basic biology to clinical promise. Leuk. Lymphoma 2018, 59, 2524–2534. [Google Scholar] [CrossRef]
- Vega, M.; Chen, Y.; Shi, Y.; Gera, J.; Lichtenstein, A. Turnover of the mTOR inhibitor, DEPTOR, and downstream AKT phosphorylation in multiple myeloma cells, is dependent on ERK1-mediated phosphorylation. J. Biol. Chem. 2022, 298, 101750. [Google Scholar] [CrossRef]
- Jiang, F.; Tang, X.; Tang, C.; Hua, Z.; Ke, M.; Wang, C.; Zhao, J.; Gao, S.; Jurczyszyn, A.; Janz, S.; et al. HNRNPA2B1 promotes multiple myeloma progression by increasing AKT3 expression via m6A-dependent stabilization of ILF3 mRNA. J. Hematol. Oncol. 2021, 14, 54. [Google Scholar] [CrossRef]
- Isa, R.; Horinaka, M.; Tsukamoto, T.; Mizuhara, K.; Fujibayashi, Y.; Taminishi-Katsuragawa, Y.; Okamoto, H.; Yasuda, S.; Kawaji-Kanayama, Y.; Matsumura-Kimoto, Y.; et al. The Rationale for the Dual-Targeting Therapy for RSK2 and AKT in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 2919. [Google Scholar] [CrossRef]
- Damiano, J.S.; Hazlehurst, L.A.; Dalton, W.S. Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 2001, 15, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Furukawa, Y. Toll-like receptor CD180 and the bone marrow microenvironment as therapeutic targets in multiple myeloma. Rinsho Ketsueki 2020, 61, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Garssen, J.; Redegeld, F. The efficacy of bortezomib in human multiple myeloma cells is enhanced by combination with omega-3 fatty acids DHA and EPA: Timing is essential. Clin. Nutr. 2021, 40, 1942–1953. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, S.; Bennett, S.; Tang, H.; Song, D.; Wood, D.; Zhan, X.; Xu, J. STAT3 and its targeting inhibitors in osteosarcoma. Cell Prolif. 2021, 54, e12974. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, W.; Han, Y.; Oyang, L.; Lin, J.; Shen, M.; Wang, J.; et al. Altered glycosylation in cancer: Molecular functions and therapeutic potential. Cancer Commun. 2024, 44, 1316–1336. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Poulose, N.; Steele, R.E.; Martin, S.E.S.; Levine, Z.G.; Duveau, D.Y.; Carelli, R.; Singh, R.; Urbanucci, A.; Loda, M.; et al. Inhibition of O-GlcNAc Transferase Renders Prostate Cancer Cells Dependent on CDK9. Mol. Cancer Res. 2020, 18, 1512–1521. [Google Scholar] [CrossRef]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-κB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef]
- Natoni, A.; Farrell, M.L.; Harris, S.; Falank, C.; Kirkham-McCarthy, L.; Macauley, M.S.; Reagan, M.R.; O’Dwyer, M. Sialyltransferase inhibition leads to inhibition of tumor cell interactions with E-selectin, VCAM1, and MADCAM1, and improves survival in a human multiple myeloma mouse model. Haematologica 2020, 105, 457–467. [Google Scholar] [CrossRef]
- Natoni, A.; Bohara, R.; Pandit, A.; O’Dwyer, M. Targeted Approaches to Inhibit Sialylation of Multiple Myeloma in the Bone Marrow Microenvironment. Front. Bioeng. Biotechnol. 2019, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- Glavey, S.V.; Manier, S.; Natoni, A.; Sacco, A.; Moschetta, M.; Reagan, M.R.; Murillo, L.S.; Sahin, I.; Wu, P.; Mishima, Y.; et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 2014, 124, 1765–1776. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Le, A.Q.; Liu, E.; Cesarano, A.; DiMeo, F.; Perna, F.; Kapur, R.; Walker, B.A.; Tran, N.T. Protein arginine methyltransferase 1 is a therapeutic vulnerability in multiple myeloma. Front. Immunol. 2023, 14, 1239614. [Google Scholar] [CrossRef]
- Mashimo, K.; Tsubaki, M.; Takeda, T.; Asano, R.; Jinushi, M.; Imano, M.; Satou, T.; Sakaguchi, K.; Nishida, S. RANKL-induced c-Src activation contributes to conventional anti-cancer drug resistance and dasatinib overcomes this resistance in RANK-expressing multiple myeloma cells. Clin. Exp. Med. 2019, 19, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, S.; Veloso, A.; Yan, C.; Oksa, L.; Alpert, E.J.; Do, D.; Hyvärinen, N.; McCarthy, K.; Adhikari, A.; Yang, Q.; et al. Therapeutic targeting of LCK tyrosine kinase and mTOR signaling in T-cell acute lymphoblastic leukemia. Blood 2022, 140, 1891–1906. [Google Scholar] [CrossRef]
- Frassanito, M.A.; De Veirman, K.; Desantis, V.; Di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I.; et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016, 30, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, L.; Szücs, K.D.; Kudla, M.; Heidenreich, J.; Jendrossek, V.; Peña-Llopis, S.; Garay, T.; Czirok, A.; Aigner, C.; Plönes, T.; et al. Nintedanib and Dasatinib Treatments Induce Protective Autophagy as a Potential Resistance Mechanism in MPM Cells. Front. Cell Dev. Biol. 2022, 10, 852812. [Google Scholar] [CrossRef]
- He, Y.; Fang, Y.; Zhang, M.; Zhao, Y.; Tu, B.; Shi, M.; Muhitdinov, B.; Asrorov, A.; Xu, Q.; Huang, Y. Remodeling “cold” tumor immune microenvironment via epigenetic-based therapy using targeted liposomes with in situ formed albumin corona. Acta Pharm. Sin. B 2022, 12, 2057–2073. [Google Scholar] [CrossRef]
- El Omari, N.; Bakrim, S.; Khalid, A.; Abdalla, A.N.; Almalki, W.H.; Lee, L.H.; Ardianto, C.; Ming, L.C.; Bouyahya, A. Molecular mechanisms underlying the clinical efficacy of panobinostat involve Stochasticity of epigenetic signaling, sensitization to anticancer drugs, and induction of cellular cell death related to cellular stresses. Biomed. Pharmacother. 2023, 164, 114886. [Google Scholar] [CrossRef]
- Renzini, A.; D’Onghia, M.; Coletti, D.; Moresi, V. Histone Deacetylases as Modulators of the Crosstalk Between Skeletal Muscle and Other Organs. Front. Physiol. 2022, 13, 706003. [Google Scholar] [CrossRef]
- Bajbouj, K.; Al-Ali, A.; Ramakrishnan, R.K.; Saber-Ayad, M.; Hamid, Q. Histone Modification in NSCLC: Molecular Mechanisms and Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 1701. [Google Scholar] [CrossRef] [PubMed]
- Parveen, R.; Harihar, D.; Chatterji, B.P. Recent histone deacetylase inhibitors in cancer therapy. Cancer 2023, 129, 3372–3380. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.R.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.; Breider, M.; et al. Rate of CRL4(CRBN) substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef] [PubMed]
- Di Bacco, A.; Bahlis, N.J.; Munshi, N.C.; Avet-Loiseau, H.; Masszi, T.; Viterbo, L.; Pour, L.; Ganly, P.; Cavo, M.; Langer, C.; et al. c-MYC expression and maturity phenotypes are associated with outcome benefit from addition of ixazomib to lenalidomide-dexamethasone in myeloma. Eur. J. Haematol. 2020, 105, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Nijhof, I.S.; Couto, S.; Levin, M.D.; Bos, G.M.J.; Broijl, A.; Klein, S.K.; Ren, Y.; Wang, M.; Koene, H.R.; et al. Cereblon loss and up-regulation of c-Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematologica 2018, 103, e368–e371. [Google Scholar] [CrossRef]
- McMillin, D.W.; Jacobs, H.M.; Delmore, J.E.; Buon, L.; Hunter, Z.R.; Monrose, V.; Yu, J.; Smith, P.G.; Richardson, P.G.; Anderson, K.C.; et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol. Cancer Ther. 2012, 11, 942–951. [Google Scholar] [CrossRef]
- Xia, M.; Wang, S.; Qi, Y.; Long, K.; Li, E.; He, L.; Pan, F.; Guo, Z.; Hu, Z. Inhibition of O-GlcNAc transferase sensitizes prostate cancer cells to docetaxel. Front. Oncol. 2022, 12, 993243. [Google Scholar] [CrossRef]
- Liu, J.; Song, T.; Zhou, W.; Xing, L.; Wang, S.; Ho, M.; Peng, Z.; Tai, Y.T.; Hideshima, T.; Anderson, K.C.; et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia 2019, 33, 171–180. [Google Scholar] [CrossRef]
- Bohl, S.R.; Schmalbrock, L.K.; Bauhuf, I.; Meyer, T.; Dolnik, A.; Szyska, M.; Blätte, T.J.; Knödler, S.; Röhner, L.; Miller, D.; et al. Comprehensive CRISPR-Cas9 screens identify genetic determinants of drug responsiveness in multiple myeloma. Blood Adv. 2021, 5, 2391–2402. [Google Scholar] [CrossRef]
- Escrivá-Fernández, J.; Cueto-Ureña, C.; Solana-Orts, A.; Lledó, E.; Ballester-Lurbe, B.; Poch, E. A CRISPR interference strategy for gene expression silencing in multiple myeloma cell lines. J. Biol. Eng. 2023, 17, 34. [Google Scholar] [CrossRef]
- Grillone, K.; Ascrizzi, S.; Cremaschi, P.; Amato, J.; Polerà, N.; Croci, O.; Rocca, R.; Riillo, C.; Conforti, F.; Graziano, R.; et al. An unbiased lncRNA dropout CRISPR-Cas9 screen reveals RP11-350G8.5 as a novel therapeutic target for multiple myeloma. Blood 2024, 144, 1705–1721. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Zhang, C.; Jiang, Y.; Liu, H.; Huang, H.; Guo, D. Therapeutic status and the prospect of CRISPR/Cas9 gene editing in multiple myeloma. Future Oncol. 2020, 16, 1125–1136. [Google Scholar] [CrossRef]
- Agathanggelou, A.; Smith, E.; Davies, N.J.; Kwok, M.; Zlatanou, A.; Oldreive, C.E.; Mao, J.; Da Costa, D.; Yadollahi, S.; Perry, T.; et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood 2017, 130, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, M.; Jiang, J.; Wan, Y.; Li, X.; Zhang, M.; Xiao, F.; Zhong, L.; Zhong, H.; Qin, Z.; et al. Targeting deubiquitinase USP7-mediated stabilization of XPO1 contributes to the anti-myeloma effects of selinexor. J. Transl. Med. 2025, 23, 62. [Google Scholar] [CrossRef]
- Baumann, V.; Lorenzer, C.; Thell, M.; Winkler, A.M.; Winkler, J. RNAi-Mediated Knockdown of Protein Expression. Methods Mol. Biol. 2017, 1654, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Dey, P.; De, A. Recent advancements in targeted protein knockdown technologies-emerging paradigms for targeted therapy. Explor. Target. Antitumor Ther. 2023, 4, 1227–1248. [Google Scholar] [CrossRef] [PubMed]
- Marschall, A.L.; Dübel, S.; Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs 2015, 7, 1010–1035. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Pang, Z.; Guo, F.; Qin, Q.; Yin, T.; Sang, Y.; Feng, C.; Li, X.; Jiang, L.; et al. Knockdown of SUMO-activating enzyme subunit 2 (SAE2) suppresses cancer malignancy and enhances chemotherapy sensitivity in small cell lung cancer. J. Hematol. Oncol. 2015, 8, 67. [Google Scholar] [CrossRef]
- Aristodemou, A.E.N.; Rueda, D.S.; Taylor, G.P.; Bangham, C.R.M. The transcriptome of HTLV-1-infected primary cells following reactivation reveals changes to host gene expression central to the proviral life cycle. PLoS Pathog. 2023, 19, e1011494. [Google Scholar] [CrossRef]
- Jia, Y.; Zhao, J.; Yu, T.; Zhang, X.; Qi, X.; Hao, T.; Jin, Z.; Zhao, X. PSMC3 promotes RNAi by maintaining AGO2 stability through USP14. Cell Mol. Biol. Lett. 2022, 27, 111. [Google Scholar] [CrossRef]
- Deng, J.; Liao, S.; Chen, C.; Han, F.; Lei, S.; Lai, X.; Ye, K.; Han, Q.; E, F.; Lu, C.; et al. Specific intracellular retention of circSKA3 promotes colorectal cancer metastasis by attenuating ubiquitination and degradation of SLUG. Cell Death Dis. 2023, 14, 750. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; He, X.Y.; Xiao, C.M.; Lin, Q.; Wang, M.Y.; Liu, C.Y.; Kong, L.Y.; Chen, Z.; Xia, Y.Z. Circular RNA-encoded oncogenic PIAS1 variant blocks immunogenic ferroptosis by modulating the balance between SUMOylation and phosphorylation of STAT1. Mol. Cancer 2024, 23, 207. [Google Scholar] [CrossRef]
- Ishida, T.; Ciulli, A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov. 2021, 26, 484–502. [Google Scholar] [CrossRef]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef]
- Ma, L.; Wang, J.; Zhang, Y.; Fang, F.; Ling, J.; Chu, X.; Zhang, Z.; Tao, Y.; Li, X.; Tian, Y.; et al. BRD4 PROTAC degrader MZ1 exerts anticancer effects in acute myeloid leukemia by targeting c-Myc and ANP32B genes. Cancer Biol. Ther. 2022, 23, 1–15. [Google Scholar] [CrossRef]
- Fiskus, W.; Mill, C.P.; Perera, D.; Birdwell, C.; Deng, Q.; Yang, H.; Lara, B.H.; Jain, N.; Burger, J.; Ferrajoli, A.; et al. BET proteolysis targeted chimera-based therapy of novel models of Richter Transformation-diffuse large B-cell lymphoma. Leukemia 2021, 35, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rong, Q.; Ye, L.; Fang, B.; Zhao, Y.; Sun, Y.; Zhou, H.; Wang, D.; He, J.; Cui, Z.; et al. Discovery of a Novel Orally Bioavailable FLT3-PROTAC Degrader for Efficient Treatment of Acute Myeloid Leukemia and Overcoming Resistance of FLT3 Inhibitors. J. Med. Chem. 2024, 67, 7197–7223. [Google Scholar] [CrossRef]
- Ahn, G.; Riley, N.M.; Kamber, R.A.; Wisnovsky, S.; Moncayo von Hase, S.; Bassik, M.C.; Banik, S.M.; Bertozzi, C.R. Elucidating the cellular determinants of targeted membrane protein degradation by lysosome-targeting chimeras. Science 2023, 382, eadf6249. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Su, L.Y.; Tian, Y.; Zheng, Q.; Cao, Y.; Yao, M.; Wang, S.; Xu, W.; Xi, C.; Clocchiatti, A.; Nie, G.; et al. Anti-tumor immunotherapy using engineered bacterial outer membrane vesicles fused to lysosome-targeting chimeras mediated by transferrin receptor. Cell Chem. Biol. 2024, 31, 1219–1230.e1215. [Google Scholar] [CrossRef]
- Malarvannan, M.; Unnikrishnan, S.; Monohar, S.; Ravichandiran, V.; Paul, D. Design and optimization strategies of PROTACs and its Application, Comparisons to other targeted protein degradation for multiple oncology therapies. Bioorg Chem. 2025, 154, 107984. [Google Scholar] [CrossRef]
- Song, J.; Hu, M.; Zhou, J.; Xie, S.; Li, T.; Li, Y. Targeted protein degradation in drug development: Recent advances and future challenges. Eur. J. Med. Chem. 2023, 261, 115839. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Q.; Xiao, H.T.; Yang, F.; Wang, Y.D.; Li, P.; Zheng, Z.G. Advancing targeted protein degradation for metabolic diseases therapy. Pharmacol. Res. 2023, 188, 106627. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cui, J.; Chen, H.; Yu, B.; Long, S. Recent progress in degradation of membrane proteins by PROTACs and alternative targeted protein degradation techniques. Eur. J. Med. Chem. 2023, 262, 115911. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Zhou, B.; Zhang, B.; Ren, H.; Zhu, L.; Zheng, G.; Ge, M.; Ge, J. Recent advances in the development of EGFR degraders: PROTACs and LYTACs. Eur. J. Med. Chem. 2022, 239, 114533. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Guenther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: Outcomes by prior treatment. Blood 2016, 127, 713–721. [Google Scholar] [CrossRef]
- Kotani, H.; Oshima, H.; Boucher, J.C.; Yamano, T.; Sakaguchi, H.; Sato, S.; Fukuda, K.; Nishiyama, A.; Yamashita, K.; Ohtsubo, K.; et al. Dual inhibition of SUMOylation and MEK conquers MYC-expressing KRAS-mutant cancers by accumulating DNA damage. J. Biomed. Sci. 2024, 31, 68. [Google Scholar] [CrossRef]
- Charliński, G.; Vesole, D.H.; Jurczyszyn, A. Rapid Progress in the Use of Immunomodulatory Drugs and Cereblon E3 Ligase Modulators in the Treatment of Multiple Myeloma. Cancers 2021, 13, 4666. [Google Scholar] [CrossRef]
- Hosen, N. Identifying and targeting multiple myeloma-specific antigens resulting from post-translational protein modifications by CAR-T cell therapies. Rinsho Ketsueki 2023, 64, 427–431. [Google Scholar] [CrossRef]
- CAR T-cell Efficacy in Solid Tumors Is Affected by N-glycosylation. Cancer Discov. 2022, 12, 598. [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef]
- Xie, Z.; Bi, C.; Chooi, J.Y.; Chan, Z.L.; Mustafa, N.; Chng, W.J. MMSET regulates expression of IRF4 in t(4;14) myeloma and its silencing potentiates the effect of bortezomib. Leukemia 2015, 29, 2347–2354. [Google Scholar] [CrossRef]
- Dolloff, N.G.; Talamo, G. Targeted therapy of multiple myeloma. Adv. Exp. Med. Biol. 2013, 779, 197–221. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Guenther, A.; Na Nakorn, T.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone: Impact of dose intensity and administration frequency on safety in the PANORAMA 1 trial. Br. J. Haematol. 2017, 179, 66–74. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Gkotzamanidou, M.; Terpou, E.; Kentepozidis, N.; Terpos, E. Targeting the Interplay between HDACs and DNA Damage Repair for Myeloma Therapy. Int. J. Mol. Sci. 2021, 22, 406. [Google Scholar] [CrossRef]
- Guo, J.; Chai, X.; Mei, Y.; Du, J.; Du, H.; Shi, H.; Zhu, J.K.; Zhang, H. Acetylproteomics analyses reveal critical features of lysine-ε-acetylation in Arabidopsis and a role of 14-3-3 protein acetylation in alkaline response. Stress. Biol. 2022, 2, 1. [Google Scholar] [CrossRef]
- Ji, Y.; Wu, Z.; Dai, Z.; Sun, K.; Wang, J.; Wu, G. Nutritional epigenetics with a focus on amino acids: Implications for the development and treatment of metabolic syndrome. J. Nutr. Biochem. 2016, 27, 1–8. [Google Scholar] [CrossRef]
- Auner, H.W.; Cenci, S. Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br. J. Haematol. 2015, 168, 14–25. [Google Scholar] [CrossRef]
- Casado, P.; Alcolea, M.P.; Iorio, F.; Rodríguez-Prados, J.C.; Vanhaesebroeck, B.; Saez-Rodriguez, J.; Joel, S.; Cutillas, P.R. Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 2013, 14, R37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lin, T.C.; Bi, X.; Lu, G.; Dawson, B.C.; Miranda, R.; Medeiros, L.J.; McNiece, I.; McCarty, N. TRIM44 promotes quiescent multiple myeloma cell occupancy and survival in the osteoblastic niche via HIF-1α stabilization. Leukemia 2019, 33, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Roy, M.; Liang, L.; Cao, W.; Hu, B.; Li, Y.; Xiao, X.; Wang, H.; Ye, M.; Sun, S.; et al. Deubiquitylase USP12 induces pro-survival autophagy and bortezomib resistance in multiple myeloma by stabilizing HMGB1. Oncogene 2022, 41, 1298–1308. [Google Scholar] [CrossRef]
- Nikesitch, N.; Ling, S.C. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97–101. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Bloem, A.C.; van der Spek, E.; Lokhorst, H.M. New treatment strategies for multiple myeloma by targeting BCL-2 and the mevalonate pathway. Curr. Pharm. Des. 2006, 12, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Tao, S.; Bi, L.; Zhang, Z.; Zhang, X. Proteomics: Addressing the challenges of multiple myeloma. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 89–95. [Google Scholar] [CrossRef]
- Kratka, K.; Sistik, P.; Olivkova, I.; Kusnierova, P.; Svagera, Z.; Stejskal, D. Mass Spectrometry-Based Proteomics in Clinical Diagnosis of Amyloidosis and Multiple Myeloma: A Review (2012–2024). J. Mass. Spectrom. 2025, 60, e5116. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).