
Precision Targeting in Metastatic Prostate Cancer: Molecular Insights to Therapeutic Frontiers



Abstract
1. Introduction
2. Molecular Pathophysiology of Metastatic Prostate Cancer
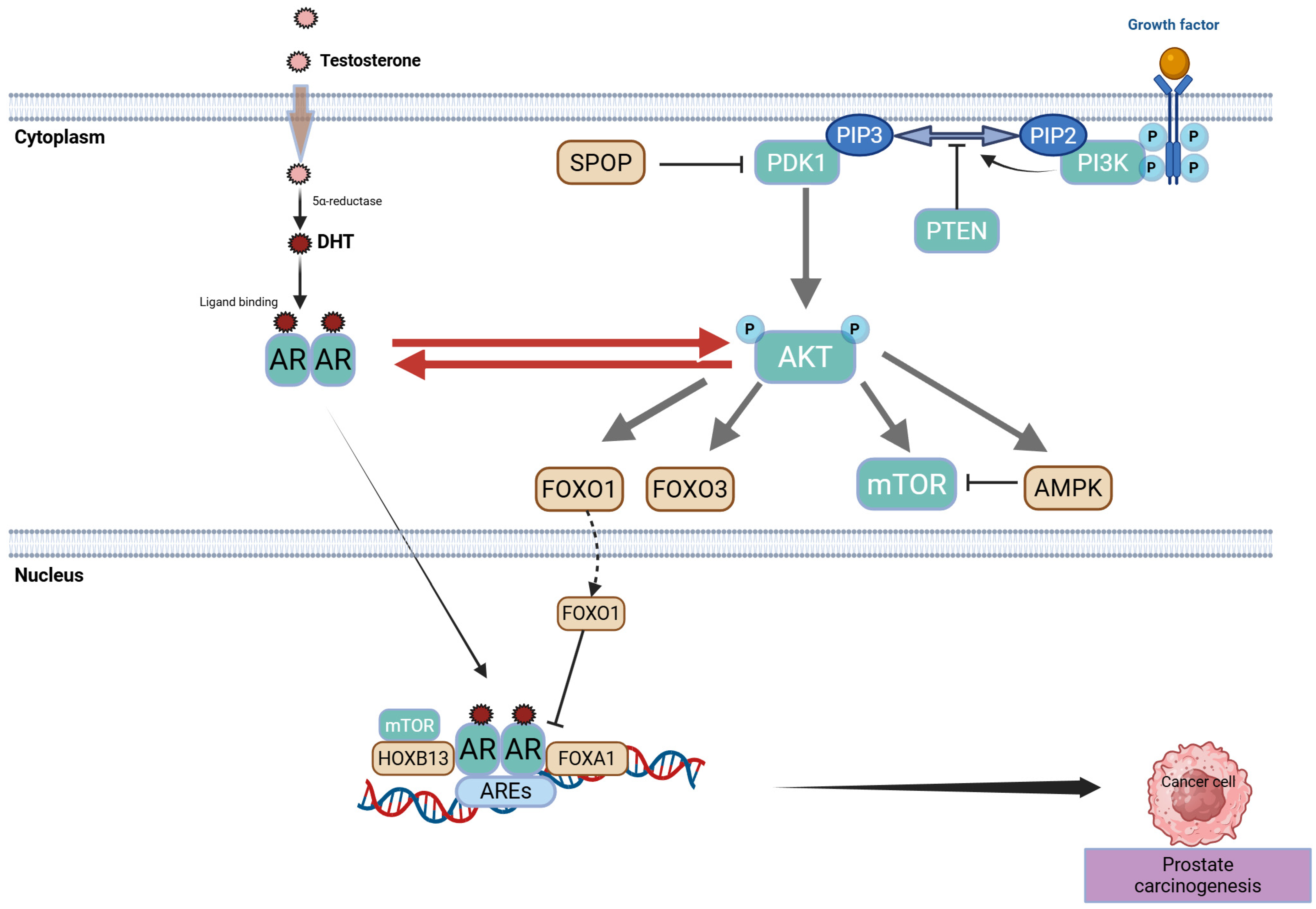
2.1. Centrality of the AR Axis
2.1.1. Historical Underpinnings and Core AR Functions
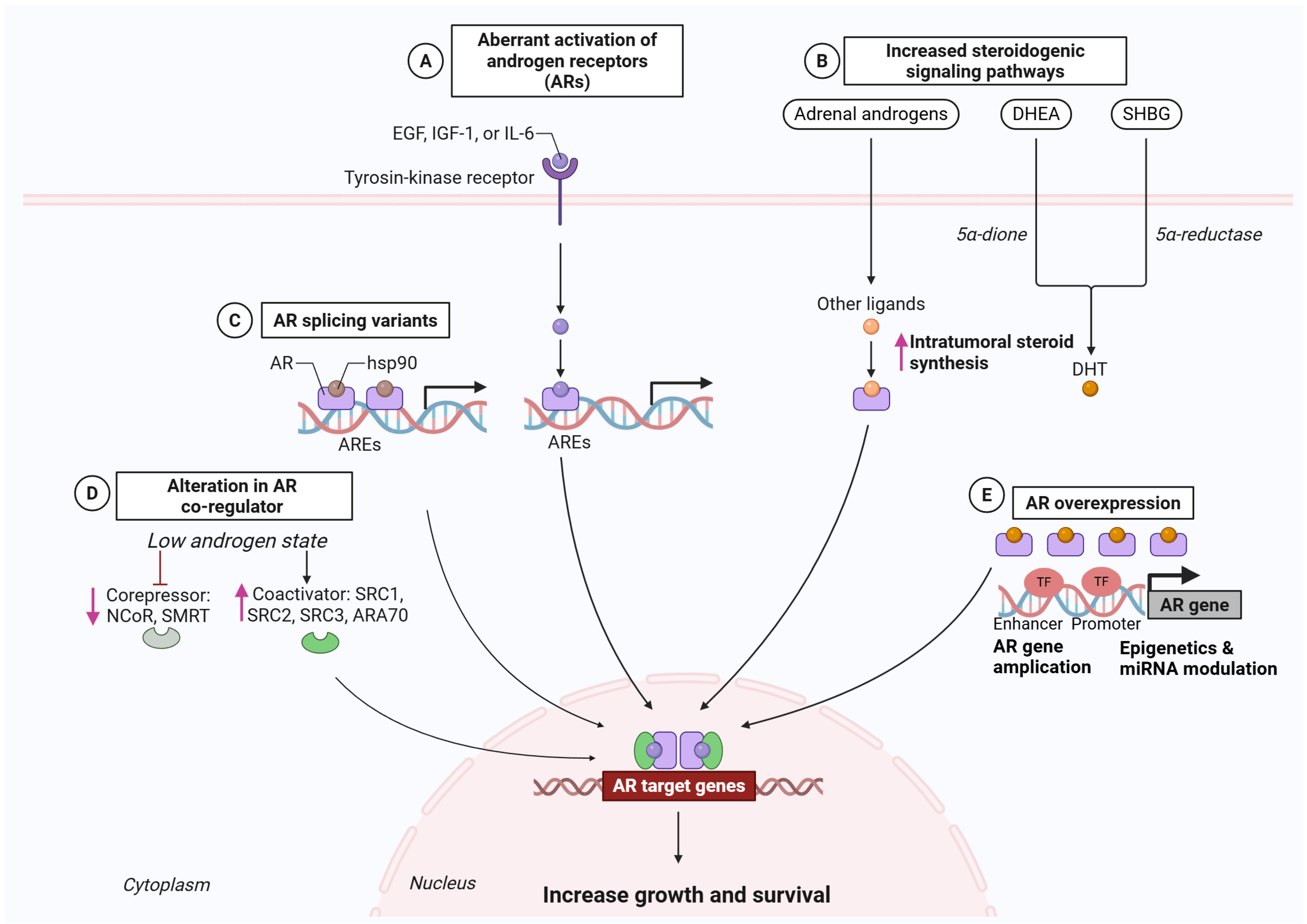
2.1.2. Routes to AR Reactivation Under Therapeutic Pressure
2.2. PARP Inhibitors and the Synthetic Lethality Paradigm
2.2.1. Historical Context and Foundational Insights
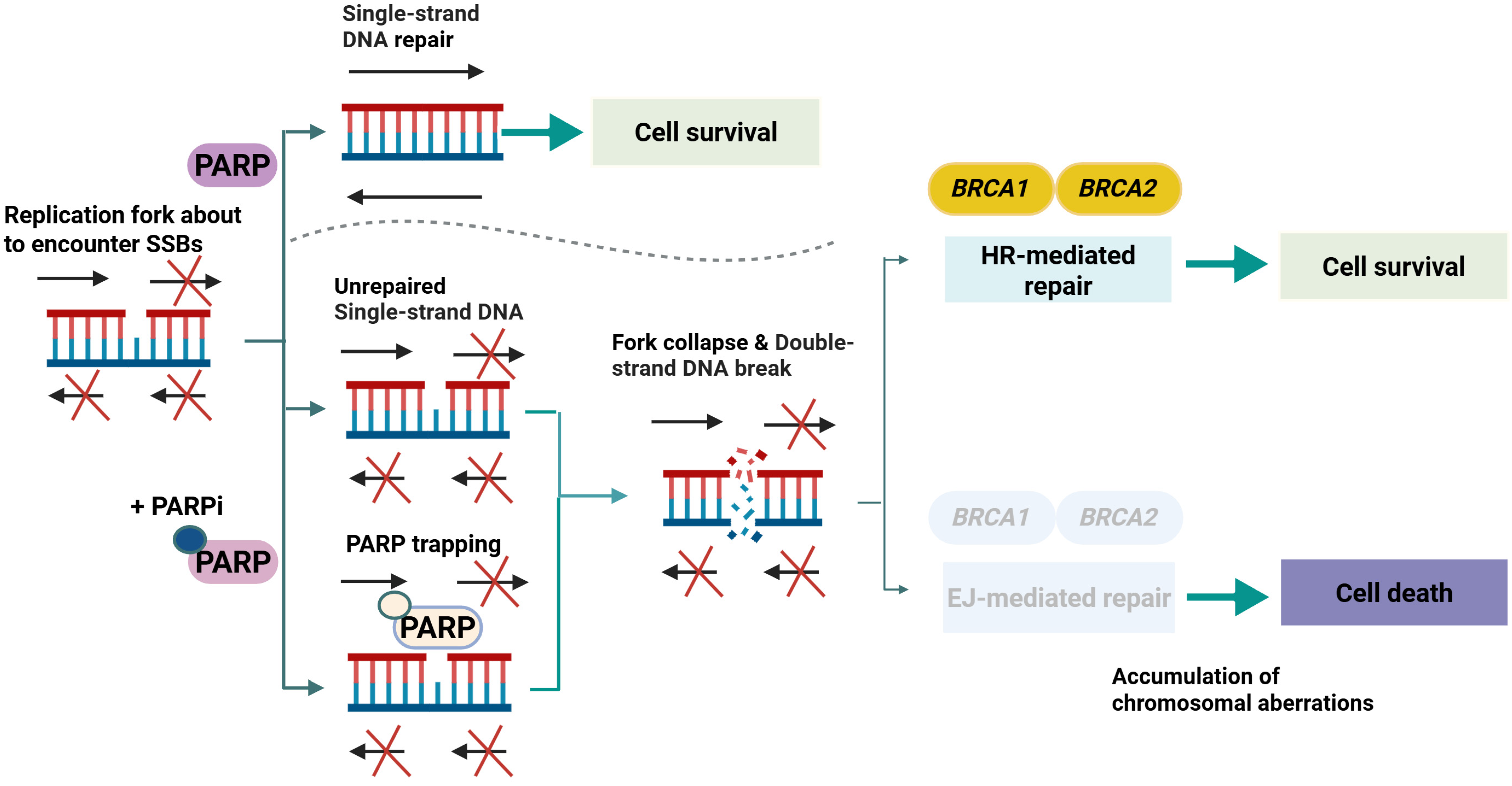
2.2.2. Core Molecular Mechanisms in Prostate Cancer
2.2.3. Adaptive and Resistance Mechanisms
2.2.4. Research Methodologies and Knowledge Gaps
2.2.5. Forward-Looking Perspectives in Molecular Pathophysiology
2.3. TMPRSS2–ERG Fusions and Oncogenic Transcription Factors (Molecular Pathophysiology Focus)
2.3.1. Historical to Current Understanding
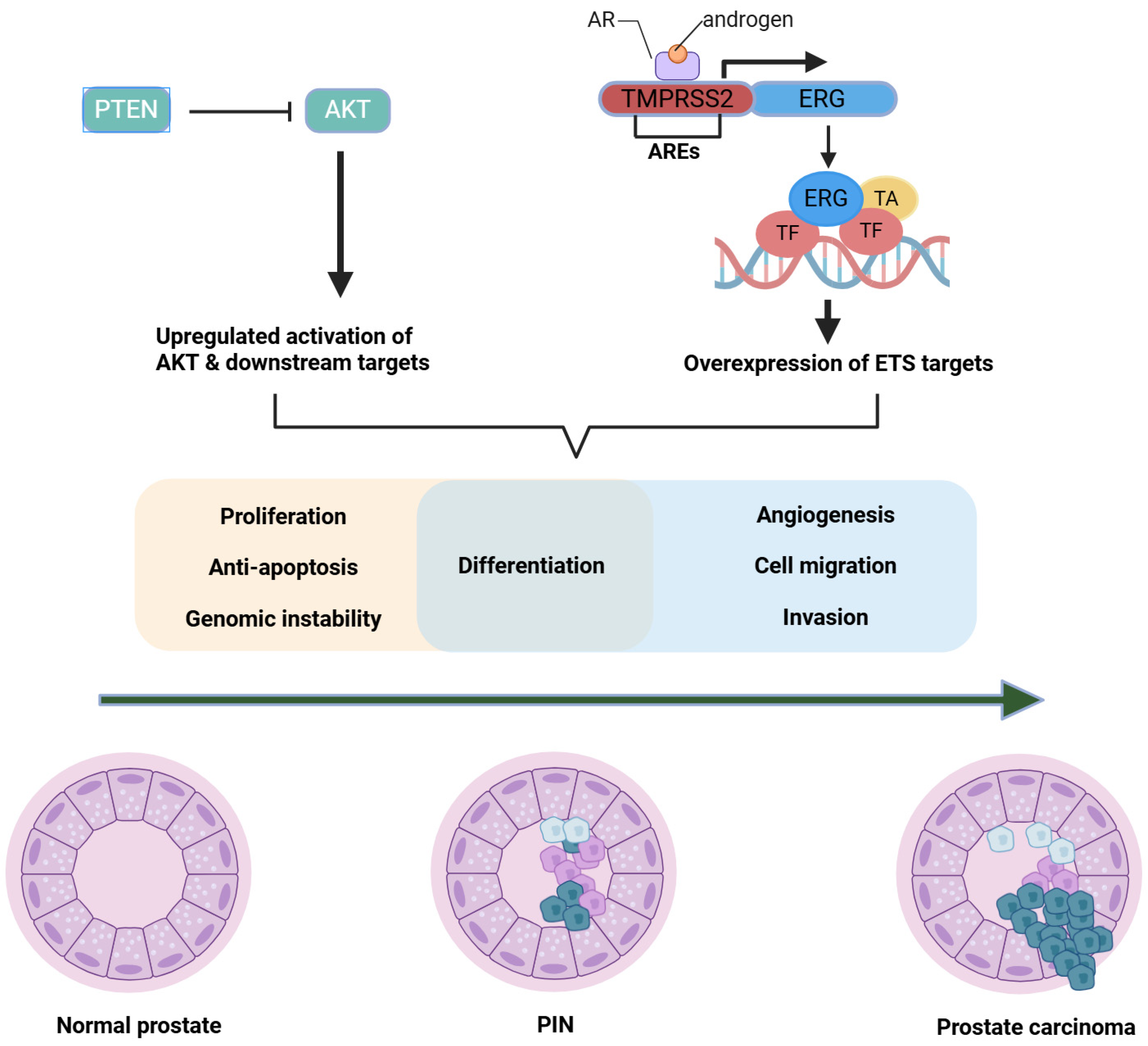
2.3.2. Mechanistic Insights
2.3.3. Methodological Limitations
2.3.4. Clinical or Scientific Significance
2.3.5. Comparisons, Divergent Findings, and Future Outlook
2.4. PTEN Loss, PI3K/AKT/mTOR Hyperactivation, and Crosstalk with AR
2.4.1. Historical to Current Perspective
2.4.2. Mechanistic Insights
2.4.3. Methodological Constraints
2.4.4. Clinical or Scientific Significance
2.4.5. Contrasting Evidence and Future Directions
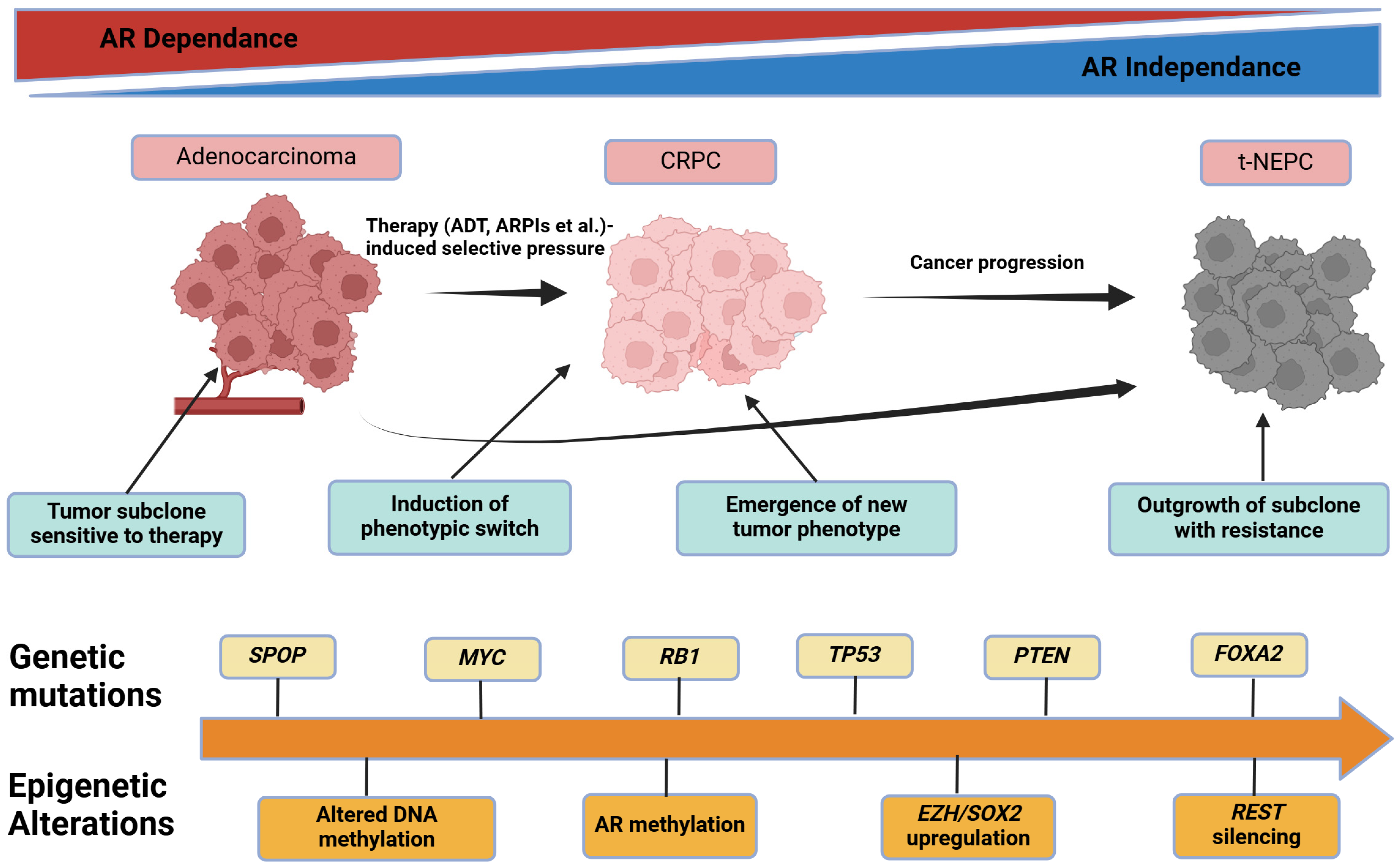
2.5. Tumor Heterogeneity, Clonal Evolution, and Lineage Plasticity
2.5.1. Historical to Current Understanding
2.5.2. Mechanistic Insights into Clonal Dynamics
2.5.3. Methodological Constraints
2.5.4. Clinical or Scientific Significance
2.5.5. Comparisons and Divergent Data
2.5.6. Future Outlook
2.6. The Tumor Microenvironment and Immune Dynamics
2.6.1. Historical to Current Understanding
2.6.2. Mechanistic Insights
2.6.3. Methodological Constraints
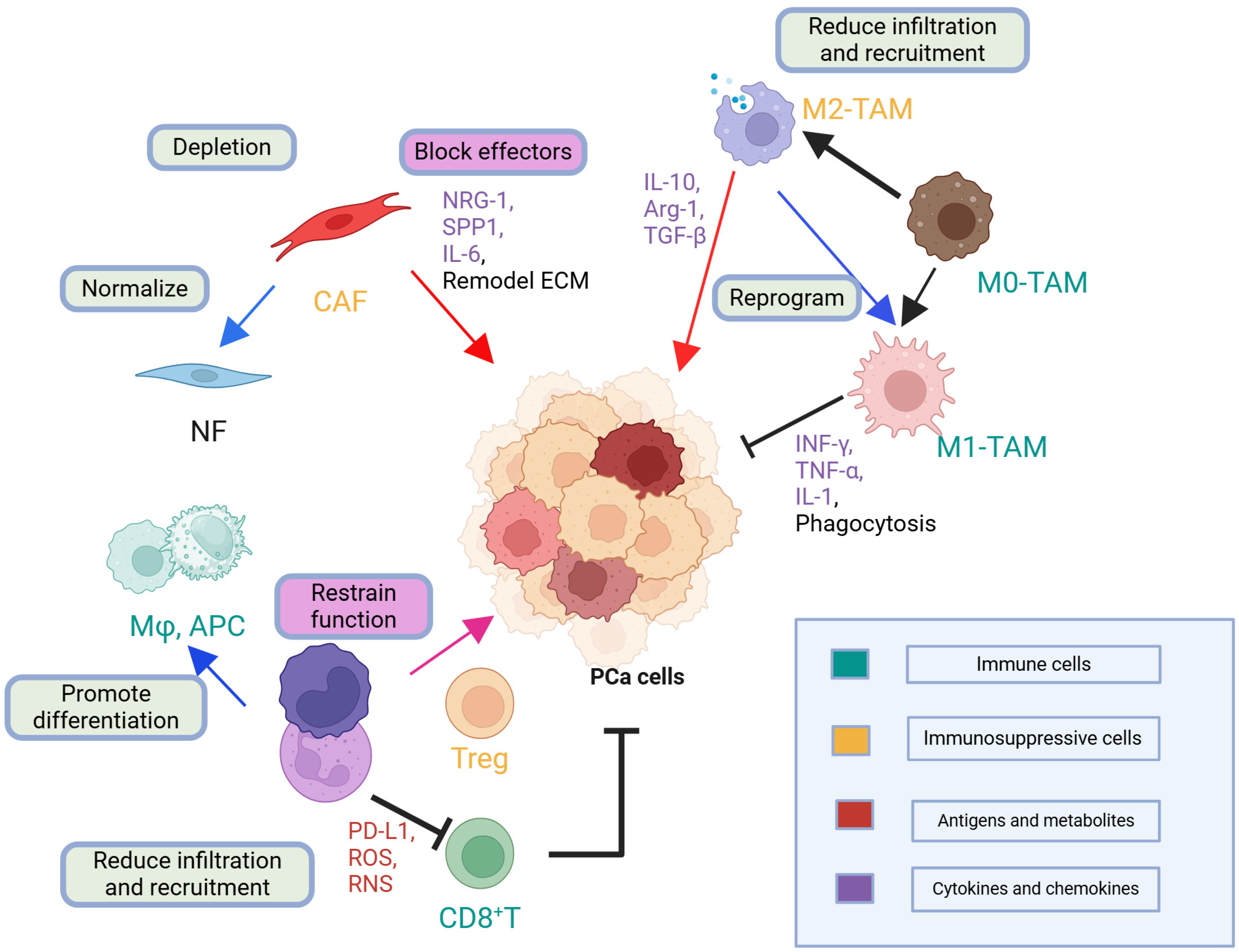
2.6.4. Clinical or Scientific Significance
2.6.5. Comparisons and Divergent Data
2.6.6. Future Outlook

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TME Component | Key Secreted Factors/Regulatory Signals | Main Function/Immune Evasion Mechanism | Clinical Implications |
|---|---|---|---|
| TAMs | IL-10, TGF-β, PD-L1 | Suppress cytotoxic T-cell responses, promote angiogenesis and tumor growth | Elevated TAMs infiltration correlates with poor prognosis; potential therapeutic target for immunomodulation |
| MDSCs | Arginase, ROS, NO, IL-10 | Inhibit T-cell function and antigen presentation, reinforce immunosuppression | High MDSCs levels linked to rapid disease progression; blocking MDSC recruitment or function may enhance immunotherapy outcomes |
| Tregs | IL-10, TGF-β | Suppress effector T-cell activity through cytokine-mediated and cell-contact mechanisms | Increased Tregs infiltration is associated with worse prognosis; Treg depletion or functional blockade may boost response to immunotherapy |
| CAFs | TGF-β, FGF, CXCL12, extracellular matrix components | Provide structural support, secrete growth factors that promote tumor invasion, and create immunosuppressive niches | FAP has emerged as a potential target; CAFs remodeling could improve T-cell infiltration in solid tumors |
| Bone Metastasis Niche (Osteoblasts/Osteoclasts) | RANKL, OPG, TGF-β | Remodel bone microenvironment to favor tumor cell growth, contribute to immune evasion through altered cytokine milieu | Central to skeletal metastases in prostate cancer; targeted RANKL inhibition (e.g., denosumab) or other bone-directed therapies may impair tumor progression |
| Endothelial Cells & Angiogenic Factors | VEGF, FGF, PDGF | Induce neovascularization that sustains tumor growth and metastasis | Anti-angiogenic strategies may enhance immunotherapy by reprogramming the tumor vasculature and improving immune cell infiltration |
3. Molecular Stratification and Diagnostic Advances
3.1. High-Resolution Molecular Profiling
3.2. Companion Diagnostics and Gene Panels
3.2.1. DDR-Focused Panels for PARP Inhibitor Selection
3.2.2. AR Variant Detection and PTEN/PI3K Panels
3.2.3. Performance Metrics of Key Molecular Biomarkers
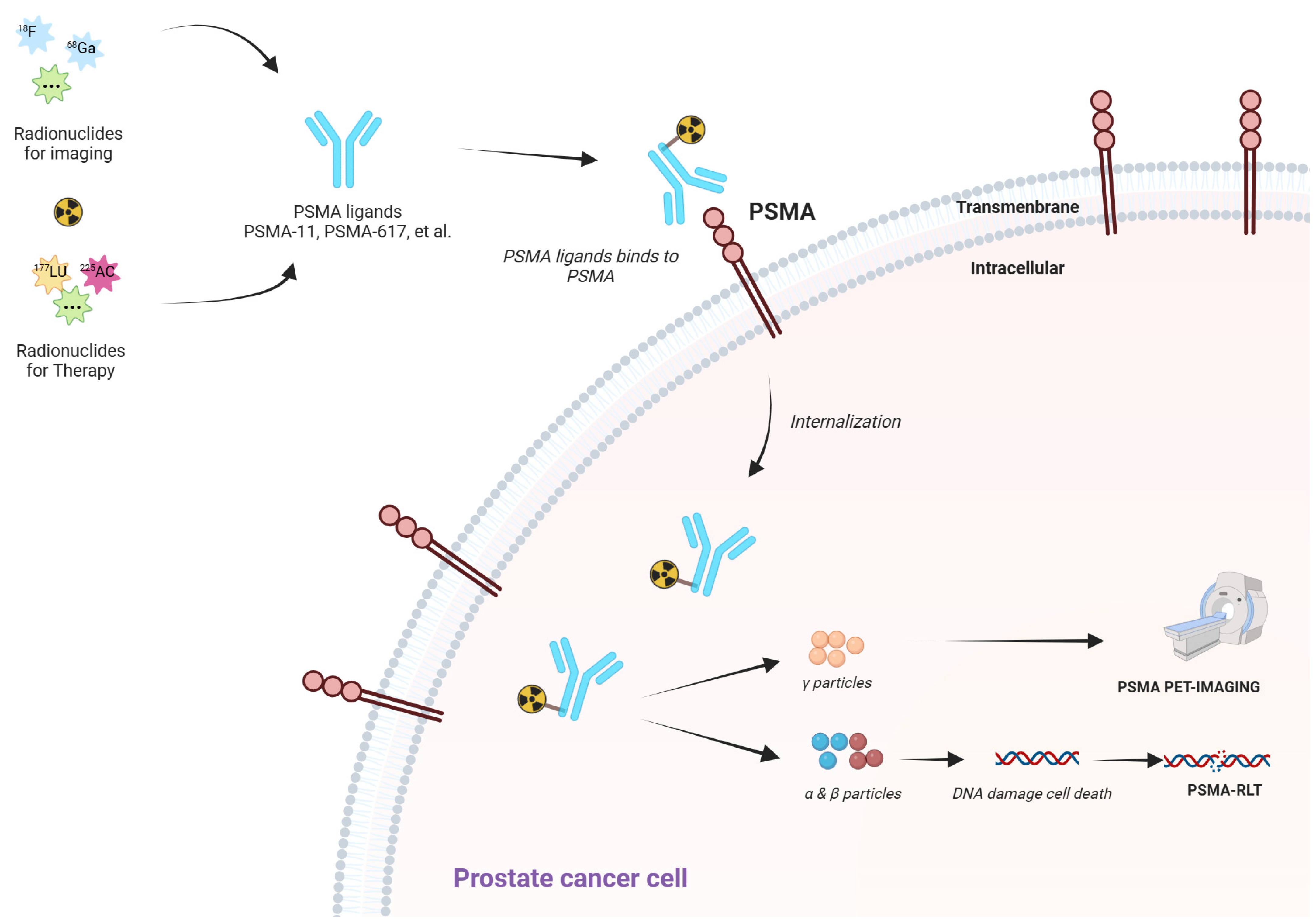
3.3. Advanced Imaging: PSMA PET-CT and Beyond
3.4. Liquid Biopsies in Metastatic Prostate Cancer
3.4.1. Historical Context and Technological Evolution
3.4.2. Methodological Approaches and Clinical Relevance
3.4.3. Critical Assessment and Significance
3.4.4. Methodological Challenges
3.4.5. Future Outlook
4. Targeted Therapeutic Approaches
4.1. AR Axis–Centric Treatments
4.2. DDR-Defect-Based Therapies: PARP Inhibitors and Beyond
4.3. Targeting PI3K/AKT/mTOR and WNT Pathways
4.4. Immuno-Oncology Approaches
4.5. PSMA-Targeted Radioligand Therapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| ARE | androgen response element |
| CAR-T | chimeric antigen receptor T |
| cfDNA | cell-free DNA |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CTC | circulating tumor cell |
| ctDNA | circulating tumor DNA |
| DBD | DNA-binding domain |
| DDR | DNA damage repair |
| DHT | testosterone–dihydrotestosterone |
| DSB | double-strand break |
| EMT | epithelial–mesenchymal transition |
| FAP | fibroblast activation protein |
| HR | homologous recombination |
| LBD | ligand-binding domain |
| mCRPC | metastatic castration-resistant prostate cancer |
| MDSC | myeloid-derived suppressor cell |
| mPCa | metastatic prostate cancer |
| NGS | next-generation sequencing |
| NTD | N-terminal transactivation domain |
| PARP | poly-ADP ribose polymerase |
| PCa | prostate cancer |
| PIP3 | phosphatidylinositol (3,4,5)-trisphosphate |
| PROTAC | proteolysis-targeting chimera |
| PSA | prostate-specific antigen |
| PSMA | prostate-specific membrane antigen |
| TAM | tumor-associated macrophage |
| t-NEPC | treatment-emergent neuroendocrine prostate cancer/treatment-induced neuroendocrine prostate cancer |
| TME | tumor microenvironment |
| Treg | regulatory T cell |
| WES/WGS | whole-exome/genome sequencing |
References
- Bergengren, O.; Pekala, K.R.; Matsoukas, K.; Fainberg, J.; Mungovan, S.F.; Bratt, O.; Bray, F.; Brawley, O.; Luckenbaugh, A.N.; Mucci, L.; et al. 2022 Update on Prostate Cancer Epidemiology and Risk Factors-A Systematic Review. Eur. Urol. 2023, 84, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wang, X.; Zhu, C.; Wang, K. The global status of research in prostate cancer bone metastasis: A bibliometric and visualized analysis. Front. Med. 2022, 9, 931422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lyu, F.; Gao, X. Advances in ferroptosis for castration-resistant prostate cancer treatment: Novel drug targets and combination therapy strategies. Prostate Cancer Prostatic Dis. 2024. [Google Scholar] [CrossRef]
- Rawat, C.; Heemers, H.V. Alternative splicing in prostate cancer progression and therapeutic resistance. Oncogene 2024, 43, 1655–1668. [Google Scholar] [CrossRef] [PubMed]
- Vitorino, R. Transforming Clinical Research: The Power of High-Throughput Omics Integration. Proteomes 2024, 12, 25. [Google Scholar] [CrossRef]
- Singhal, U.; Nallandhighal, S.; Tosoian, J.J.; Hu, K.; Pham, T.M.; Stangl-Kremser, J.; Liu, C.-J.; Karim, R.; Plouffe, K.R.; Morgan, T.M.; et al. Integrative multi-region molecular profiling of primary prostate cancer in men with synchronous lymph node metastasis. Nat. Commun. 2024, 15, 4341. [Google Scholar] [CrossRef]
- Taylor, A.K.; Kosoff, D.; Emamekhoo, H.; Lang, J.M.; Kyriakopoulos, C.E. PARP inhibitors in metastatic prostate cancer. Front. Oncol. 2023, 13, 1159557. [Google Scholar] [CrossRef]
- Kwon, W.-A. PARP Inhibitors in the Treatment of Prostate Cancer: From Scientific Rationale to Clinical Development. World J. Mens. Health 2024, 42, 290–303. [Google Scholar] [CrossRef]
- Seed, G.; Beije, N.; Yuan, W.; Bertan, C.; Goodall, J.; Lundberg, A.; Tyler, M.; Figueiredo, I.; Pereira, R.; Baker, C.; et al. Elucidating acquired PARP inhibitor resistance in advanced prostate cancer. Cancer Cell 2024, 42, 2113–2123.e2114. [Google Scholar] [CrossRef]
- Srivastava, T.P.; Ajmeriya, S.; Goel, I.; Talukdar, J.; Srivastava, A.; Parshad, R.; Deo, S.V.S.; Mathur, S.R.; Gogia, A.; Rai, A.; et al. Prognostic role of Androgen Receptor splice variant 7 (AR-V7) in the pathogenesis of breast cancer. BMC Cancer 2024, 24, 1398. [Google Scholar] [CrossRef]
- Mena, E.; Lindenberg, L.; Choyke, P. The Impact of PSMA PET/CT Imaging in Prostate Cancer Radiation Treatment. Semin. Nucl. Med. 2022, 52, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, J.; Zeng, H. Our Current Understanding of the Heterogeneity in Prostate Cancer and Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 1526. [Google Scholar] [CrossRef] [PubMed]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1426. [Google Scholar] [CrossRef]
- Woodcock, D.J.; Riabchenko, E.; Taavitsainen, S.; Kankainen, M.; Gundem, G.; Brewer, D.S.; Ellonen, P.; Lepistö, M.; Golubeva, Y.A.; Warner, A.C.; et al. Prostate cancer evolution from multilineage primary to single lineage metastases with implications for liquid biopsy. Nat. Commun. 2020, 11, 5070. [Google Scholar] [CrossRef]
- Bruni, S.; Mercogliano, M.F.; Mauro, F.L.; Cordo Russo, R.I.; Schillaci, R. Cancer immune exclusion: Breaking the barricade for a successful immunotherapy. Front. Oncol. 2023, 13, 1135456. [Google Scholar] [CrossRef]
- Tiwari, A.; Oravecz, T.; Dillon, L.A.; Italiano, A.; Audoly, L.; Fridman, W.H.; Clifton, G.T. Towards a consensus definition of immune exclusion in cancer. Front. Immunol. 2023, 14, 1084887. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, M.; Wu, L.; Yang, H.; Yao, Y.; Yang, Q.; Du, J.; Liu, L.; Li, Y.; Bai, Y. Stromal cells in the tumor microenvironment: Accomplices of tumor progression? Cell Death Dis. 2023, 14, 587. [Google Scholar] [CrossRef]
- Clifton, G.T.; Rothenberg, M.; Ascierto, P.A.; Begley, G.; Cecchini, M.; Eder, J.P.; Ghiringhelli, F.; Italiano, A.; Kochetkova, M.; Li, R.; et al. Developing a definition of immune exclusion in cancer: Results of a modified Delphi workshop. J. Immunother. Cancer 2023, 11, e006773. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef]
- Kaizer, A.M.; Belli, H.M.; Ma, Z.; Nicklawsky, A.G.; Roberts, S.C.; Wild, J.; Wogu, A.F.; Xiao, M.; Sabo, R.T. Recent innovations in adaptive trial designs: A review of design opportunities in translational research. J. Clin. Transl. Sci. 2023, 7, e125. [Google Scholar] [CrossRef]
- Duan, X.-P.; Qin, B.-D.; Jiao, X.-D.; Liu, K.; Wang, Z.; Zang, Y.-S. New clinical trial design in precision medicine: Discovery, development and direction. Signal Transduct. Target. Ther. 2024, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A.; Moore, K.N.; Colombo, N.; Ray-Coquard, I.; Pignata, S. The forefront of ovarian cancer therapy: Update on PARP inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, V.; Morrison, G.J.; Falcone, M.; Quinn, D.I.; Goldkorn, A. Integration of Liquid Biopsies in Clinical Management of Metastatic Prostate Cancer. Curr. Oncol. Rep. 2022, 24, 1287–1298. [Google Scholar] [CrossRef]
- Trujillo, B.; Wu, A.; Wetterskog, D.; Attard, G. Blood-based liquid biopsies for prostate cancer: Clinical opportunities and challenges. Br. J. Cancer 2022, 127, 1394–1402. [Google Scholar] [CrossRef]
- Benadada, F.; Saad, F.; Delouya, G.; Taussky, D. Charles Brenton Huggins: A historical review of the Nobel laureate’s pioneering discoveries. Cancer 2024, 130, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, W.; Xiao, Y.-T.; Huang, H.; Gu, D.; Ren, S. Targeting signaling pathways in prostate cancer: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 198. [Google Scholar] [CrossRef]
- Jacob, A.; Raj, R.; Allison, D.B.; Myint, Z.W. Androgen Receptor Signaling in Prostate Cancer and Therapeutic Strategies. Cancers 2021, 13, 5417. [Google Scholar] [CrossRef]
- Ge, Q.; Li, J.; Yang, F.; Tian, X.; Zhang, M.; Hao, Z.; Liang, C.; Meng, J. Molecular classifications of prostate cancer: Basis for individualized risk stratification and precision therapy. Ann. Med. 2023, 55, 2279235. [Google Scholar] [CrossRef]
- Li, C.; Cheng, D.; Li, P. Androgen receptor dynamics in prostate cancer: From disease progression to treatment resistance. Front. Oncol. 2025, 15, 1542811. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Han, D.; Labaf, M.; Zhao, Y.; Owiredu, J.; Zhang, S.; Patel, K.; Venkataramani, K.; Steinfeld, J.S.; Han, W.; Li, M.; et al. Androgen receptor splice variants drive castration-resistant prostate cancer metastasis by activating distinct transcriptional programs. J. Clin. Investig. 2024, 134, e168649. [Google Scholar] [CrossRef] [PubMed]
- Moigne, R.L.; Mawji, N.R.; Banuelos, C.A.; Wang, J.; Jian, K.; Andersen, R.J.; Sadar, M.D.; Zhou, H.-J.; Virsik, P. Abstract 1292: A new generation of N-terminal domain androgen receptor inhibitors, with improved pharmaceutical properties, in castration-resistant prostate cancer models. Cancer Res. 2019, 79, 1292. [Google Scholar] [CrossRef]
- Hung, C.L.; Liu, H.H.; Fu, C.W.; Yeh, H.H.; Hu, T.L.; Kuo, Z.K.; Lin, Y.C.; Jhang, M.R.; Hwang, C.S.; Hsu, H.C.; et al. Targeting androgen receptor and the variants by an orally bioavailable Proteolysis Targeting Chimeras compound in castration resistant prostate cancer. EBioMedicine 2023, 90, 104500. [Google Scholar] [CrossRef] [PubMed]
- Gokbayrak, B.; Altintas, U.B.; Lingadahalli, S.; Morova, T.; Huang, C.-C.F.; Ersoy Fazlioglu, B.; Pak Lok Yu, I.; Kalkan, B.M.; Cejas, P.; Kung, S.H.Y.; et al. Identification of selective SWI/SNF dependencies in enzalutamide-resistant prostate cancer. Commun. Biol. 2025, 8, 169. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, H.; Hao, M. Interactions between key genes and pathways in prostate cancer progression and therapy resistance. Front. Oncol. 2025, 15, 1467540. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, C.; Zhang, Y.; Shi, C. Application of Organoid Models in Prostate Cancer Research. Front. Oncol. 2021, 11, 736431. [Google Scholar] [CrossRef]
- Fang, S.; Zhe, S.; Lin, H.-M.; Azad, A.A.; Fettke, H.; Kwan, E.M.; Horvath, L.; Mak, B.; Zheng, T.; Du, P.; et al. Multi-Omic Integration of Blood-Based Tumor-Associated Genomic and Lipidomic Profiles Using Machine Learning Models in Metastatic Prostate Cancer. JCO Clin. Cancer Inform. 2023, 7, e2300057. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Catalano, M.; Generali, D.; Gatti, M.; Riboli, B.; Paganini, L.; Nesi, G.; Roviello, G. DNA repair deficiency as circulating biomarker in prostate cancer. Front. Oncol. 2023, 13, 1115241. [Google Scholar] [CrossRef]
- Gong, X.; Liu, C.; Tang, H.; Wu, S.; Yang, Q. Application and research progress of synthetic lethality in the development of anticancer therapeutic drugs. Front. Oncol. 2024, 14, 1460412. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Barnicle, A.; Adelman, C.A.; Armenia, J.; Kang, J.; Barrett, J.C.; Harrington, E.A. Impact of DNA damage repair alterations on prostate cancer progression and metastasis. Front. Oncol. 2023, 13, 1162644. [Google Scholar] [CrossRef] [PubMed]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)—Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Castro, E.; Aragón, I.M.; Cendón, Y.; Cattrini, C.; López-Casas, P.P.; Olmos, D. Genetic aberrations in DNA repair pathways: A cornerstone of precision oncology in prostate cancer. Br. J. Cancer 2021, 124, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, D.; Haro-Silerio, J.I.; Yap, T.A.; Ngoi, N. PARP inhibitors: Enhancing efficacy through rational combinations. Br. J. Cancer 2023, 129, 904–916. [Google Scholar] [CrossRef]
- Herberts, C.; Wyatt, A.W.; Nguyen, P.L.; Cheng, H.H. Genetic and Genomic Testing for Prostate Cancer: Beyond DNA Repair. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390384. [Google Scholar] [CrossRef]
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis. 2020, 23, 24–37. [Google Scholar] [CrossRef]
- Qian, J.; Liao, G.; Chen, M.; Peng, R.-W.; Yan, X.; Du, J.; Huang, R.; Pan, M.; Lin, Y.; Gong, X.; et al. Advancing cancer therapy: New frontiers in targeting DNA damage response. Front. Pharmacol. 2024, 15, 1474337. [Google Scholar] [CrossRef]
- Walmsley, C.S.; Jonsson, P.; Cheng, M.L.; McBride, S.; Kaeser, C.; Vargas, H.A.; Laudone, V.; Taylor, B.S.; Kappagantula, R.; Baez, P.; et al. Convergent evolution of BRCA2 reversion mutations under therapeutic pressure by PARP inhibition and platinum chemotherapy. npj Precis. Oncol. 2024, 8, 34. [Google Scholar] [CrossRef]
- St John, J.; Powell, K.; Conley-Lacomb, M.K.; Chinni, S.R. TMPRSS2–ERG Fusion Gene Expression in Prostate Tumor Cells and Its Clinical and Biological Significance in Prostate Cancer Progression. J. Cancer Sci. Ther. 2012, 4, 94–101. [Google Scholar] [CrossRef]
- Lorenzin, F.; Demichelis, F. Past, Current, and Future Strategies to Target ERG Fusion-Positive Prostate Cancer. Cancers 2022, 14, 1118. [Google Scholar] [CrossRef]
- Scaravilli, M.; Koivukoski, S.; Latonen, L. Androgen-Driven Fusion Genes and Chimeric Transcripts in Prostate Cancer. Front. Cell Dev. Biol. 2021, 9, 623809. [Google Scholar] [CrossRef] [PubMed]
- van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef]
- He, J.; Zhang, C.; Ozkan, A.; Feng, T.; Duan, P.; Wang, S.; Yang, X.; Xie, J.; Liu, X. Patient-derived tumor models and their distinctive applications in personalized drug therapy. Mechanobiol. Med. 2023, 1, 100014. [Google Scholar] [CrossRef]
- García-Perdomo, H.A.; Chaves, M.J.; Osorio, J.C.; Sanchez, A. Association between TMPRSS2: ERG fusion gene and the prostate cancer: Systematic review and meta-analysis. Cent. Eur. J. Urol. 2018, 71, 410–419. [Google Scholar] [CrossRef]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef]
- Nam, R.K.; Sugar, L.; Yang, W.; Srivastava, S.; Klotz, L.H.; Yang, L.Y.; Stanimirovic, A.; Encioiu, E.; Neill, M.; Loblaw, D.A.; et al. Expression of the TMPRSS2: ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br. J. Cancer 2007, 97, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Turnham, D.J.; Bullock, N.; Dass, M.S.; Staffurth, J.N.; Pearson, H.B. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells 2020, 9, 2342. [Google Scholar] [CrossRef]
- Hashemi, M.; Taheriazam, A.; Daneii, P.; Hassanpour, A.; Kakavand, A.; Rezaei, S.; Hejazi, E.S.; Aboutalebi, M.; Gholamrezaie, H.; Saebfar, H.; et al. Targeting PI3K/Akt signaling in prostate cancer therapy. J. Cell Commun. Signal 2023, 17, 423–443. [Google Scholar] [CrossRef] [PubMed]
- Raith, F.; O’Donovan, D.H.; Lemos, C.; Politz, O.; Haendler, B. Addressing the Reciprocal Crosstalk between the AR and the PI3K/AKT/mTOR Signaling Pathways for Prostate Cancer Treatment. Int. J. Mol. Sci. 2023, 24, 2289. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, M.; Mai, F.; Li, X.; Wang, W.; Huang, Q.; Du, X.; Ding, W.; Li, Y.; Barwick, B.G.; et al. Interruption of KLF5 acetylation promotes PTEN-deficient prostate cancer progression by reprogramming cancer-associated fibroblasts. J. Clin. Investig. 2024, 134, e175949. [Google Scholar] [CrossRef]
- Gupta, S.; To, T.M.; Graf, R.; Kadel, E.E.; Reilly, N.; Albarmawi, H. Real-World Overall Survival and Treatment Patterns by PTEN Status in Metastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2024, 8, e2300562. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Degan, S.; Wang, Y.; Cohen, J.; Ku, S.Y.; Goodrich, D.W.; Gelman, I.H. PTEN-regulated PI3K-p110 and AKT isoform plasticity controls metastatic prostate cancer progression. Oncogene 2024, 43, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Mateos, J.; Cresswell, G.D.; Trahearn, N.; Webb, K.; Sakr, C.; Lampis, A.; Stuttle, C.; Corbishley, C.M.; Stavrinides, V.; Zapata, L.; et al. Tumor evolution metrics predict recurrence beyond 10 years in locally advanced prostate cancer. Nat. Cancer 2024, 5, 1334–1351. [Google Scholar] [CrossRef]
- Hasan, A.M.M.; Cremaschi, P.; Wetterskog, D.; Jayaram, A.; Wong, S.Q.; Williams, S.; Pasam, A.; Trigos, A.; Trujillo, B.; Grist, E.; et al. Copy number architectures define treatment-mediated selection of lethal prostate cancer clones. Nat. Commun. 2023, 14, 4823. [Google Scholar] [CrossRef]
- Imamura, J.; Ganguly, S.; Muskara, A.; Liao, R.S.; Nguyen, J.K.; Weight, C.; Wee, C.E.; Gupta, S.; Mian, O.Y. Lineage plasticity and treatment resistance in prostate cancer: The intersection of genetics, epigenetics, and evolution. Front. Endocrinol. 2023, 14, 1191311. [Google Scholar] [CrossRef]
- Weber, Z.T.; Collier, K.A.; Tallman, D.; Forman, J.; Shukla, S.; Asad, S.; Rhoades, J.; Freeman, S.; Parsons, H.A.; Williams, N.O.; et al. Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer. Genome Med. 2021, 13, 89. [Google Scholar] [CrossRef]
- Hong, M.K.H.; Macintyre, G.; Wedge, D.C.; Van Loo, P.; Patel, K.; Lunke, S.; Alexandrov, L.B.; Sloggett, C.; Cmero, M.; Marass, F.; et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 2015, 6, 6605. [Google Scholar] [CrossRef]
- Ge, R.; Wang, Z.; Cheng, L. Tumor microenvironment heterogeneity an important mediator of prostate cancer progression and therapeutic resistance. npj Precis. Oncol. 2022, 6, 31. [Google Scholar] [CrossRef]
- Peter, M.R.; Bilenky, M.; Davies, A.; Isserlin, R.; Bader, G.D.; Fleshner, N.E.; Hirst, M.; Zoubeidi, A.; Bapat, B. Distinct DNA methylation patterns associated with treatment resistance in metastatic castration resistant prostate cancer. Sci. Rep. 2021, 11, 6630. [Google Scholar] [CrossRef]
- Kumaraswamy, A.; Duan, Z.; Flores, D.; Zhang, C.; Sehrawat, A.; Hu, Y.M.; Swaim, O.A.; Rodansky, E.; Storck, W.K.; Kuleape, J.A.; et al. LSD1 promotes prostate cancer reprogramming by repressing TP53 signaling independently of its demethylase function. JCI Insight 2023, 8, e167440. [Google Scholar] [CrossRef] [PubMed]
- Koushyar, S.; Meniel, V.S.; Phesse, T.J.; Pearson, H.B. Exploring the Wnt Pathway as a Therapeutic Target for Prostate Cancer. Biomolecules 2022, 12, 309. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, S.; Han, S.W. Utilizing Plasma Circulating Tumor DNA Sequencing for Precision Medicine in the Management of Solid Cancers. Cancer Res. Treat. 2023, 55, 367–384. [Google Scholar] [CrossRef]
- Lin, M.; Sun, X.; Lv, L. New insights and options into the mechanisms and effects of combined targeted therapy and immunotherapy in prostate cancer. Mol. Ther. Oncolytics 2023, 29, 91–106. [Google Scholar] [CrossRef]
- Wang, H.-C.; Xia, R.; Chang, W.-H.; Hsu, S.-W.; Wu, C.-T.; Chen, C.-H.; Shih, T.-C. Improving cancer immunotherapy in prostate cancer by modulating T cell function through targeting the galectin-1. Front. Immunol. 2024, 15, 1372956. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, X.; Zhang, X.; Jiang, S.; Zhang, T.; Li, G.; Lu, Z.; Zhang, D.; Wang, S.; Qin, C. Immune Microenvironment and Response in Prostate Cancer Using Large Population Cohorts. Front. Immunol. 2021, 12, 686809. [Google Scholar] [CrossRef]
- Hirz, T.; Mei, S.; Sarkar, H.; Kfoury, Y.; Wu, S.; Verhoeven, B.M.; Subtelny, A.O.; Zlatev, D.V.; Wszolek, M.W.; Salari, K.; et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat. Commun. 2023, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Novysedlak, R.; Guney, M.; Al Khouri, M.; Bartolini, R.; Koumbas Foley, L.; Benesova, I.; Ozaniak, A.; Novak, V.; Vesely, S.; Pacas, P.; et al. The Immune Microenvironment in Prostate Cancer: A Comprehensive Review. Oncology 2024, 9, 1–25. [Google Scholar] [CrossRef]
- van Genderen, M.N.G.; Kneppers, J.; Zaalberg, A.; Bekers, E.M.; Bergman, A.M.; Zwart, W.; Eduati, F. Agent-based modeling of the prostate tumor microenvironment uncovers spatial tumor growth constraints and immunomodulatory properties. npj Syst. Biol. Appl. 2024, 10, 20. [Google Scholar] [CrossRef]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Angappulige, D.H.; Mahajan, N.P.; Mahajan, K. Epigenetic underpinnings of tumor-immune dynamics in prostate cancer immune suppression. Trends Cancer 2024, 10, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Palano, M.T.; Gallazzi, M.; Cucchiara, M.; Dehò, F.; Capogrosso, P.; Bruno, A.; Mortara, L. The tumor innate immune microenvironment in prostate cancer: An overview of soluble factors and cellular effectors. Explor. Target. Anti-Tumor Ther. 2022, 3, 694–718. [Google Scholar] [CrossRef]
- Stone, L. Singling out the immune-suppressive TME in prostate cancer. Nat. Rev. Urol. 2023, 20, 199. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Liu, Y.; Guo, J.; Dou, M.; Zhang, X.; Hu, L.; Chen, J. Progression in immunotherapy for advanced prostate cancer. Front. Oncol. 2023, 13, 1126752. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nagata, M.; Hachiya, T.; Wakita, H.; Ikehata, Y.; Takahashi, K.; China, T.; Shimizu, F.; Lu, J.; Jin, Y.; et al. Increased circulating polymorphonuclear myeloid-derived suppressor cells are associated with prognosis of metastatic castration-resistant prostate cancer. Front. Immunol. 2024, 15, 1372771. [Google Scholar] [CrossRef]
- Lyu, A.; Fan, Z.; Clark, M.; Lea, A.; Luong, D.; Setayesh, A.; Starzinski, A.; Wolters, R.; Arias-Badia, M.; Allaire, K.; et al. Evolution of myeloid-mediated immunotherapy resistance in prostate cancer. Nature 2025, 637, 1207–1217. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Rodrigues, D.N.; Gurel, B.; Clarke, M. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2022, 18, 45–55. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Kim, R.; Kim, S.; Oh, B.B.-L.; Yu, W.S.; Kim, C.W.; Hur, H.; Son, S.-Y.; Yang, M.J.; Cho, D.S.; Ha, T.; et al. Clinical application of whole-genome sequencing of solid tumors for precision oncology. Exp. Mol. Med. 2024, 56, 1856–1868. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Mizuno, K.; Beltran, H. Future directions for precision oncology in prostate cancer. Prostate 2022, 82 (Suppl. S1), S86–S96. [Google Scholar] [CrossRef]
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Vandekerkhove, G.; Giri, V.N.; Halabi, S.; McNair, C.; Hamade, K.; Bitting, R.L.; Wyatt, A.W. Toward Informed Selection and Interpretation of Clinical Genomic Tests in Prostate Cancer. JCO Precis. Oncol. 2024, 8, e2300654. [Google Scholar] [CrossRef]
- Cabral, B.P.; Braga, L.A.M.; Syed-Abdul, S.; Mota, F.B. Future of Artificial Intelligence Applications in Cancer Care: A Global Cross-Sectional Survey of Researchers. Curr. Oncol. 2023, 30, 3432–3446. [Google Scholar] [CrossRef]
- Sowalsky, A.G.; Figueiredo, I.; Lis, R.T.; Coleman, I.; Gurel, B.; Bogdan, D.; Yuan, W.; Russo, J.W.; Bright, J.R.; Whitlock, N.C.; et al. Assessment of Androgen Receptor Splice Variant-7 as a Biomarker of Clinical Response in Castration-Sensitive Prostate Cancer. Clin. Cancer Res. 2022, 28, 3509–3525. [Google Scholar] [CrossRef]
- Zhuang, H.-H.; Qu, Q.; Teng, X.-Q.; Dai, Y.-H.; Qu, J. Superenhancers as master gene regulators and novel therapeutic targets in brain tumors. Exp. Mol. Med. 2023, 55, 290–303. [Google Scholar] [CrossRef]
- Song, H.; Weinstein, H.N.W.; Allegakoen, P.; Wadsworth, M.H.; Xie, J.; Yang, H.; Castro, E.A.; Lu, K.L.; Stohr, B.A.; Feng, F.Y.; et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. Nat. Commun. 2022, 13, 141. [Google Scholar] [CrossRef]
- Ruprecht, N.A.; Kennedy, J.D.; Bansal, B.; Singhal, S.; Sens, D.; Maggio, A.; Doe, V.; Hawkins, D.; Campbel, R.; O’Connell, K.; et al. Transcriptomics and epigenetic data integration learning module on Google Cloud. Brief. Bioinform. 2024, 25, bbae352. [Google Scholar] [CrossRef]
- Xu, J.; Yang, P.; Xue, S.; Sharma, B.; Sanchez-Martin, M.; Wang, F.; Beaty, K.A.; Dehan, E.; Parikh, B. Translating cancer genomics into precision medicine with artificial intelligence: Applications, challenges and future perspectives. Hum. Genet. 2019, 138, 109–124. [Google Scholar] [CrossRef]
- Kiviaho, A.; Eerola, S.K.; Kallio, H.M.L.; Andersen, M.K.; Hoikka, M.; Tiihonen, A.M.; Salonen, I.; Spotbeen, X.; Giesen, A.; Parker, C.T.A.; et al. Single cell and spatial transcriptomics highlight the interaction of club-like cells with immunosuppressive myeloid cells in prostate cancer. Nat. Commun. 2024, 15, 9949. [Google Scholar] [CrossRef] [PubMed]
- Villette, C.C.; Dupuy, N.; Brightman, F.A.; Zimmermann, A.; Lignet, F.; Zenke, F.T.; Terranova, N.; Bolleddula, J.; El Bawab, S.; Chassagnole, C. Semi-mechanistic efficacy model for PARP + ATR inhibitors—Application to rucaparib and talazoparib in combination with gartisertib in breast cancer PDXs. Br. J. Cancer 2025, 132, 481–491. [Google Scholar] [CrossRef]
- Ragupathi, A.; Singh, M.; Perez, A.M.; Zhang, D. Targeting the BRCA1/2 deficient cancer with PARP inhibitors: Clinical outcomes and mechanistic insights. Front. Cell Dev. Biol. 2023, 11, 1133472. [Google Scholar] [CrossRef]
- Miller, K.J.; Henry, I.; Maylin, Z.; Smith, C.; Arunachalam, E.; Pandha, H.; Asim, M. A compendium of Androgen Receptor Variant 7 target genes and their role in Castration Resistant Prostate Cancer. Front. Oncol. 2023, 13, 1129140. [Google Scholar] [CrossRef]
- Fenton, S.E.; VanderWeeler, D.J.; Rebbeck, T.R.; Chen, D.L. Advancing Prostate Cancer Care: Treatment Approaches to Precision Medicine, Biomarker Innovations, and Equitable Access. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e433138. [Google Scholar] [CrossRef] [PubMed]
- Vidović, V.; Davidov, I.; Ružić, Z.; Erdeljan, M.; Galfi Vukomanović, A.; Blagojević, B. Androgen Receptors in Human Breast Cancer and Female Canine Mammary Tumors. Molecules 2025, 30, 1411. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Mukherjee, S.; Saha, N.; Yarravarapu, N.; Lone, S.N.; Masoodi, T.; Chauhan, R.; Maacha, S.; Bagga, P.; et al. Integration of CRISPR/Cas9 with artificial intelligence for improved cancer therapeutics. J. Transl. Med. 2022, 20, 534. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Nakayama, H.; Miyazawa, Y.; Arai, S.; Koike, H.; Matsui, H.; Shibata, Y.; Ito, K.; Suzuki, K. Ratio of the expression levels of androgen receptor splice variant 7 to androgen receptor in castration refractory prostate cancer. Oncol. Lett. 2021, 22, 831. [Google Scholar] [CrossRef]
- Mian, O.Y.; Tendulkar, R.D.; Abazeed, M.E. The evolving role of molecular profiling in prostate cancer: Basal and luminal subtyping transcends tissue of origin. Transl. Cancer Res. 2017, 6, S1441–S1445. [Google Scholar] [CrossRef]
- Hofman, M.S.; Lawrentschuk, N.; Francis, R.J.; Tang, C.; Vela, I.; Thomas, P.; Rutherford, N.; Martin, J.M.; Frydenberg, M.; Shakher, R.; et al. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): A prospective, randomised, multicentre study. Lancet 2020, 395, 1208–1216. [Google Scholar] [CrossRef]
- Fendler, W.P.; Calais, J.; Eiber, M.; Flavell, R.R.; Mishoe, A.; Feng, F.Y.; Nguyen, H.G.; Reiter, R.E.; Rettig, M.B.; Okamoto, S.; et al. Assessment of 68Ga-PSMA-11 PET Accuracy in Localizing Recurrent Prostate Cancer: A Prospective Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Lawhn-Heath, C.; Flavell, R.R.; Behr, S.C.; Yohannan, T.; Greene, K.L.; Feng, F.; Carroll, P.R.; Hope, T.A. Single-Center Prospective Evaluation of (68)Ga-PSMA-11 PET in Biochemical Recurrence of Prostate Cancer. AJR Am. J. Roentgenol. 2019, 213, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.L.; Nyame, Y.A.; Mitchell, E. Disparities in prostate cancer. J. Natl. Med. Assoc. 2023, 115, S38–S45. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ketteler, S.; Bagheri, S.; Ebrahimifard, A.; Luster, M.; Librizzi, D.; Yousefi, B.H. Diagnostic efficacy of [99mTc]Tc-PSMA SPECT/CT for prostate cancer: A meta-analysis. BMC Cancer 2024, 24, 982. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. (68)Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef]
- He, W.; Huang, W.; Zhang, L.; Wu, X.; Zhang, S.; Zhang, B. Radiogenomics: Bridging the gap between imaging and genomics for precision oncology. MedComm 2024, 5, e722. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Hetzheim, H.; Kübler, W.; Kratochwil, C.; Giesel, F.L.; Hope, T.A.; Eder, M.; Eisenhut, M.; Kopka, K.; Haberkorn, U. Radiation dosimetry of (68)Ga-PSMA-11 (HBED-CC) and preliminary evaluation of optimal imaging timing. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1611–1620. [Google Scholar] [CrossRef]
- Parekh, V.; Jacobs, M.A. Radiomics: A new application from established techniques. Expert. Rev. Precis. Med. Drug Dev. 2016, 1, 207–226. [Google Scholar] [CrossRef]
- von Stauffenberg, F.; Poyet, C.; Beintner-Skawran, S.; Maurer, A.; Schmid, F.A. Current Clinical Applications of PSMA-PET for Prostate Cancer Diagnosis, Staging, and Treatment. Cancers 2024, 16, 4263. [Google Scholar] [CrossRef]
- Dorff, T.B.; Blanchard, M.S.; Adkins, L.N.; Luebbert, L.; Leggett, N.; Shishido, S.N.; Macias, A.; Del Real, M.M.; Dhapola, G.; Egelston, C.; et al. PSCA-CAR T cell therapy in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2024, 30, 1636–1644. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive review of CRISPR-based gene editing: Mechanisms, challenges, and applications in cancer therapy. Mol. Cancer 2024, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Broughton, P.; Abdollahi, H.; Valafar, H.; Blenda, A.V. Integrating Omics Data and AI for Cancer Diagnosis and Prognosis. Cancers 2024, 16, 2448. [Google Scholar] [CrossRef]
- Allen, T.A. The Role of Circulating Tumor Cells as a Liquid Biopsy for Cancer: Advances, Biology, Technical Challenges, and Clinical Relevance. Cancers 2024, 16, 1377. [Google Scholar] [CrossRef]
- Gu, X.; Wei, S.; Lv, X. Circulating tumor cells: From new biological insights to clinical practice. Signal Transduct. Target. Ther. 2024, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Urabe, F.; Sumiyoshi, T.; Tashiro, K.; Goto, T.; Kimura, T.; Kobayashi, T. Prostate cancer and liquid biopsies: Clinical applications and challenges. Int. J. Urol. 2024, 31, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Lin, H.; Zhu, Y.; Huang, D.; Lai, M.; Xi, X.; Huang, J.; Zhang, W.; Zhong, T. Research progress of CTC, ctDNA, and EVs in cancer liquid biopsy. Front. Oncol. 2024, 14, 1303335. [Google Scholar] [CrossRef]
- Sartor, O. Circulating Tumor DNA Biomarkers for Response Assessment in Prostate Cancer. Clin. Cancer Res. 2023, 29, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xia, C.; Xia, C. Biology and function of exosomes in tumor immunotherapy. Biomed. Pharmacother. 2023, 169, 115853. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, J.; Liu, Y.; Shi, Z.; Xuan, Z.; Yang, K.; Xu, C.; Bai, Y.; Fu, M.; Xiao, Q.; et al. The Crucial Role of AR-V7 in Enzalutamide-Resistance of Castration-Resistant Prostate Cancer. Cancers 2022, 14, 4877. [Google Scholar] [CrossRef]
- Crocetto, F.; Russo, G.; Di Zazzo, E.; Pisapia, P.; Mirto, B.F.; Palmieri, A.; Pepe, F.; Bellevicine, C.; Russo, A.; La Civita, E.; et al. Liquid Biopsy in Prostate Cancer Management-Current Challenges and Future Perspectives. Cancers 2022, 14, 3272. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J. Current and Future Perspectives of Cell-Free DNA in Liquid Biopsy. Curr. Issues Mol. Biol. 2022, 44, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-C.T.; Pan, I.-W.; Teich, N. Global Challenges in Access to and Implementation of Precision Oncology: The Health Care Manager and Health Economist Perspective. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 429–437. [Google Scholar] [CrossRef]
- Lipkova, J.; Chen, R.J.; Chen, B.; Lu, M.Y.; Barbieri, M.; Shao, D.; Vaidya, A.J.; Chen, C.; Zhuang, L.; Williamson, D.F.K.; et al. Artificial intelligence for multimodal data integration in oncology. Cancer Cell 2022, 40, 1095–1110. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Chen, Z. Emerging role of exosomes in cancer therapy: Progress and challenges. Mol. Cancer 2025, 24, 13. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid biopsy in cancer: Current status, challenges and future prospects. Signal Transduct. Target. Ther. 2024, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Bastos, D.A.; Soares, A.; Schutz, F.A.B.; Cronemberger, E.; de Almeida Luz, M.; Martins, S.; Muniz, D.Q.B.; Cárcano, F.M.; Smaletz, O.; Peixoto, F.A.; et al. Androgen Receptor Pathway Inhibitor Therapy for Advanced Prostate Cancer: Secondary Analysis of a Randomized Clinical Trial. JAMA Netw. Open 2025, 8, e2454253. [Google Scholar] [CrossRef]
- Bourlon, M.T.; Valdez, P.; Castro, E. Development of PARP inhibitors in advanced prostate cancer. Ther. Adv. Med. Oncol. 2024, 16, 17588359231221337. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Wang, H.; Agarwal, N.; Smith, D.C.; Schweizer, M.T.; Stein, M.N.; Assikis, V.; Twardowski, P.W.; Flaig, T.W.; Szmulewitz, R.Z.; et al. TRANSFORMER: A Randomized Phase II Study Comparing Bipolar Androgen Therapy Versus Enzalutamide in Asymptomatic Men With Castration-Resistant Metastatic Prostate Cancer. J. Clin. Oncol. 2021, 39, 1371–1382. [Google Scholar] [CrossRef]
- Markowski, M.C.; Taplin, M.-E.; Aggarwal, R.; Sena, L.A.; Wang, H.; Qi, H.; Lalji, A.; Sinibaldi, V.; Carducci, M.A.; Paller, C.J.; et al. Bipolar androgen therapy plus nivolumab for patients with metastatic castration-resistant prostate cancer: The COMBAT phase II trial. Nat. Commun. 2024, 15, 14. [Google Scholar] [CrossRef]
- Grewal, K.; Grewal, K.; Tabbara, I.A. PARP Inhibitors in Prostate Cancer. Anticancer. Res. 2021, 41, 551–556. [Google Scholar] [CrossRef]
- Chen, Q.H.; Munoz, E.; Ashong, D. Insight into Recent Advances in Degrading Androgen Receptor for Castration-Resistant Prostate Cancer. Cancers 2024, 16, 663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ming, A.; Wang, J.; Chen, W.; Fang, Z. PROTACs targeting androgen receptor signaling: Potential therapeutic agents for castration-resistant prostate cancer. Pharmacol. Res. 2024, 205, 107234. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Abrahamse, H. Optimizing CRISPR/Cas9 precision: Mitigating off-target effects for safe integration with photodynamic and stem cell therapies in cancer treatment. Biomed. Pharmacother. 2024, 180, 117516. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.R.; Luciano, F.C.; Anaya, B.J.; Ongoren, B.; Kara, A.; Molina, G.; Ramirez, B.I.; Sánchez-Guirales, S.A.; Simon, J.A.; Tomietto, G.; et al. Artificial Intelligence (AI) Applications in Drug Discovery and Drug Delivery: Revolutionizing Personalized Medicine. Pharmaceutics 2024, 16, 1328. [Google Scholar] [CrossRef]
- Hamid, A.A.; Sayegh, N.; Tombal, B.; Hussain, M.; Sweeney, C.J.; Graff, J.N.; Agarwal, N. Metastatic Hormone-Sensitive Prostate Cancer: Toward an Era of Adaptive and Personalized Treatment. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390166. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, D.K.; Mishra, D.; Singh, R.P. Targeting DNA repair for cancer treatment: Lessons from PARP inhibitor trials. Oncol. Res. 2023, 31, 405–421. [Google Scholar] [CrossRef]
- Jain, R.; Kumar, A.; Sharma, A.; Sahoo, R.K.; Sharma, A.; Seth, A.; Nayak, B.; Shamim, S.A.; Kaushal, S.; Kp, H.; et al. Carboplatin in Patients With Metastatic Castration-Resistant Prostate Cancer Harboring Somatic or Germline Homologous Recombination Repair Gene Mutations: Phase II Single-Arm Trial. JMIR Res. Protoc. 2024, 13, e54086. [Google Scholar] [CrossRef]
- Gleave, M.; Belanger, E.; Scurll, J.; Oo, H.Z.; Nappi, L.; Beltran, H.; Wyatt, A.W.; Mannas, M.; Black, P.C.; Zoubeidi, A.; et al. Genomic alterations and their pathologic responses in high-risk localized prostate cancer (HRLPC) in subprotocol 1 of the Genomic Umbrella Neoadjuvant study (GUNS). J. Clin. Oncol. 2025, 43, 403. [Google Scholar] [CrossRef]
- Azad, A.; Agarwal, N.; Armstrong, A.J.; Hellmis, E.; Schlürmann, F.; Sugimoto, M.; Ürün, Y.; Xing, N.; Aregay, M.; Lima, J.; et al. Phase III, double-blind, placebo-controlled, 2-cohort, randomized study of saruparib (AZD5305) in combination with androgen receptor pathway inhibitors in patients with metastatic hormone-sensitive prostate cancer with and without homologous recombination repair mutation (EvoPAR-Prostate01). J. Clin. Oncol. 2025, 43, TPS279. [Google Scholar] [CrossRef]
- Fizazi, K.; George, D.J.; Santis, M.D.; Clarke, N.; Fay, A.P.; Uemura, H.; Grinsted, L.; Rooney, C.; Verheijen, R.B.; Anjum, R.; et al. A phase III trial of capivasertib and abiraterone versus placebo and abiraterone in patients with de novo metastatic hormone-sensitive prostate cancer characterized by PTEN deficiency (CAPItello-281). J. Clin. Oncol. 2021, 39, TPS178. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Yasuda, T.; Uchihara, T.; Yasuda-Yoshihara, N.; Tan, B.J.Y.; Yonemura, A.; Semba, T.; Yamasaki, J.; Komohara, Y.; Ohnishi, K.; et al. Stromal Reprogramming through Dual PDGFRα/β Blockade Boosts the Efficacy of Anti–PD-1 Immunotherapy in Fibrotic Tumors. Cancer Res. 2023, 83, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr. -Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.B.; Reimers, M.A.; Perera, C.; Abida, W.; Chou, J.; Feng, F.Y.; Antonarakis, E.S.; McKay, R.R.; Pachynski, R.K.; Zhang, J.; et al. Evaluating Immune Checkpoint Blockade in Metastatic Castration-Resistant Prostate Cancers with Deleterious CDK12 Alterations in the Phase 2 IMPACT Trial. Clin. Cancer Res. 2024, 30, 3200–3210. [Google Scholar] [CrossRef]
- Choudhury, A.D.; Kwak, L.; Cheung, A.; Allaire, K.M.; Marquez, J.; Yang, D.D.; Tripathi, A.; Kilar, J.M.; Flynn, M.; Maynard, B.; et al. Randomized Phase II Study Evaluating the Addition of Pembrolizumab to Radium-223 in Metastatic Castration-resistant Prostate Cancer. Cancer Immunol. Res. 2024, 12, 704–718. [Google Scholar] [CrossRef]
- Bhatia, V.; Kamat, N.V.; Pariva, T.E.; Wu, L.-T.; Tsao, A.; Sasaki, K.; Sun, H.; Javier, G.; Nutt, S.; Coleman, I.; et al. Targeting advanced prostate cancer with STEAP1 chimeric antigen receptor T cell and tumor-localized IL-12 immunotherapy. Nat. Commun. 2023, 14, 2041. [Google Scholar] [CrossRef]
- Zhao, L.; Pang, Y.; Zhou, Y.; Chen, J.; Fu, H.; Guo, W.; Xu, W.; Xue, X.; Su, G.; Sun, L.; et al. Antitumor efficacy and potential mechanism of FAP-targeted radioligand therapy combined with immune checkpoint blockade. Signal Transduct. Target. Ther. 2024, 9, 142. [Google Scholar] [CrossRef]
- Handa, S.; Hans, B.; Goel, S.; Bashorun, H.O.; Dovey, Z.; Tewari, A. Immunotherapy in prostate cancer: Current state and future perspectives. Ther. Adv. Urol. 2020, 12, 1756287220951404. [Google Scholar] [CrossRef]
- Nakamura, H.; Arihara, Y.; Takada, K. Targeting STEAP1 as an anticancer strategy. Front. Oncol. 2023, 13, 1285661. [Google Scholar] [CrossRef]
- Kelly, W.K.; Danila, D.C.; Lin, C.C.; Lee, J.L.; Matsubara, N.; Ward, P.J.; Armstrong, A.J.; Pook, D.; Kim, M.; Dorff, T.B.; et al. Xaluritamig, a STEAP1 × CD3 XmAb 2+1 Immune Therapy for Metastatic Castration-Resistant Prostate Cancer: Results from Dose Exploration in a First-in-Human Study. Cancer Discov. 2024, 14, 76–89. [Google Scholar] [CrossRef]
- Calais, J.; Murthy, V.; Voter, A.; Nguyen, K.; Allen-Auerbach, M.S.; Caputo, S.; Ledet, E.M.; Akerele, O.; Tuchayi, A.M.; Heath, C.L.; et al. Efficacy and toxicity of [177Lu]Lu-PSMA-617 for metastatic castration-resistant prostate cancer in a real-world setting: Results from the U.S. Expanded Access Program and comparisons with phase 3 VISION data. J. Clin. Oncol. 2024, 42, e17061. [Google Scholar] [CrossRef]
- Besiroglu, H.; Kadihasanoglu, M. The Safety and Efficacy of Targeted Alpha Therapy, Ac-225 Prostate-Specific Membrane Antigen, in Patients With Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis. Prostate 2025, 85, e24857. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.P.; Kostos, L.; Alipour, R.; Jackson, P.; McInstosh, L.; Emmerson, B.; Haskali, M.B.; Xie, J.; Medhurst, E.; Ravi, R.; et al. Clinical Trial Protocol for VIOLET: A Single-Center, Phase I/II Trial Evaluation of Radioligand Treatment in Patients with Metastatic Castration-Resistant Prostate Cancer with [161Tb]Tb-PSMA-I&T. J. Nucl. Med. 2024, 65, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Kiess, A.P.; Nordquist, L.; Gupta, S.; George, D.J.; Messmann, R.A.; Kantoff, P.W.; Nauseef, J.T.; Bander, N.H.; Antonarakis, E.S. CONVERGE-01: Dosimetry, randomized dose optimization, dose escalation, and efficacy of ac-225 rosopatamab tetraxetan in participants with PSMA-positive castration-resistant prostate cancer. J. Clin. Oncol. 2025, 43, TPS289. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Phase I/II Study of [225Ac]Ac-PSMA-R2 in PSMA-Positive Prostate Cancer, with/Without Prior 177Lu-PSMA RLT. Identifier: NCT05983198. Updated [31 January 2025]. Available online: https://clinicaltrials.gov/study/NCT05983198 (accessed on 28 February 2025).
- Sandhu, S.; Subramaniam, S.; Hofman, M.S.; Stockler, M.R.; Martin, A.J.; Pokorski, I.; Goh, J.C.; Pattison, D.A.; Dhiantravan, N.; Gedye, C.; et al. Evolution: Phase II study of radionuclide 177Lu-PSMA-617 therapy versus 177Lu-PSMA-617 in combination with ipilimumab and nivolumab for men with metastatic castration-resistant prostate cancer (mCRPC.; ANZUP 2001). J. Clin. Oncol. 2023, 41, TPS271. [Google Scholar] [CrossRef]
- Kostos, L.K.; Buteau, J.P.; Yeung, T.; Xie, S.; Iulio, J.D.; Cardin, A.; Owen, K.; Fettke, H.; Chin, K.Y.; Emmerson, B.; et al. AlphaBet: A phase I/II trial evaluating the combination of radium-223 and [177Lu]Lu-PSMA-I&T in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2023, 41, TPS280. [Google Scholar] [CrossRef]
- Ruigrok, E.A.M.; Verkaik, N.S.; de Blois, E.; de Ridder, C.; Stuurman, D.; Roobol, S.J.; Van Gent, D.C.; de Jong, M.; Van Weerden, W.M.; Nonnekens, J. Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 8037. [Google Scholar] [CrossRef]
- Giunta, E.F.; Brighi, N.; Gurioli, G.; Matteucci, F.; Paganelli, G.; De Giorgi, U. 177Lu-PSMA therapy in metastatic prostate cancer: An updated review of prognostic and predictive biomarkers. Cancer Treat. Rev. 2024, 125, 102699. [Google Scholar] [CrossRef]
- Perrone, E.; Giordano, A.; Calcagni, M.L.; Leccisotti, L.; Moretti, R.; Eismant, A.; Ghai, K.; Parkar, T.; Mishra, A.; Heidenreich, A.; et al. Long-Term Safety and Survival Outcomes of [(225)Ac]Ac-PSMA (Prostate-Specific Membrane Antigen) and [(225)Ac]Ac-/[(177)Lu]Lu-PSMA (TANDEM) Radioligand Therapy (PRLT) in Metastatic Castration-Resistant Prostate Cancer. Cancers 2025, 17, 405. [Google Scholar] [CrossRef]
- Wang, H.; Li, G.; Zhao, J.; Eiber, M.; Tian, R. Current status of PSMA-targeted imaging and therapy. Front. Oncol. 2024, 13, 1230251. [Google Scholar] [CrossRef]
- Marshall, C.H.; Antonarakis, E.S. Emerging treatments for metastatic castration-resistant prostate cancer: Immunotherapy, PARP inhibitors, and PSMA-targeted approaches. Cancer Treat. Res. Commun. 2020, 23, 100164. [Google Scholar] [CrossRef] [PubMed]
- Karimzadeh, A.; Lehnert, W.; Koehler, D.; Shenas, F.; Kisters, A.; Apostolova, I.; Klutmann, S.; Adam, G.; Sauer, M. Overview of selected completed prospective studies on PSMA-targeted radioligand therapy with [177Lu]Lu-PSMA-617 in metastatic castration-resistant prostate cancer. Rofo 2025. [Google Scholar] [CrossRef] [PubMed]
- Hachem, S.; Yehya, A.; El Masri, J.; Mavingire, N.; Johnson, J.R.; Dwead, A.M.; Kattour, N.; Bouchi, Y.; Kobeissy, F.; Rais-Bahrami, S.; et al. Contemporary Update on Clinical and Experimental Prostate Cancer Biomarkers: A Multi-Omics-Focused Approach to Detection and Risk Stratification. Biology 2024, 13, 762. [Google Scholar] [CrossRef]
- Bono, J.d.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Sartor, O.; Bono, J.d.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e483. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide and Overall Survival in Prostate Cancer. Eur. Urol. 2021, 79, 150–158. [Google Scholar] [CrossRef]
| Molecular Target or Genetic Alteration | Key Mechanism/Function | Clinical Features | Clinical Utility |
|---|---|---|---|
| AR Amplification/AR Splice Variants (e.g., AR-V7) | Sustained AR signaling under low-androgen conditions; ligand-independent activation | Poor response or resistance to AR-targeted therapies; commonly seen in mCRPC | Predicts resistance to enzalutamide or abiraterone; potential biomarker for treatment selection |
| PTEN Loss | Hyperactivation of the PI3K/AKT/mTOR pathway; cross-talk with AR signaling | Associated with high-grade tumors and aggressive clinical course | May guide PI3K/AKT/mTOR inhibitor-based combination trials; potential prognostic indicator |
| DDR Defects (e.g., BRCA2, ATM) | Impaired DNA repair and increased genomic instability; vulnerability to PARP inhibition | More aggressive behavior if untreated; better response to PARP inhibitors | Companion diagnostics for PARP inhibitors; synthetic lethality-based therapy targeting |
| TMPRSS2–ERG Fusion | ETS transcription factor (ERG) overexpression; promotes invasion, EMT, and genomic instability | High prevalence in localized prostate cancer; variable association with outcomes in mPCa | Potential prognostic marker in combination with other alterations (e.g., PTEN) |
| PI3K/AKT/mTOR Mutations | Aberrant cell proliferation and survival; metabolic reprogramming | Often co-occurs with AR pathway alterations; contributes to therapeutic resistance | Under investigation in clinical trials targeting AKT and mTOR; potential combination strategy with AR inhibitors |
| TP53/RB1 Co-mutations | Disruption of cell-cycle checkpoints; may facilitate lineage plasticity or neuroendocrine differentiation | Common in advanced mPCa; associated with poor prognosis | Emerging biomarker for early switch to chemotherapy or combination therapies |
| Diagnostic Panel/Biomarker | Testing Method | Clinical Significance | Limitations/Considerations |
|---|---|---|---|
| DDR-Focused Panel (BRCA1/2, ATM, etc.) | Targeted NGS or expanded gene panels Germline vs. somatic testing | Identifies candidates for PARP inhibitors and platinum-based therapies May inform familial genetic risk | Cost and limited access in some regions May miss epigenetic alterations |
| AR Splice Variants (e.g., AR-V7) | RT-PCR or ddPCR on CTCs Tissue-based RNA assays | Predicts resistance to enzalutamide or abiraterone Can guide switch to chemotherapy or other targeted agents | Variable sensitivity depending on assay Not yet universally available or standardized |
| PTEN/PI3K/AKT | IHC, FISH Targeted sequencing for hotspot mutation | Potential biomarker for AKT/mTOR inhibitors May correlate with disease aggressiveness | Limited predictive validation in some trials Reimbursement issues in certain regions |
| TP53/RB1 | Targeted NGS or WES/WGS IHC for protein loss | Associated with poor prognosis May indicate early progression toward neuroendocrine differentiation | Rarely used in routine practice Data interpretation can be complex (co-occurring events) |
| TMPRSS2–ERG Fusion | FISH, RT-PCR, or RNA-seq | Possible prognostic marker when combined with other aberrations (e.g., PTEN) | Prognostic impact remains debated May not be actionable with current therapies |
| Biomarker | Common Test Method (s) | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Notes/References |
|---|---|---|---|---|---|---|
| AR-V7 (Circulating Tumor Cells) | RT-PCR or ddPCR of isolated CTCs | ~60–80 | ~70–90 | ~65–85 | ~70–90 | Varies by platform and cutoff used. Higher PPV for resistance to AR inhibitors [104,108]. Not standardized across all labs. |
| BRCA1/2 & DDR Panel (Genomic Testing) | NGS panels (targeted, WES) | ~85–95 | ~95–99 | ~90–95 | ~85–95 | Typically, high analytical accuracy for well-validated panels [6,89]. Clinical performance depends on presence of true driver mutations. |
| PTEN Loss (tissue-based) | IHC or FISH | ~70–80 | ~80–90 | ~60–85 | ~75–90 | Studies show variability in PTEN detection [52]. Co-occurring alterations (e.g., TP53) may influence true clinical impact. |
| TP53/RB1 (tissue-based) | Targeted NGS, WES/WGS, IHC for protein | ~70–90 | ~85–95 | ~80 | ~85 | Highly dependent on sample purity and co-mutations [109]; associated with poor prognosis |
| Mismatch Repair Defects (e.g., MSH2, MLH1) | IHC for mismatch repair proteins, NGS | ~85–95 | ~85–95 | ~75–90 | ~90 | Predictive for PD-1 checkpoint inhibition; modest incidence in PCa [6] |
| TMPRSS2–ERG Fusion (Tissue-Based Assays) | FISH, RT-PCR, or RNA-seq in tumor tissue | ~50–70 | ~80–95 | ~70–90 | ~60–80 | Variable positivity rates in localized vs. metastatic PCa [49,56]. Prognostic value still debated. |
| Modality | Specimen Characteristics | Analytical Techniques | Clinical Applications | Advantages | Limitations |
|---|---|---|---|---|---|
| ctDNA | Cell-free DNA fragments shed by tumor cells Detected in plasma or serum | Targeted/whole-exome NGS ddPCR | Real-time monitoring of tumor burden Detection of actionable mutations (e.g., BRCA2) | Minimally invasive Repeat sampling feasible Reflects genomic heterogeneity | Low abundance in early disease Sensitivity depends on tumor fraction Assay costs and standardization issues |
| CTCs | Intact, viable tumor cells in the bloodstream May be enriched via immunomagnetic or size-based separation methods | Immunophenotyping Single-cell genomics/transcriptomics | Prognostic biomarker (CTC count) AR-V7 status for therapy guidance Potential ex vivo drug testing | Allows morphological and molecular analyses Provides insight into specific cell populations | Rare cells, labor-intensive Limited sensitivity in low-volume disease Heterogeneity among different CTC populations |
| Exosomes and Extracellular Vesicles | Nano-scale vesicles containing proteins, RNA, and DNA Released by tumor and stromal cells into bodily fluids | RNA-seq, proteomics Nanoparticle tracking Advanced mass spectrometry | May reveal early resistance signatures Potential biomarkers for immune and stromal interactions | Reflects active secretory pathways Can capture tumor–stromal communication | Isolation protocols not standardized Complexity of vesicle subtypes Data interpretation is challenging |
| Treatment or Combination | Primary Target/Mechanism | Trial Phase | Patient Population | Key Outcomes | Current Status | Reference |
|---|---|---|---|---|---|---|
| Olaparib vs. Abiraterone/Enzalutamide (PROfound) | PARP inhibition (DDR deficiency) | Phase III | mCRPC with HRR gene alterations (e.g., BRCA1/2) | Improved radiographic PFS and OS in biomarker-selected patients | Approved for HRR-mutated mCRPC | [175] |
| Ipatasertib + Abiraterone (IPATential150) | AKT inhibitor + AR axis blockade | Phase III | mCRPC, particularly with PTEN loss | Prolonged PFS in the PTEN-loss subgroup | Ongoing or completed; subset analyses continuing | [151] |
| 177Lu-PSMA-617 + Standard of Care (VISION) | PSMA-targeted radioligand therapy | Phase III | Heavily pretreated mCRPC | Improved OS and PFS vs. standard care | Approved in multiple regions | [176] |
| Nivolumab + Ipilimumab (CheckMate 650) | Dual immune checkpoint blockade (PD-1, CTLA-4) | Phase II | mCRPC, previously treated | Moderate objective response; significant immune-related toxicity | Further refinement of combination strategies needed | [177] |
| Pembrolizumab (KEYNOTE-199) | PD-1 immune checkpoint blockade | Phase II | mCRPC with prior treatments | Modest response rates; better outcomes in MSI-H or DNA repair defects | Investigational in selected biomarker-defined subgroups | [178] |
| Apalutamide (SPARTAN) | Next-generation AR antagonist | Phase III | nmCRPC (non-metastatic CRPC) | Significantly improved metastasis-free survival (MFS) | Approved for nmCRPC | [179] |
| Therapeutic Strategy | Mechanism of Action | Associated Biomarkers | Advantages | Disadvantages/Limitations | Reference |
|---|---|---|---|---|---|
| AR-Targeted Therapies (e.g., Abiraterone, Enzalutamide, Apalutamide, Darolutamide) | Second-generation agents block AR function by inhibiting ligand binding (enzalutamide) or androgen biosynthesis (abiraterone) | AR-V7 and other splice variants AR amplification, LBD mutations | Mainstay for mCSPC/mCRPC with proven OS benefits Generally well-tolerated compared to chemotherapy Can be combined with other targeted agents | Inevitable resistance (AR mutations/splice variants) Cross-resistance among agents Limited efficacy in NEPC | [29,136,137,140,141,142,143] |
| PARP Inhibitors (e.g., Olaparib, Rucaparib) | Exploit synthetic lethality in tumors with deficient DNA repair (e.g., BRCA2) | BRCA1/2, ATM, PALB2, or other DDR gene alterations | Markedly improved outcomes in BRCA2-mutant mCRPC Potential synergy with AR pathway inhibition or chemo Established companion diagnostics for biomarker-driven selection | Resistance via reversion mutations or upregulation of alternative repair pathways Limited efficacy in non-BRCA/ATM-altered tumors Class-related toxicities (myelosuppression, GI events) High cost and variable global availability | [46,146,147] |
| Immunotherapy (Checkpoint Inhibitors, Vaccines, Bispecific T cell Engagers) | Enhances immune-mediated tumor recognition and destruction (e.g., PD-1/PD-L1 or CTLA-4 blockade) BiTEs (e.g., STEAP1-targeting) redirect T cells to tumor cells Vaccines stimulate specific T cell responses | MMR deficiency, MSI-H Emerging markers (e.g., tumor mutation burden, PD-L1 expression, STEAP1 expression) | Durable responses in a subset of patients with high TMB/MSI-H Potential synergy with radiation or targeted therapies Bispecific T cell engagers can be highly potent, bypassing the need for prior T cell priming | Prostate cancer is typically immunologically “cold” Limited responses outside biomarker-selected populations Immune-related adverse events (e.g., colitis, dermatitis) BiTE therapies can cause cytokine release syndrome | [154,155,156,157,158,159] |
| PSMA-Targeted Radioligand Therapy (e.g., 177Lu-PSMA-617) | Delivers cytotoxic radiation specifically to PSMA-expressing tumor cells | PSMA PET imaging for target expression | Offers both diagnostic (PSMA PET-CT) and therapeutic potential (“theranostics”) Demonstrated survival advantage in heavily pretreated mCRPC (VISION trial) Can target micro-metastatic disease | Not all PCa lesions overexpress PSMA (e.g., neuroendocrine variants) Salivary gland toxicity and xerostomia, especially with alpha-emitters (225Ac) Requires specialized facilities for radiopharmaceutical handling Cost and limited access in some regions | [161,162,167,169,171] |
| PI3K/AKT/mTOR Inhibitors (e.g., Ipatasertib, Capivasertib) | Blocks downstream signaling of PI3K/AKT/mTOR axis, often hyperactivated due to PTEN loss | PTEN status PIK3CA, AKT mutations | Potential synergy with AR blockade May delay disease progression in PTEN-deficient mCRPC Encouraging early-phase trial results in combination regimens | On-target metabolic toxicities (hyperglycemia, rash) Relatively modest single-agent activity Patient selection crucial to avoid unnecessary toxicity | [150,151,152,153] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, W.-A.; Joung, J.Y. Precision Targeting in Metastatic Prostate Cancer: Molecular Insights to Therapeutic Frontiers. Biomolecules 2025, 15, 625. https://doi.org/10.3390/biom15050625
Kwon W-A, Joung JY. Precision Targeting in Metastatic Prostate Cancer: Molecular Insights to Therapeutic Frontiers. Biomolecules. 2025; 15(5):625. https://doi.org/10.3390/biom15050625
Chicago/Turabian StyleKwon, Whi-An, and Jae Young Joung. 2025. "Precision Targeting in Metastatic Prostate Cancer: Molecular Insights to Therapeutic Frontiers" Biomolecules 15, no. 5: 625. https://doi.org/10.3390/biom15050625
APA StyleKwon, W.-A., & Joung, J. Y. (2025). Precision Targeting in Metastatic Prostate Cancer: Molecular Insights to Therapeutic Frontiers. Biomolecules, 15(5), 625. https://doi.org/10.3390/biom15050625