

Matrix Metalloproteinases: Pathophysiologic Implications and Potential Therapeutic Targets in Cardiovascular Disease

 , ,
, ,  , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract

1. Introduction
2. Structure, Activation, and Function of MMPs
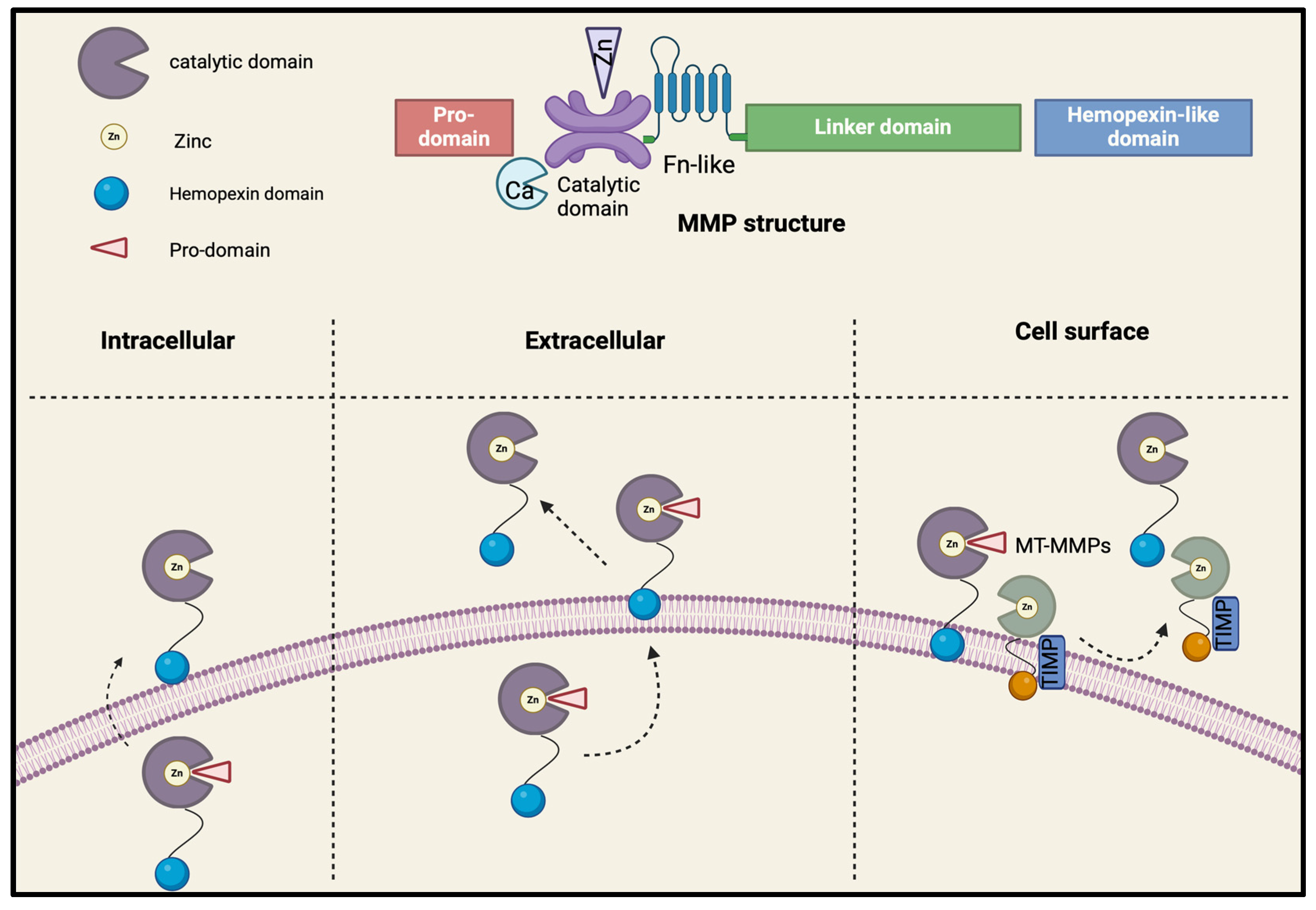
2.1. Structure
2.2. Activation and Inhibition
2.3. MMPs Tissue Expression
3. MMPs’ Interplay in Cardiovascular Diseases
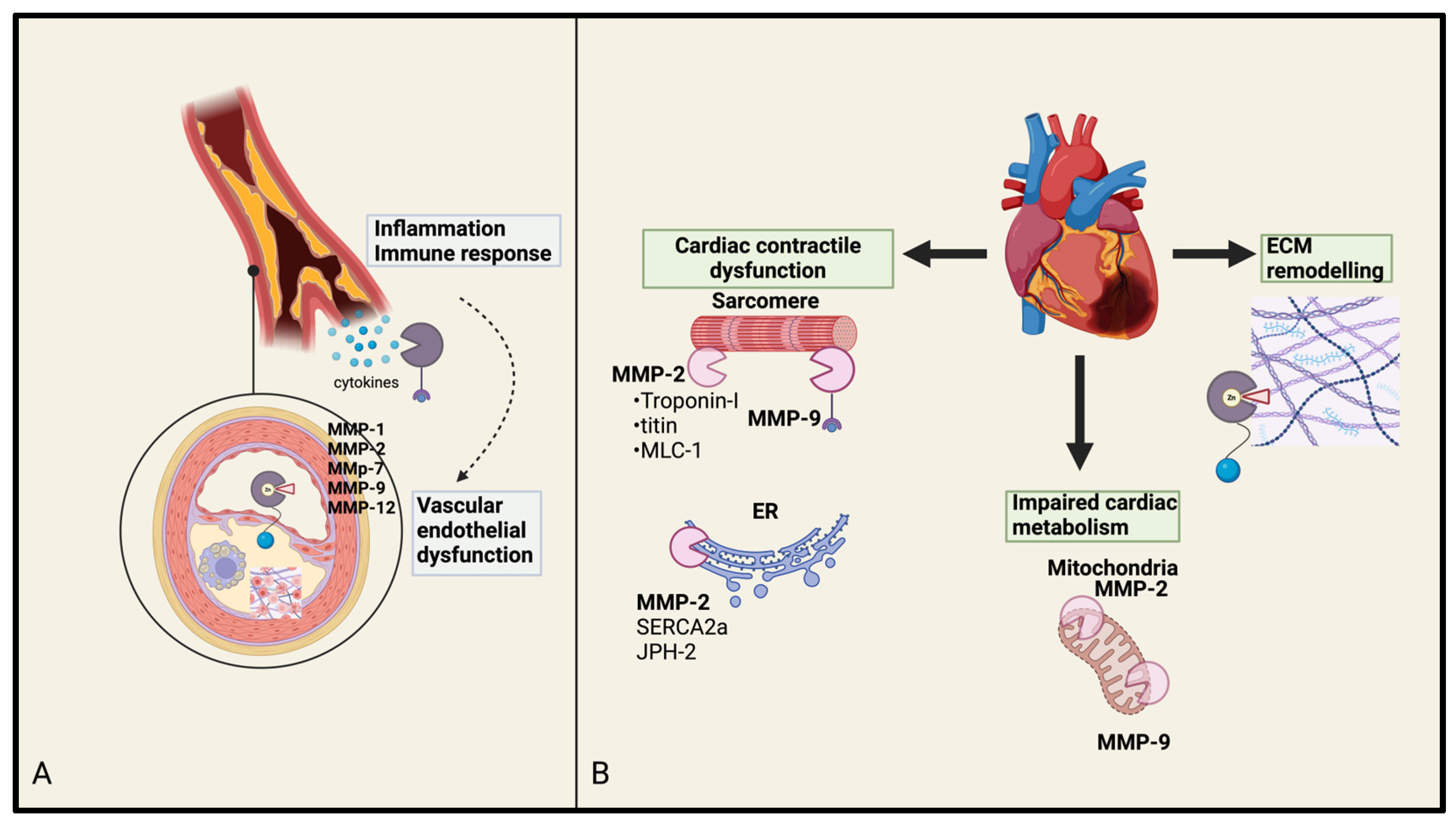
3.1. MMPs in Atherosclerosis and Coronary Artery Disease
3.2. MMPs in Myocardial Infarction
3.3. MMPs in Heart Failure
3.4. MMPs in Valvular Heart Disease: Focus on Aortic Stenosis
4. Potential Therapeutic Targets
4.1. MMP Inhibitors
4.2. Gene Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amin, M.; Pushpakumar, S.; Muradashvili, N.; Kundu, S.; Tyagi, S.C.; Sen, U. Regulation and Involvement of Matrix Metalloproteinases in Vascular Diseases. Front. Biosci. (Landmark Ed.) 2016, 21, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Arkadash, V.; Yosef, G.; Shirian, J.; Cohen, I.; Horev, Y.; Grossman, M.; Sagi, I.; Radisky, E.S.; Shifman, J.M.; Papo, N. Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution. J. Biol. Chem. 2017, 292, 3481–3495. [Google Scholar] [CrossRef]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef] [PubMed]
- Bassiouni, W.; Ali, M.A.M.; Schulz, R. Multifunctional Intracellular Matrix Metalloproteinases: Implications in Disease. FEBS J. 2021, 288, 7162–7182. [Google Scholar] [CrossRef]
- Lindsey, M.L. Assigning Matrix Metalloproteinase Roles in Ischaemic Cardiac Remodelling. Nat. Rev. Cardiol. 2018, 15, 471–479. [Google Scholar] [CrossRef]
- Yuan, W.; Zhang, J.; Huo, R.; Hou, C.; Yang, J.; Wang, T. Intraperitoneal Injection of Human Ferritin Heavy Chain Attenuates the Atherosclerotic Process in APOE-Knockout Mice. J. Cardiovasc. Dev. Dis. 2023, 10, 309. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Chen, Y.; Waqar, A.B.; Nishijima, K.; Ning, B.; Kitajima, S.; Matsuhisa, F.; Chen, L.; Liu, E.; Koike, T.; Yu, Y.; et al. Macrophage-Derived MMP-9 Enhances the Progression of Atherosclerotic Lesions and Vascular Calcification in Transgenic Rabbits. J. Cell Mol. Med. 2020, 24, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Ielapi, N.; Minici, R.; Bevacqua, E.; Ciranni, S.; Cristodoro, L.; Torcia, G.; Di Taranto, M.D.; Bracale, U.M.; Andreucci, M.; et al. Metalloproteinases between History, Health, Disease, and the Complex Dimension of Social Determinants of Health. J. Vasc. Dis. 2023, 2, 282–298. [Google Scholar] [CrossRef]
- Crascì, L.; Lauro, M.R.; Puglisi, G.; Panico, A. Natural Antioxidant Polyphenols on Inflammation Management: Anti-Glycation Activity vs Metalloproteinases Inhibition. Crit. Rev. Food Sci. Nutr. 2018, 58, 893–904. [Google Scholar] [CrossRef]
- Ahmed, A.; Ahmed, S.; Arvidsson, M.; Bouzina, H.; Lundgren, J.; Rådegran, G. Prolargin and Matrix Metalloproteinase-2 in Heart Failure after Heart Transplantation and Their Association with Haemodynamics. ESC Heart Fail. 2019, 7, 223. [Google Scholar] [CrossRef]
- Aminuddin, A.; Cheong, S.S.; Roos, N.A.C.; Ugusman, A. Smoking and Unstable Plaque in Acute Coronary Syndrome: A Systematic Review of The Role of Matrix Metalloproteinases. Int. J. Med. Sci. 2023, 20, 482–492. [Google Scholar] [CrossRef]
- Belo, V.A.; Guimarães, D.A.; Castro, M.M. Matrix Metalloproteinase 2 as a Potential Mediator of Vascular Smooth Muscle Cell Migration and Chronic Vascular Remodeling in Hypertension. J. Vasc. Res. 2015, 52, 221–231. [Google Scholar] [CrossRef]
- Baghirova, S.; Hughes, B.G.; Poirier, M.; Kondo, M.Y.; Schulz, R. Nuclear Matrix Metalloproteinase-2 in the Cardiomyocyte and the Ischemic-Reperfused Heart. J. Mol. Cell. Cardiol. 2016, 94, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.A.; Williams, H.; George, S.J. Evidence for the Involvement of Matrix-Degrading Metalloproteinases (MMPs) in Atherosclerosis. Prog. Mol. Biol. Transl. Sci. 2017, 147, 197–237. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Benko, C.; Gill, S.E.; Dufour, A. The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals 2020, 14, 31. [Google Scholar] [CrossRef]
- Kobusiak-Prokopowicz, M.; Krzysztofik, J.; Kaaz, K.; Jolda-Mydlowska, B.; Mysiak, A. MMP-2 and TIMP-2 in Patients with Heart Failure and Chronic Kidney Disease. Open Med. 2018, 13, 237–246. [Google Scholar] [CrossRef]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Lee, T.Y.T.; Mahmud, Z.; Schulz, R. Junctophilin-2 Is a Target of Matrix Metalloproteinase-2 in Myocardial Ischemia–Reperfusion Injury. Basic Res. Cardiol. 2019, 114, 42. [Google Scholar] [CrossRef]
- Prado, A.F.; Batista, R.I.M.; Tanus-Santos, J.E.; Gerlach, R.F. Matrix Metalloproteinases and Arterial Hypertension: Role of Oxidative Stress and Nitric Oxide in Vascular Functional and Structural Alterations. Biomolecules 2021, 11, 585. [Google Scholar] [CrossRef]
- Bräuninger, H.; Krüger, S.; Bacmeister, L.; Nyström, A.; Eyerich, K.; Westermann, D.; Lindner, D. Matrix Metalloproteinases in Coronary Artery Disease and Myocardial Infarction. Basic Res. Cardiol. 2023, 118, 18. [Google Scholar] [CrossRef] [PubMed]
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. The Complex Role of Matrix Metalloproteinase-2 (MMP-2) in Health and Disease. Int. J. Mol. Sci. 2024, 25, 13691. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Tscheuschler, A.; Laschinski, P.; Uffelmann, X.; Discher, P.; Fuchs, J.; Kreibich, M.; Peyronnet, R.; Kari, F.A. A Potential Key Mechanism in Ascending Aortic Aneurysm Development: Detection of a Linear Relationship between MMP-14/TIMP-2 Ratio and Active MMP-2. PLoS ONE 2019, 14, e0212859. [Google Scholar] [CrossRef]
- Młynarczyk, G.; Tokarzewicz, A.; Gudowska-Sawczuk, M.; Mroczko, B.; Novák, V.; Novák, A.; Mitura, P.; Romanowicz, L. MMP-14 Exhibits Greater Expression, Content and Activity Compared to MMP-15 in Human Renal Carcinoma. Int. J. Mol. Sci. 2024, 25, 8107. [Google Scholar] [CrossRef]
- Mariaule, V.; Kriaa, A.; Soussou, S.; Rhimi, S.; Boudaya, H.; Hernandez, J.; Maguin, E.; Lesner, A.; Rhimi, M. Digestive Inflammation: Role of Proteolytic Dysregulation. Int. J. Mol. Sci. 2021, 22, 2817. [Google Scholar] [CrossRef]
- Boumiza, S.; Chahed, K.; Tabka, Z.; Jacob, M.-P.; Norel, X.; Ozen, G. MMPs and TIMPs Levels Are Correlated with Anthropometric Parameters, Blood Pressure, and Endothelial Function in Obesity. Sci. Rep. 2021, 11, 20052. [Google Scholar] [CrossRef]
- Yamamoto, K.; Okano, H.; Miyagawa, W.; Visse, R.; Shitomi, Y.; Santamaria, S.; Dudhia, J.; Troeberg, L.; Strickland, D.K.; Hirohata, S.; et al. MMP-13 Is Constitutively Produced in Human Chondrocytes and Co-Endocytosed with ADAMTS-5 and TIMP-3 by the Endocytic Receptor LRP1. Matrix Biol. 2016, 56, 57–73. [Google Scholar] [CrossRef]
- Kudelski, J.; Młynarczyk, G.; Darewicz, B.; Bruczko-Goralewska, M.; Romanowicz, L. Dominative Role of MMP-14 over MMP-15 in Human Urinary Bladder Carcinoma on the Basis of Its Enhanced Specific Activity. Medicine 2020, 99, e19224. [Google Scholar] [CrossRef]
- Gan, J.; Eisen, C.; Smider, V. Identification of Potential Antibody Epitopes in MMP-15. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kaminski, A.R.; Moore, E.T.; Daseke, M.J.; Valerio, F.M.; Flynn, E.R.; Lindsey, M.L. The Compendium of Matrix Metalloproteinase Expression in the Left Ventricle of Mice Following Myocardial Infarction. Am. J. Physiol.-Heart Circ. Physiol. 2020, 318, H706–H714. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Iwanaga, S.; Mochizuki, S.; Okamoto, H.; Ogawa, S.; Okada, Y. Targeted Deletion or Pharmacological Inhibition of MMP-2 Prevents Cardiac Rupture after Myocardial Infarction in Mice. J. Clin. Investig. 2005, 115, 599–609. [Google Scholar] [CrossRef]
- Matsusaka, H.; Ikeuchi, M.; Matsushima, S.; Ide, T.; Kubota, T.; Feldman, A.M.; Takeshita, A.; Sunagawa, K.; Tsutsui, H. Selective Disruption of MMP-2 Gene Exacerbates Myocardial Inflammation and Dysfunction in Mice with Cytokine-Induced Cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2005, 289, H1858–H1864. [Google Scholar] [CrossRef]
- Luttun, A.; Lutgens, E.; Manderveld, A.; Maris, K.; Collen, D.; Carmeliet, P.; Moons, L. Loss of Matrix Metalloproteinase-9 or Matrix Metalloproteinase-12 Protects Apolipoprotein E–Deficient Mice Against Atherosclerotic Media Destruction but Differentially Affects Plaque Growth. Circulation 2004, 109, 1408–1414. [Google Scholar] [CrossRef]
- Amor, M.; Bianco, V.; Buerger, M.; Lechleitner, M.; Vujić, N.; Dobrijević, A.; Akhmetshina, A.; Pirchheim, A.; Schwarz, B.; Pessentheiner, A.R.; et al. Genetic Deletion of MMP12 Ameliorates Cardiometabolic Disease by Improving Insulin Sensitivity, Systemic Inflammation, and Atherosclerotic Features in Mice. Cardiovasc. Diabetol. 2023, 22, 327. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.M.; Buda, S.J.; Fiotta, N.; Xiong, W.; Griener, T.; Shapiro, S.; Baxter, B.T. MMP-12 Has a Role in Abdominal Aortic Aneurysms in Mice. Surgery 2005, 137, 457–462. [Google Scholar] [CrossRef]
- Ben Braiek, A.; Chahed, H.; Dumont, F.; Abdelhak, F.; Hichem, D.; Gamra, H.; Baudin, B. Identification of Biomarker Panels as Predictors of Severity in Coronary Artery Disease. J. Cell. Mol. Med. 2021, 25, 1518–1530. [Google Scholar] [CrossRef]
- Kondapalli, M.S.; Galimudi, R.K.; Gundapaneni, K.K.; Padala, C.; Cingeetham, A.; Gantala, S.; Ali, A.; Shyamala, N.; Sahu, S.K.; Nallari, P.; et al. MMP 1 Circulating Levels and Promoter Polymorphism in Risk Prediction of Coronary Artery Disease in Asymptomatic First Degree Relatives. Gene 2016, 595, 115–120. [Google Scholar] [CrossRef]
- Akbari, T.; Kazemi Fard, T.; Fadaei, R.; Rostami, R.; Moradi, N.; Movahedi, M.; Fallah, S. Evaluation of MMP-9, IL-6, TNF-α Levels and Peripheral Blood Mononuclear Cells Genes Expression of MMP-9 and TIMP-1 in Iranian Patients with Coronary Artery Disease. J. Cardiovasc. Thorac. Res. 2023, 15, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ezhov, M.; Safarova, M.; Afanasieva, O.; Mitroshkin, M.; Matchin, Y.; Pokrovsky, S. Matrix Metalloproteinase 9 as a Predictor of Coronary Atherosclerotic Plaque Instability in Stable Coronary Heart Disease Patients with Elevated Lipoprotein(a) Levels. Biomolecules 2019, 9, 129. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.R.; Nascimento, L.D.; Gerlach, R.F.; Rodrigues, K.E.; Prado, A.F. Matrix Metalloproteinase 2 as a Pharmacological Target in Heart Failure. Pharmaceuticals 2022, 15, 920. [Google Scholar] [CrossRef]
- Matilla, L.; Roncal, C.; Ibarrola, J.; Arrieta, V.; García-Peña, A.; Fernández-Celis, A.; Navarro, A.; Álvarez, V.; Gainza, A.; Orbe, J.; et al. A Role for MMP-10 (Matrix Metalloproteinase-10) in Calcific Aortic Valve Stenosis. ATVB 2020, 40, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Meng, X.; Li, F.; Venardos, N.; Fullerton, D.; Jaggers, J. MMP-12–Induced Pro-Osteogenic Responses in Human Aortic Valve Interstitial Cells. J. Surg. Res. 2019, 235, 44–51. [Google Scholar] [CrossRef]
- Ebert, S.; Zang, L.; Ismail, N.; Otabil, M.; Fröhlich, A.; Egea, V.; Ács, S.; Hoeberg, M.; Berres, M.-L.; Weber, C.; et al. Tissue Inhibitor of Metalloproteinases-1 Interacts with CD74 to Promote AKT Signaling, Monocyte Recruitment Responses, and Vascular Smooth Muscle Cell Proliferation. Cells 2023, 12, 1899. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.K.; Wang, Y.; Flynn, L.K.; Turner, S.E.; Rade, J.J.; Kimmelstiel, C.D.; Gurbel, P.A.; Bliden, K.P.; Covic, L.; Kuliopulos, A. Deficiency of MMP1a (Matrix Metalloprotease 1a) Collagenase Suppresses Development of Atherosclerosis in Mice: Translational Implications for Human Coronary Artery Disease. ATVB 2021, 41, e265–e279. [Google Scholar] [CrossRef]
- Lahdentausta, L.; Leskelä, J.; Winkelmann, A.; Tervahartiala, T.; Sorsa, T.; Pesonen, E.; Pussinen, P.J. Serum MMP-9 Diagnostics, Prognostics, and Activation in Acute Coronary Syndrome and Its Recurrence. J. Cardiovasc. Trans. Res. 2018, 11, 210–220. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Feng, Y.; Dong, G.; Wang, Y.; Yang, J. The Role of Matrix Metalloproteinase-9 in Atherosclerotic Plaque Instability. Mediat. Inflamm. 2020, 2020, 3872367. [Google Scholar] [CrossRef]
- Kremastiotis, G.; Handa, I.; Jackson, C.; George, S.; Johnson, J. Disparate Effects of MMP and TIMP Modulation on Coronary Atherosclerosis and Associated Myocardial Fibrosis. Sci. Rep. 2021, 11, 23081. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wei, R.; Wang, L.; Lu, J.; Liu, H.; Zhang, W. Correlations of MMP-1, MMP-3, and MMP-12 with the Degree of Atherosclerosis, Plaque Stability and Cardiovascular and Cerebrovascular Events. Exp. Ther. Med. 2017, 15, 1994. [Google Scholar] [CrossRef]
- Cavusoglu, E.; Marmur, J.D.; Hegde, S.; Yanamadala, S.; Batuman, O.A.; Chopra, V.; Ay, G.; Eng, C. Relation of Baseline Plasma MMP-1 Levels to Long-Term All-Cause Mortality in Patients with Known or Suspected Coronary Artery Disease Referred for Coronary Angiography. Atherosclerosis 2015, 239, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M.; Greif, M.; Broedl, U.C.; Lebherz, C.; Laubender, R.P.; Becker, A.; Von Ziegler, F.; Tittus, J.; Reiser, M.; Becker, C.; et al. MMP-1 Serum Levels Predict Coronary Atherosclerosis in Humans. Cardiovasc. Diabetol. 2009, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Falcinelli, E.; Momi, S.; Petito, E.; Sebastiano, M. Platelets and Matrix Metalloproteinases: A Bidirectional Interaction with Multiple Pathophysiologic Implications. Hamostaseologie 2021, 41, 136–145. [Google Scholar] [CrossRef]
- Abbas, A.; Aukrust, P.; Russell, D.; Krohg-Sørensen, K.; Almås, T.; Bundgaard, D.; Bjerkeli, V.; Sagen, E.L.; Michelsen, A.E.; Dahl, T.B.; et al. Matrix Metalloproteinase 7 Is Associated with Symptomatic Lesions and Adverse Events in Patients with Carotid Atherosclerosis. PLoS ONE 2014, 9, e84935. [Google Scholar] [CrossRef]
- Williams, H.; Johnson, J.L.; Jackson, C.L.; White, S.J.; George, S.J. MMP-7 Mediates Cleavage of N-Cadherin and Promotes Smooth Muscle Cell Apoptosis. Cardiovasc. Res. 2010, 87, 137–146. [Google Scholar] [CrossRef]
- LaFramboise, W.A.; Dhir, R.; Kelly, L.A.; Petrosko, P.; Krill-Burger, J.M.; Sciulli, C.M.; Lyons-Weiler, M.A.; Chandran, U.R.; Lomakin, A.; Masterson, R.V.; et al. Serum Protein Profiles Predict Coronary Artery Disease in Symptomatic Patients Referred for Coronary Angiography. BMC Med. 2012, 10, 157. [Google Scholar] [CrossRef]
- Kaptoge, S.; Seshasai, S.R.K.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.O.; et al. Inflammatory Cytokines and Risk of Coronary Heart Disease: New Prospective Study and Updated Meta-Analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; George, S.J.; Newby, A.C.; Jackson, C.L. Divergent Effects of Matrix Metalloproteinases 3, 7, 9, and 12 on Atherosclerotic Plaque Stability in Mouse Brachiocephalic Arteries. Proc. Natl. Acad. Sci. USA 2005, 102, 15575–15580. [Google Scholar] [CrossRef]
- Deng, H.; Li, Q.; Zhu, D. Therapeutic Effects of Allopurinol on the Function of Left Ventricular and Activity of Matrix Metalloproteinase Enzymes (MMPs) in Patients with Chronic Heart Failure. Cell. Mol. Biol. 2022, 68, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Kassiri, Z. Biology of Tissue Inhibitor of Metalloproteinase 3 (TIMP3), and Its Therapeutic Implications in Cardiovascular Pathology. Front. Physiol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Giannakos, E.; Vardali, E.; Bartekova, M.; Fogarassyova, M.; Barancik, M.; Radosinska, J. Changes in Activities of Circulating MMP-2 and MMP-9 in Patients Suffering from Heart Failure in Relation to Gender, Hypertension and Treatment: A Cross-Sectional Study. Physiol. Res. 2016, 65 (Suppl. S1), S149–S152. [Google Scholar] [CrossRef]
- Fang, L.; Murphy, A.J.; Dart, A.M. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front. Pharmacol. 2017, 8, 186. [Google Scholar] [CrossRef]
- Nandi, S.S.; Katsurada, K.; Sharma, N.M.; Anderson, D.R.; Mahata, S.K.; Patel, K.P. MMP9 Inhibition Increases Autophagic Flux in Chronic Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1414–H1437. [Google Scholar] [CrossRef]
- Rodrigues, K.E.; Pontes, M.H.B.; Cantão, M.B.S.; Prado, A.F. The Role of Matrix Metalloproteinase-9 in Cardiac Remodeling and Dysfunction and as a Possible Blood Biomarker in Heart Failure. Pharmacol. Res. 2024, 206, 107285. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Myocardial Injury, Repair, and Remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Katsi, V.; Magkas, N.; Antonopoulos, A.; Trantalis, G.; Toutouzas, K.; Tousoulis, D. Aortic Valve: Anatomy and Structure and the Role of Vasculature in the Degenerative Process. Acta Cardiol. 2021, 76, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-J.; Razavian, M.; Challa, A.A.; Nie, L.; Golestani, R.; Zhang, J.; Ye, Y.; Russell, K.S.; Robinson, S.P.; Heistad, D.D.; et al. Multimodality and Molecular Imaging of Matrix Metalloproteinase Activation in Calcific Aortic Valve Disease. J. Nucl. Med. 2015, 56, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, P.W.G.F.G.; Moreira, J.L.D.S.; Silva, L.L.D.; Oliveira, B.F.; Da Silva, J.A.; Pinto, S.S.A.; Costa, L.L.F.D.; Pinheiro, S.D.F.L. Endogenous Synthesis of Collagen and Its Relation to Aging: A Systematic Review / Síntese Endógena Do Colágeno e Sua Relação Com o Envelhecimento: Uma Revisão Sistemática. IDonline 2022, 16, 495–514. [Google Scholar] [CrossRef]
- Park, J.Y.; Ryu, S.K.; Choi, J.W.; Kim, M.H.; Jun, J.H.; Rha, S.; Park, S.; Kim, H.J.; Choi, B.G.; Noh, Y.; et al. Association of Inflammation, Myocardial Fibrosis and Cardiac Remodelling in Patients with Mild Aortic Stenosis as Assessed by Biomarkers and Echocardiography. Clin. Exp. Pharmacol. Physiol. 2014, 41, 185–191. [Google Scholar] [CrossRef]
- Drăgan, A.; Mateescu, A.D. Novel Biomarkers and Advanced Cardiac Imaging in Aortic Stenosis: Old and New. Biomolecules 2023, 13, 1661. [Google Scholar] [CrossRef]
- Lurins, J.; Lurina, D.; Svirskis, S.; Nora-Krukle, Z.; Tretjakovs, P.; Mackevics, V.; Lejnieks, A.; Rapisarda, V.; Baylon, V. Impact of Several Proinflammatory and Cell Degradation Factors in Patients with Aortic Valve Stenosis. Exp. Ther. Med. 2019, 17, 2433–2442. [Google Scholar] [CrossRef]
- Bäz, L.; Dannberg, G.; Grün, K.; Westphal, J.; Möbius-Winkler, S.; Jung, C.; Pfeil, A.; Schulze, P.C.; Franz, M. Serum Biomarkers of Cardiovascular Remodelling Reflect Extra-Valvular Cardiac Damage in Patients with Severe Aortic Stenosis. Int. J. Mol. Sci. 2020, 21, 4174. [Google Scholar] [CrossRef]
- Liang, X.; Li, Y.; Wang, P.; Liu, H. Key Regulators of Vascular Calcification in Chronic Kidney Disease: Hyperphosphatemia, BMP2, and RUNX2. PeerJ 2024, 12, e18063. [Google Scholar] [CrossRef]
- Dharmarajan, S.; Speer, M.Y.; Pierce, K.; Lally, J.; Leaf, E.M.; Lin, M.-E.; Scatena, M.; Giachelli, C.M. Role of Runx2 in Calcific Aortic Valve Disease in Mouse Models. Front. Cardiovasc. Med. 2021, 8, 687210. [Google Scholar] [CrossRef] [PubMed]
- Matilla, L.; Garaikoetxea, M.; Arrieta, V.; García-Peña, A.; Fernández-Celis, A.; Navarro, A.; Gainza, A.; Álvarez, V.; Sádaba, R.; Jover, E.; et al. Sex-Differences in Aortic Stenosis: Mechanistic Insights and Clinical Implications. Front. Cardiovasc. Med. 2022, 9, 818371. [Google Scholar] [CrossRef]
- Di Nubila, A.; Dilella, G.; Simone, R.; Barbieri, S.S. Vascular Extracellular Matrix in Atherosclerosis. Int. J. Mol. Sci. 2024, 25, 12017. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tay, F.R.; Yiu, C.K.Y. The Past, Present and Future Perspectives of Matrix Metalloproteinase Inhibitors. Pharmacol. Ther. 2020, 207, 107465. [Google Scholar] [CrossRef]
- Atkinson, G.; Bianco, R.; Di Gregoli, K.; Johnson, J.L. The Contribution of Matrix Metalloproteinases and Their Inhibitors to the Development, Progression, and Rupture of Abdominal Aortic Aneurysms. Front. Cardiovasc. Med. 2023, 10, 1248561. [Google Scholar] [CrossRef]
- Bassiouni, W.; Mahmud, Z.; Simmen, T.; Seubert, J.M.; Schulz, R. MMP-2 Inhibition Attenuates ER Stress-Mediated Cell Death during Myocardial Ischemia-Reperfusion Injury by Preserving IRE1α. J. Mol. Cell. Cardiol. 2025, 198, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Sergi, C.; Keschrumrus, V.; Churko, J.M.; Granzier, H.; Schulz, R. MMP Inhibitors Attenuate Doxorubicin Cardiotoxicity by Preventing Intracellular and Extracellular Matrix Remodelling. Cardiovasc. Res. 2021, 117, 188–200. [Google Scholar] [CrossRef]
- Gömöri, K.; Szabados, T.; Kenyeres, É.; Pipis, J.; Földesi, I.; Siska, A.; Dormán, G.; Ferdinandy, P.; Görbe, A.; Bencsik, P. Cardioprotective Effect of Novel Matrix Metalloproteinase Inhibitors. Int. J. Mol. Sci. 2020, 21, 6990. [Google Scholar] [CrossRef]
- Bencsik, P.; Kupai, K.; Görbe, A.; Kenyeres, É.; Varga, Z.V.; Pálóczi, J.; Gáspár, R.; Kovács, L.; Weber, L.; Takács, F.; et al. Development of Matrix Metalloproteinase-2 Inhibitors for Cardioprotection. Front. Pharmacol. 2018, 9, 296. [Google Scholar] [CrossRef]
- Wang, L.L.; Chung, J.J.; Li, E.C.; Uman, S.; Atluri, P.; Burdick, J.A. Injectable and Protease-Degradable Hydrogel for siRNA Sequestration and Triggered Delivery to the Heart. J. Control. Release 2018, 285, 152–161. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Qin, X.; Liu, M.; Sun, W. Morin, a Matrix Metalloproteinase 9 Inhibitor, Attenuates Endothelial-to-Mesenchymal Transition in Atherosclerosis by Downregulating Notch-1 Signaling. J. Integr. Med. 2024, 22, 683–695. [Google Scholar] [CrossRef]
- Alam, A.; Subhan, N. Morin, a Promising Phytochemicals for the Treatment of Diabetes and Dyslipidemia. 2023. Available online: https://www.researchgate.net/profile/Md-Alam-44/publication/376315680_Morin_a_promising_phytochemicals_for_the_treatment_of_diabetes_and_dyslipidemia/links/6573093bfc4b416622a852ca/Morin-a-promising-phytochemicals-for-the-treatment-of-diabetes-and-dyslipidemia.pdf (accessed on 21 February 2025).
- Eckhouse, S.R.; Purcell, B.P.; McGarvey, J.R.; Lobb, D.; Logdon, C.B.; Doviak, H.; O’Neill, J.W.; Shuman, J.A.; Novack, C.P.; Zellars, K.N.; et al. Local Hydrogel Release of Recombinant TIMP-3 Attenuates Adverse Left Ventricular Remodeling After Experimental Myocardial Infarction. Sci. Transl. Med. 2014, 6, 223ra21. [Google Scholar] [CrossRef]
- Paghdar, S.; Khan, T.M.; Patel, N.P.; Chandrasekaran, S.; De Sousa, J.F.M.; Tsouklidis, N. Doxycycline Therapy for Abdominal Aortic Aneurysm: Inhibitory Effect on Matrix Metalloproteinases. Cureus 2021, 13, e14966. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.L.; Duryee, M.J.; Hall, J.H.; Thiele, G.M.; Mikuls, T.R.; Klassen, L.W.; Zimmerman, M.C.; Anderson, D.R. Relevance of the Antioxidant Properties of Methotrexate and Doxycycline to Their Treatment of Cardiovascular Disease. Pharmacol. Ther. 2020, 205, 107413. [Google Scholar] [CrossRef]
- Parente, J.M.; Blascke De Mello, M.M.; Silva, P.H.L.D.; Omoto, A.C.M.; Pernomian, L.; Oliveira, I.S.D.; Mahmud, Z.; Fazan, R.; Arantes, E.C.; Schulz, R.; et al. MMP Inhibition Attenuates Hypertensive Eccentric Cardiac Hypertrophy and Dysfunction by Preserving Troponin I and Dystrophin. Biochem. Pharmacol. 2021, 193, 114744. [Google Scholar] [CrossRef]
- Skrzypiec-Spring, M.; Urbaniak, J.; Sapa-Wojciechowska, A.; Pietkiewicz, J.; Orda, A.; Karolko, B.; Danielewicz, R.; Bil-Lula, I.; Woźniak, M.; Schulz, R.; et al. Matrix Metalloproteinase-2 Inhibition in Acute Ischemia-Reperfusion Heart Injury—Cardioprotective Properties of Carvedilol. Pharmaceuticals 2021, 14, 1276. [Google Scholar] [CrossRef] [PubMed]
- Mendes, A.S.; Blascke De Mello, M.M.; Parente, J.M.; Omoto, A.C.M.; Neto-Neves, E.M.; Fazan, R.; Tanus-Santos, J.E.; Castro, M.M. Verapamil Decreases Calpain-1 and Matrix Metalloproteinase-2 Activities and Improves Hypertension-Induced Hypertrophic Cardiac Remodeling in Rats. Life Sci. 2020, 244, 117153. [Google Scholar] [CrossRef]
- Olejarz, W.; Łacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- Almeida, S.O.; Budoff, M. Effect of Statins on Atherosclerotic Plaque. Trends Cardiovasc. Med. 2019, 29, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Henein, M.Y.; Vancheri, S.; Longo, G.; Vancheri, F. The Role of Inflammation in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 12906. [Google Scholar] [CrossRef] [PubMed]
- Skrzypiec-Spring, M.; Sapa-Wojciechowska, A.; Rak-Pasikowska, A.; Kaczorowski, M.; Bil-Lula, I.; Hałoń, A.; Szeląg, A. The Protective Effect of Simvastatin on the Systolic Function of the Heart in the Model of Acute Ischemia and Reperfusion Is Due to Inhibition of the RhoA Pathway and Independent of Reduction of MMP-2 Activity. Biomolecules 2022, 12, 1291. [Google Scholar] [CrossRef] [PubMed]
- Murad, H. Effect of Eucalyptol on Matrix Metalloproteinase-9 and Its Tissue Inhibitor in Hypertensive Rats. Bioinformation 2023, 19, 562–564. [Google Scholar] [CrossRef]
- Du, H.; Li, X.; Zhao, W.; Jiang, N. The Difference between Sacubitril Valsartan and Valsartan on Vascular Endothelial Function, APN, MMP-9, and BNP Levels in Patients with Hypertension and Chronic Heart Failure. J. Healthc. Eng. 2022, 2022, 9494981. [Google Scholar] [CrossRef]
- Lindsey, M.L.; De Castro Brás, L.E. Matrix Metalloproteinase-9-Dependent Mechanisms of Reduced Contractility and Increased Stiffness in the Aging Heart. In Fibrosis in Disease; Molecular and Translational Medicine; Willis, M.S., Yates, C.C., Schisler, J.C., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 335–347. ISBN 978-3-319-98142-0. [Google Scholar]
- Korzeń, D.; Sierka, O.; Dąbek, J. Transcriptional Activity of Metalloproteinase 9 (MMP-9) and Tissue Metalloproteinase 1 (TIMP-1) Genes as a Diagnostic and Prognostic Marker of Heart Failure Due to Ischemic Heart Disease. Biomedicines 2023, 11, 2776. [Google Scholar] [CrossRef]
- Nappi, F.; Iervolino, A.; Avtaar Singh, S.S.; Chello, M. MicroRNAs in Valvular Heart Diseases: Biological Regulators, Prognostic Markers and Therapeutical Targets. Int. J. Mol. Sci. 2021, 22, 12132. [Google Scholar] [CrossRef]
- Scrimgeour, N.R.; Wrobel, A.; Pinho, M.J.; Høydal, M.A. microRNA-451a Prevents Activation of Matrix Metalloproteinases 2 and 9 in Human Cardiomyocytes during Pathological Stress Stimulation. Am. J. Physiol.-Cell Physiol. 2020, 318, C94–C102. [Google Scholar] [CrossRef]
- Guenther, C.M.; Brun, M.J.; Bennett, A.D.; Ho, M.L.; Chen, W.; Zhu, B.; Lam, M.; Yamagami, M.; Kwon, S.; Bhattacharya, N.; et al. Protease-Activatable Adeno-Associated Virus Vector for Gene Delivery to Damaged Heart Tissue. Mol. Ther. 2019, 27, 611–622. [Google Scholar] [CrossRef]
- Brown, D.L.; Desai, K.K.; Vakili, B.A.; Nouneh, C.; Lee, H.-M.; Golub, L.M. Clinical and Biochemical Results of the Metalloproteinase Inhibition with Subantimicrobial Doses of Doxycycline to Prevent Acute Coronary Syndromes (MIDAS) Pilot Trial. ATVB 2004, 24, 733–738. [Google Scholar] [CrossRef]
- Buckley, L.F.; Agha, A.M.; Dorbala, P.; Claggett, B.L.; Yu, B.; Hussain, A.; Nambi, V.; Chen, L.Y.; Matsushita, K.; Hoogeveen, R.C.; et al. MMP-2 Associates with Incident Heart Failure and Atrial Fibrillation: The ARIC Study. Circ. Heart Fail. 2023, 16, e010849. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and Atherosclerosis: Signaling Pathways and Therapeutic Intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, Y.; Liu, Y.; Gao, Y.; San, T.; Li, X.; Song, S.; Yan, B.; Zhao, Z. Advances in Application of Single-Cell RNA Sequencing in Cardiovascular Research. Front. Cardiovasc. Med. 2022, 9, 905151. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Refs. | MMP Family | MMP Members | Main Substrates | Biological Function |
|---|---|---|---|---|
| [4,7,9] | Collagenases | MMP-1, MMP-8, MMP-13, MMP-18 | Fibrillar collagens (I, II, III) | Collagen breakdown in tissue remodeling |
| [4,7,9] | Gelatinases | MMP-2, MMP-9 | Denatured collagens, gelatin | Degradation of basement membrane |
| [4,7,9] | Stromelysins | MMP-3, MMP-10, MMP-11 | ECM proteins, proteoglycans | Tissue remodeling, inflammation |
| [4,7,9] | Matrilysins | MMP-7, MMP-26 | Laminin, elastin, fibronectin | Wound healing |
| [4,7,9] | MT-MMPs | MMP-14, MMP-15, MMP-16, MMP-17 | ECM components, other MMPs | Cell invasion, pericellular proteolysis |
| [4,7,9] | Other MMPs | MMP-19, MMP-20 | Elastin, dentin matrix proteins | Vascular remodeling |
| Refs. | Cardiovascular Disease | MMPs Involved | Target |
|---|---|---|---|
| [8,36,37,38] | Atherosclerosis | MMP-1, MMP-2, MMP-3, MMP-9 | Plaque degradation, vascular inflammation |
| [19,21,39] | Myocardial Infarction | MMP-2, MMP-7, MMP-9 | ECM breakdown |
| [30,40,41] | Heart Failure | MMP-2, MMP-9, MMP-14 | Myocardial remodeling, fibrosis |
| [42,43] | Aortic Stenosis | MMP-1, MMP-9, MMP-12 | Valve calcification, ECM degradation |
| MMP Inhibitor Type | Examples | Mechanism of Action | Limitations |
|---|---|---|---|
| Synthetic inhibitors [76,77,78] | Batimastat, Marimastat | Zinc-chelation, broad-spectrum inhibition | Toxicity, off-target effects |
| Synthetic inhibitors [79,80] | ONO-4817 | Selective MMP-2 inhibitor | Limited impact on other MMPs |
| Synthetic inhibitors [81,82] | MMPI-1154 and MMPI-1260 | Selective MMP-2 inhibitor | |
| Non-selective β-adrenergic antagonist [90] | Carvedilol | Suppress MMP-2 activity | |
| Calcium channel blocker [41] | Verapamil | Inhibit MMP-2 activity | |
| Tetracyclines [78,87] | Doxycycline | Downregulation of MMP expression | Limited specificity |
| Statins [41,92,93,95] | Atorvastatin | Reducing MMP-9 levels | |
| Atorvastatin, Rosuvastatin, and Pravastatin | Reduced serum MMP-2 levels | Limited specificity, potential drug interactions, short duration | |
| Atorvastatin, Simvastatin, and Lovastatin | Inhibit activation of MMP-2 and MMP-9 | ||
| ACEi [96,98] | Lisinopril | Suppression activity on MMP-9 | Dose-dependent effects |
| Natural compounds [84,85] | Morin | Antioxidant-mediated MMP suppression | Low bioavailability |
| Gene therapy [102] | AAV9-based vectors, miRNA-451a | Inhibits MMP-2/MMP-9 expression | Indirect MMP inhibition, stability, and uptake variability |
| Ref. | Cardiovascular Disease | Subjects | Notable Effects |
|---|---|---|---|
| [44] | Atherosclerosis and coronary artery disease | Human Peripheral Blood Monocytes (PBMCs) | TIMP-1–CD74 axis in inflammatory/atherogenic responses |
| [45] | ApoE−/− mice deficient in human MMP1 ortholog, MMP1a. | Role for MMP1a in atherosclerotic lesion development | |
| [46] | Age- and gender-matched case–control study | MMP-9 and the MMP-9/TIMP-1 molar ratio may be valuable in acute coronary syndrome diagnosis and prognosis | |
| [36] | 472 patients with CAD | Combination of MMP-9, TIMP-2, and Apo-CIII values (‘CAD aggravation panel’) characterizes theseverity of CAD | |
| [48] | ApoE-deficient mice with concomitant deletion of MMP-7, MMP-9, MMP-12, or TIMP-1 | MMP-7 deficiency increased incidence of sudden deathMMP-12 deficiency promoted survivalMMP-9 or TIMP-1 deficiency had no effect | |
| [49] | Serum protein levels of MMP-1, MMP-3, and MMP-12 from patients with carotid atherosclerosis (CAS) | MMP-1, MMP-3, and MMP-12 were significantly increased and had significantly positive correlations with the occurrence of CAS | |
| [37] | Serum concentration of MMP 1 from 300 CAD patients | MMP 1 serum levels and polymorphism as potential prognostic markers for future cardiovascular events | |
| [50] | 364 male patients | Elevated levels of MMP-1 are associated with an increased risk of long-term all-causemortality | |
| [8] | MMP-9 transgenic (Tg) rabbits | Macrophage-derived MMP-9 facilitates the infiltration of monocyte/macrophages, enhancing the progression of atherosclerosis | |
| [39] | Blood samples from 32 subjects with stable coronary heart disease (CHD) and elevated lipoprotein(a) (Lp(a) | MMP-9 is a strong independent predictor ofatherosclerotic plaque instability in stable CHD patients | |
| [38] | 132 patients who underwent coronaryangiography | High levels of TNF-α and IL-6 could influence the MMP-9/TIMP-1 balance and lipid metabolism, leading to plaque formation/rupture | |
| [19] | Myocardial Infarction | Male Sprague–Dawley rats | Degradation of JPH-2 by MMP-2 is an early consequence of myocardial IR injury |
| [103] | 50 patients | Pro-MMP-9 activity reduced by 50% after Doxycycline 20 mg twice daily | |
| [14] | IR injury | Conditioned media from human fibrosarcoma HT1080 cellSprague–Dawley neonatal ratsventricular cardiomyocytes (NRVMs) | Nuclear MMP-2 activity indicates lamin proteolysis |
| [11] | Heart failure | Humans | MMP-2 may reflect aberrant ECMremodeling involved in the pathophysiology of HF and associated pulmonary hypertension |
| [60] | Venous blood samples from patients with HF | Potential inhibitory effect ofantihypertensive treatment on pro-MMP-2 activity | |
| [18] | Venous blood sample from 101 patients with chronic HF | Elevated levels of MMP-2 and TIMP-2were found in serum from patients with chronic kidney diseaseSerum levels of MMP-2 were correlated with the degree of kidney failure | |
| [62] | Male Sprague Dawley rats | Beneficial outcome of MMP-9 inhibition on pathological cardiac remodeling | |
| [104] | 4693 participants from atherosclerosis risk in community study | Higher MMP-2 levels were associated with HF and diastolic dysfunction | |
| [42] | Valvular heart disease | Blood samples from patients undergoing valvereplacement | MMP-10 plays a central role in calcificationin AS through Akt phosphorylation |
| [67] | ApoE−/− mice fed a Western diet (WD) | MMP-targeted imaging detects valvular inflammation and remodeling in CAVD | |
| [43] | Human aortic valve interstitial cells (AVICs) | MMP-12 induces pro-osteogenic responses in AVICs by activation of p38 MAPK signaling pathway | |
| [71] | Venous blood samples from patients with aortic stenosis | MMP-1 levels could indicate the development ofcalcinosis in severe stenosis | |
| [72] | Patients with aortic stenosis | MMP-9 alterations reflect the switch of extra-valvular cardiac damage from left ventricular to left atrial involvement | |
| [75] | 238 patients with severe aortic stenosis undergoing surgical valve replacement | Women exhibited increased MMP-1 and decreased TIMP-2 expression |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanase, D.M.; Valasciuc, E.; Anton, I.-B.; Gosav, E.M.; Dima, N.; Cucu, A.I.; Costea, C.F.; Floria, D.E.; Hurjui, L.L.; Tarniceriu, C.C.; et al. Matrix Metalloproteinases: Pathophysiologic Implications and Potential Therapeutic Targets in Cardiovascular Disease. Biomolecules 2025, 15, 598. https://doi.org/10.3390/biom15040598
Tanase DM, Valasciuc E, Anton I-B, Gosav EM, Dima N, Cucu AI, Costea CF, Floria DE, Hurjui LL, Tarniceriu CC, et al. Matrix Metalloproteinases: Pathophysiologic Implications and Potential Therapeutic Targets in Cardiovascular Disease. Biomolecules. 2025; 15(4):598. https://doi.org/10.3390/biom15040598
Chicago/Turabian StyleTanase, Daniela Maria, Emilia Valasciuc, Ioana-Bianca Anton, Evelina Maria Gosav, Nicoleta Dima, Andrei Ionut Cucu, Claudia Florida Costea, Diana Elena Floria, Loredana Liliana Hurjui, Claudia Cristina Tarniceriu, and et al. 2025. "Matrix Metalloproteinases: Pathophysiologic Implications and Potential Therapeutic Targets in Cardiovascular Disease" Biomolecules 15, no. 4: 598. https://doi.org/10.3390/biom15040598
APA StyleTanase, D. M., Valasciuc, E., Anton, I.-B., Gosav, E. M., Dima, N., Cucu, A. I., Costea, C. F., Floria, D. E., Hurjui, L. L., Tarniceriu, C. C., Ciocoiu, M., & Floria, M. (2025). Matrix Metalloproteinases: Pathophysiologic Implications and Potential Therapeutic Targets in Cardiovascular Disease. Biomolecules, 15(4), 598. https://doi.org/10.3390/biom15040598