
Modeling Necroptotic and Pyroptotic Signaling in Saccharomyces cerevisiae


Abstract
1. Introduction: SMOC Assembly, Innate Immunity, and Cell Death
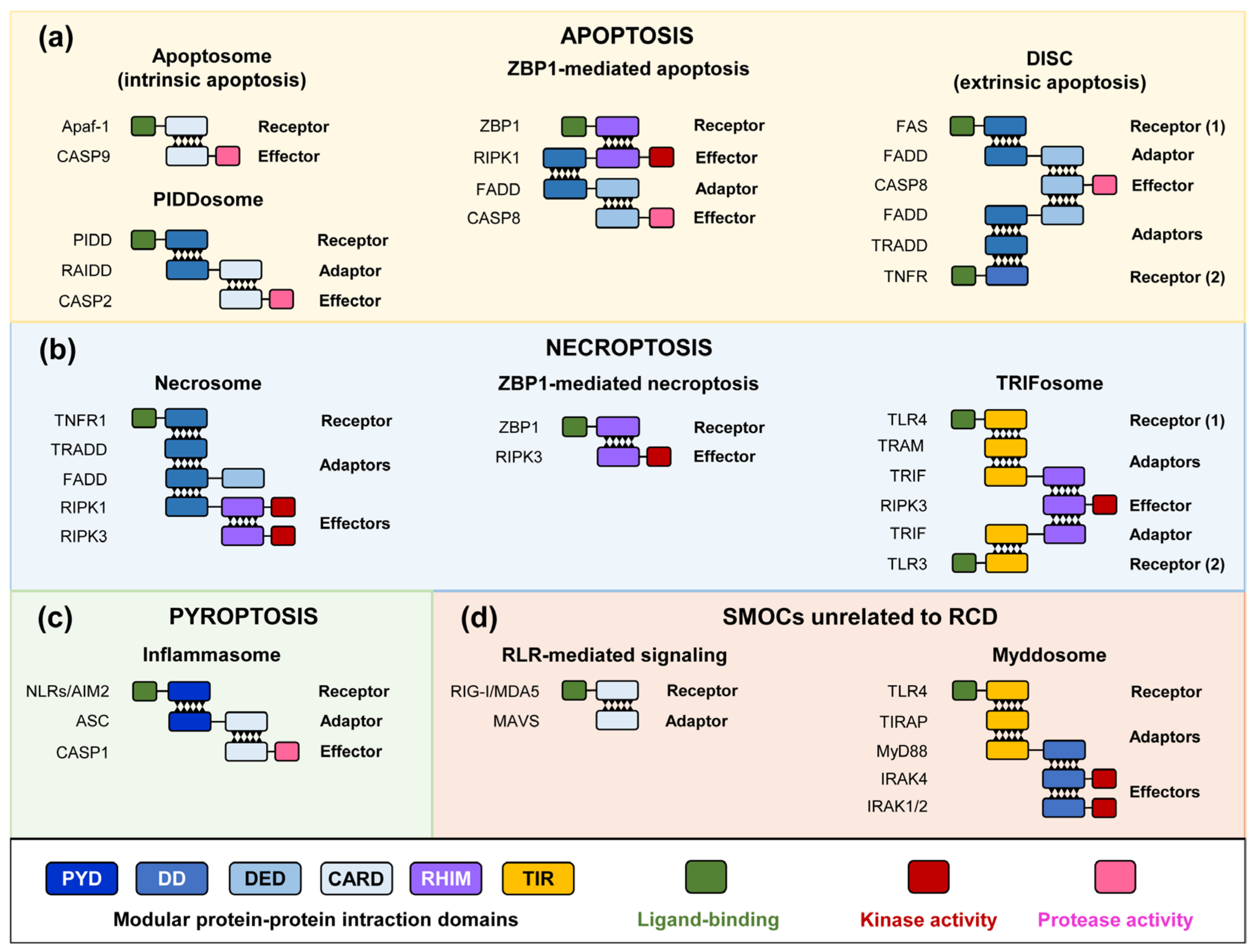
2. RCD Pathways and SMOC Assembly
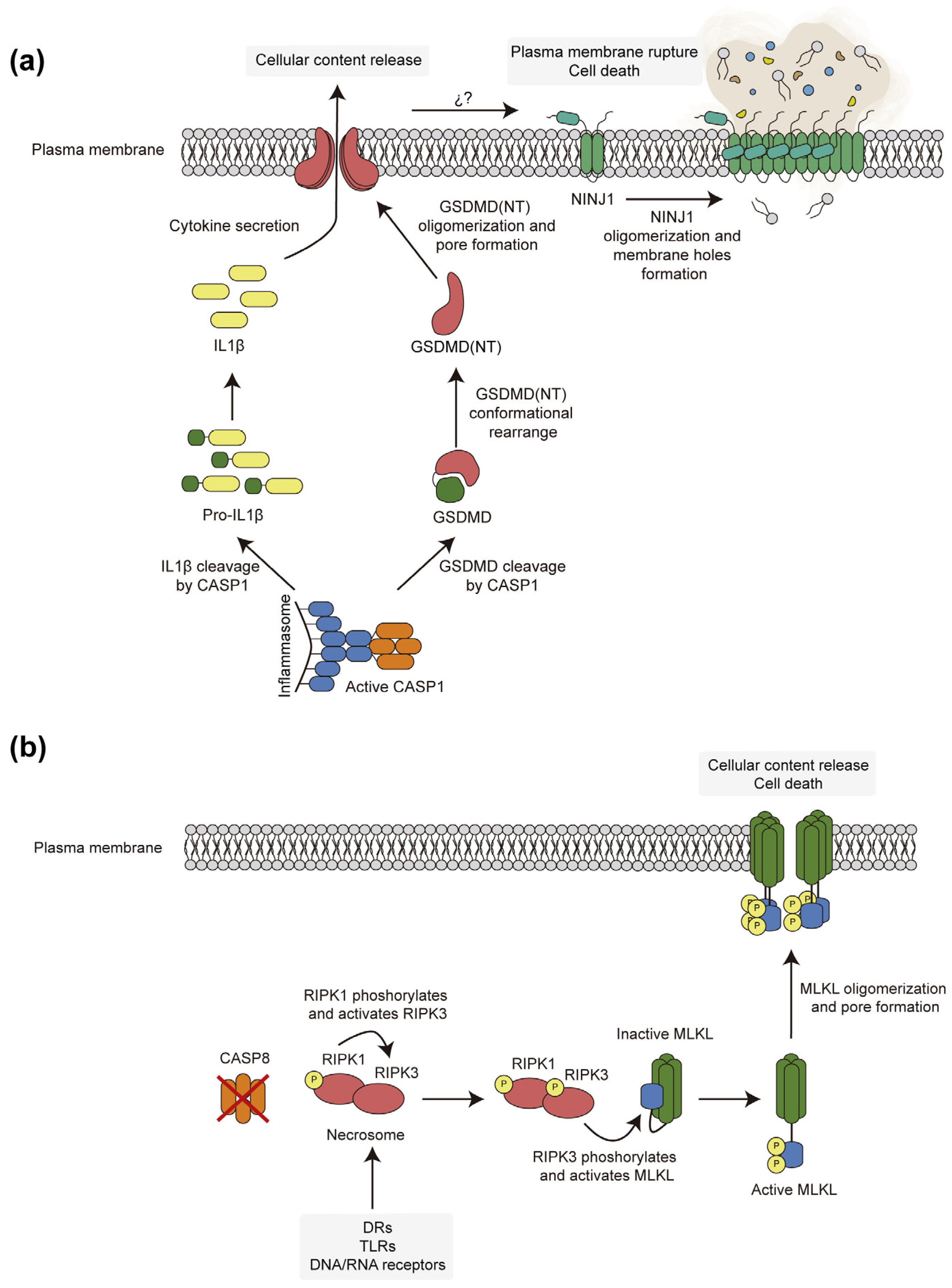
3. Pore-Forming Executors of Cell Death: Pyroptosis vs. Necroptosis
4. Modeling SMOC Assembly, Necroptosis, and Pyroptosis in Yeast
4.1. Heterologous Expression of Caspases in S. cerevisae
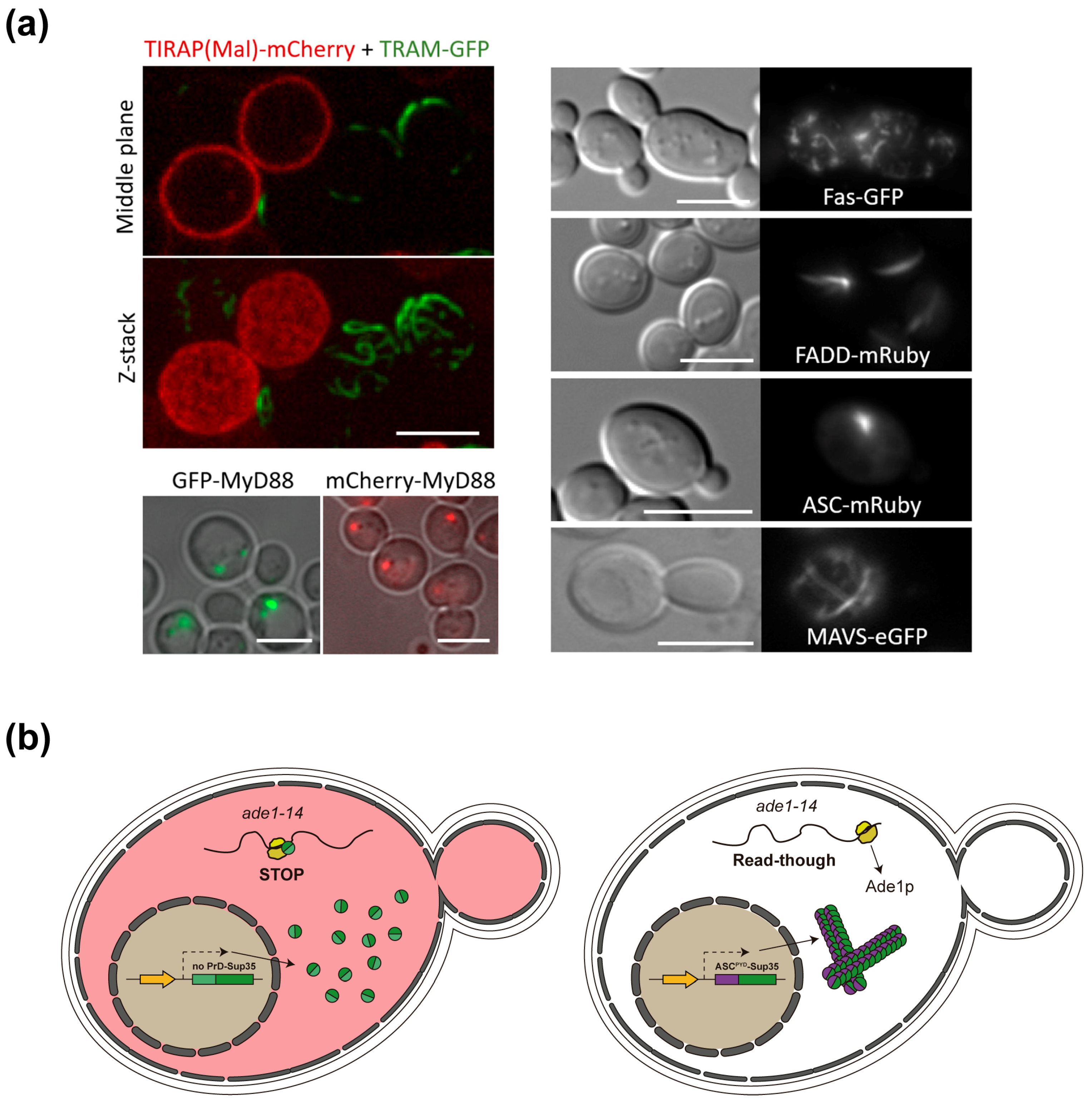
4.2. Yeast-Based Models for Inflammasome and Necrosome Assembly
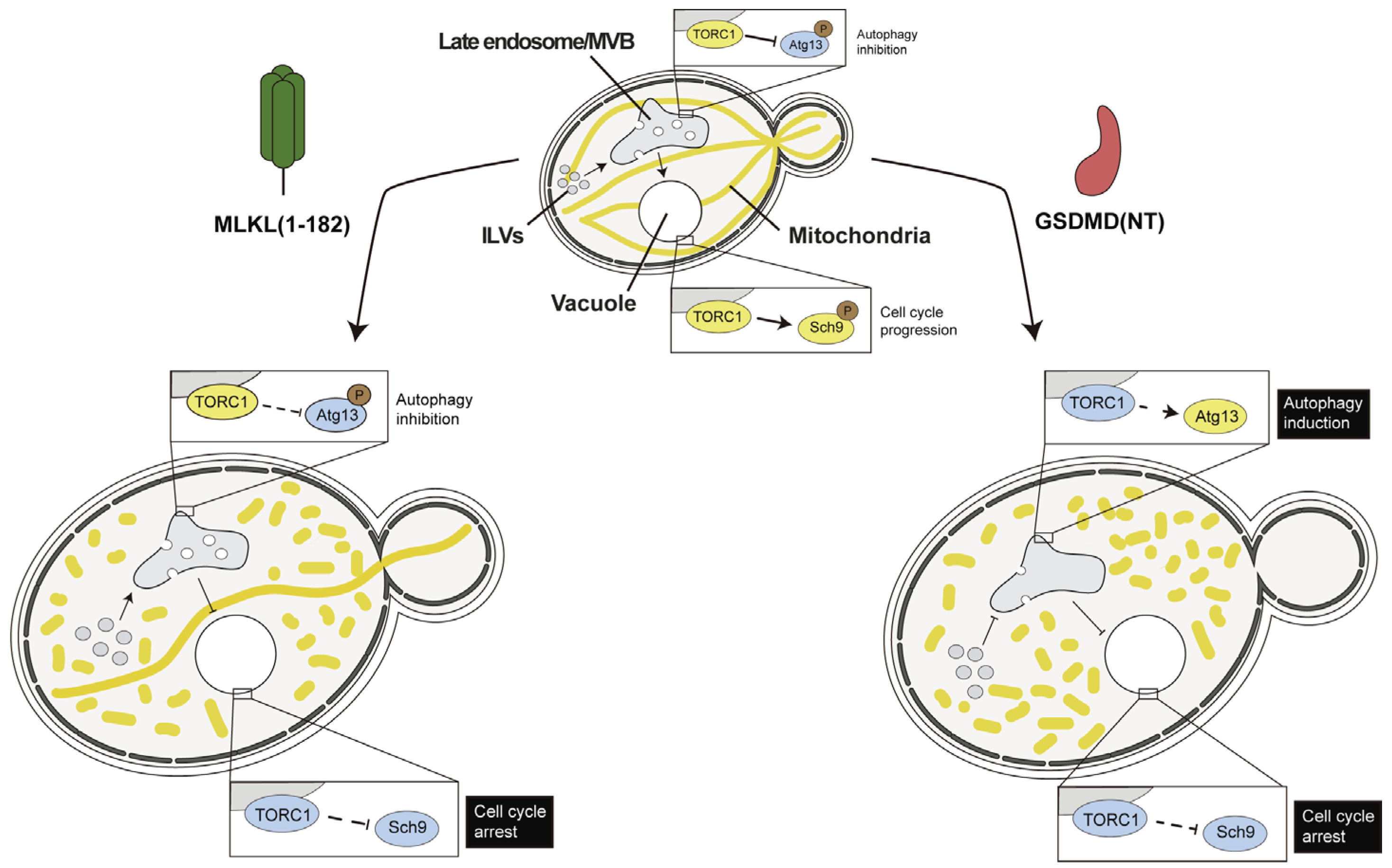
4.3. Necroptotic and Pyroptotic Pore-Forming Cell Death Executors Kill Yeast Cells by Means Other than Cell Lysis
4.4. ASC and Other SMOC Adaptor Proteins in Yeast: A Prion-Like Model
5. Caveats and Challenges of SIGNALING by Cooperative Assembly Formation (SCAF) Yeast-Based Models
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riera Romo, M. Cell Death as Part of Innate Immunity: Cause or Consequence? Immunology 2021, 163, 399–415. [Google Scholar] [CrossRef]
- Lee, E.; Song, C.-H.; Bae, S.-J.; Ha, K.-T.; Karki, R. Regulated Cell Death Pathways and Their Roles in Homeostasis, Infection, Inflammation, and Tumorigenesis. Exp. Mol. Med. 2023, 55, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Magupalli, V.G.; Wu, H. SMOCs: Supramolecular Organizing Centres That Control Innate Immunity. Nat. Rev. Immunol. 2014, 14, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Sušjan-Leite, P.; Ramuta, T.Ž.; Boršić, E.; Orehek, S.; Hafner-Bratkovič, I. Supramolecular Organizing Centers at the Interface of Inflammation and Neurodegeneration. Front. Immunol. 2022, 13, 940969. [Google Scholar] [CrossRef]
- Vajjhala, P.R.; Ve, T.; Bentham, A.; Stacey, K.J.; Kobe, B. The Molecular Mechanisms of Signaling by Cooperative Assembly Formation in Innate Immunity Pathways. Mol. Immunol. 2017, 86, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Nanson, J.D.; Rahaman, M.H.; Ve, T.; Kobe, B. Regulation of Signaling by Cooperative Assembly Formation in Mammalian Innate Immunity Signalosomes by Molecular Mimics. Semin. Cell Dev. Biol. 2020, 99, 96–114. [Google Scholar] [CrossRef]
- Nanson, J.D.; Kobe, B.; Ve, T. Death, TIR, and RHIM: Self-Assembling Domains Involved in Innate Immunity and Cell-Death Signaling. J. Leukoc. Biol. 2019, 105, 363–375. [Google Scholar] [CrossRef]
- Ha, H.J. Assembly of Platforms for Signal Transduction in the New Era: Dimerization, Helical Filament Assembly, and Beyond. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Cui, J.; Zhao, S.; Li, Y.; Zhang, D.; Wang, B.; Xie, J.; Wang, J. Regulated Cell Death: Discovery, Features and Implications for Neurodegenerative Diseases. Cell. Commun. Signal. 2021, 19, 120. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Sun, G. Death and Survival from Executioner Caspase Activation. Semin. Cell Dev. Biol. 2024, 156, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H. Structural Features of Caspase-Activating Complexes. Int. J. Mol. Sci. 2012, 13, 4807–4818. [Google Scholar] [CrossRef]
- Weiler, E.S.; Szabo, T.G.; Garcia-Carpio, I.; Villunger, A. PIDD1 in Cell Cycle Control, Sterile Inflammation and Cell Death. Biochem. Soc. Trans. 2022, 50, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Brown-Suedel, A.N.; Bouchier-Hayes, L. Caspase-2 Substrates: To Apoptosis, Cell Cycle Control, and Beyond. Front. Cell Dev. Biol. 2020, 8, 610022. [Google Scholar] [CrossRef]
- De Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrín, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, R.; Ouyang, Y.; Gu, W.; Xiao, T.; Yang, H.; Tang, L.; Wang, H.; Xiang, B.; Chen, P. Pyroptosis in Health and Disease: Mechanisms, Regulation and Clinical Perspective. Sig Transduct Target Ther 2024, 9, 245. [Google Scholar] [CrossRef]
- Horne, C.R.; Samson, A.L.; Murphy, J.M. The Web of Death: The Expanding Complexity of Necroptotic Signaling. Trends. Cell. Biol. 2023, 33, 162–174. [Google Scholar] [CrossRef]
- Petrie, E.J.; Czabotar, P.E.; Murphy, J.M. The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem. Sci. 2019, 44, 53–63. [Google Scholar] [CrossRef]
- Orning, P.; Lien, E. Multiple Roles of Caspase-8 in Cell Death, Inflammation, and Innate Immunity. J. Leukoc. Biol. 2021, 109, 121–141. [Google Scholar] [CrossRef]
- Li, M.; Beg, A.A. Induction of Necrotic-Like Cell Death by Tumor Necrosis Factor Alpha and Caspase Inhibitors: Novel Mechanism for Killing Virus-Infected Cells. J. Virol. 2000, 74, 7470–7477. [Google Scholar] [CrossRef]
- Upton, J.W.; Chan, F.K.-M. Staying Alive: Cell Death in Antiviral Immunity. Mol. Cell 2014, 54, 273–280. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.-G. Cleavage of the Death Domain Kinase RIP by Caspase-8 Prompts TNF-Induced Apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed Lineage Kinase Domain-like Is a Key Receptor Interacting Protein 3 Downstream Component of TNF-Induced Necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Dai, Y.; Wan, X.; Hu, X.; Zhao, W.; Ban, X.; Wan, H.; Huang, K.; Zhang, Q.; Xiong, K. ZBP1-Mediated Necroptosis: Mechanisms and Therapeutic Implications. Molecules 2022, 28, 52. [Google Scholar] [CrossRef]
- Bryant, C.E. Rethinking Toll-like Receptor Signalling. Curr. Opin. Immunol. 2024, 91, 102460. [Google Scholar] [CrossRef]
- Pereira, M.; Gazzinelli, R.T. Regulation of Innate Immune Signaling by IRAK Proteins. Front. Immunol. 2023, 14, 1133354. [Google Scholar] [CrossRef]
- Lin, K.-M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. IRAK-1 Bypasses Priming and Directly Links TLRs to Rapid NLRP3 Inflammasome Activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-Dependent TLR Signaling, Its Functions in Host Defense and Inflammation, and Its Potential as a Therapeutic Target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef]
- Gao, J.; Xiong, A.; Liu, J.; Li, X.; Wang, J.; Zhang, L.; Liu, Y.; Xiong, Y.; Li, G.; He, X. PANoptosis: Bridging Apoptosis, Pyroptosis, and Necroptosis in Cancer Progression and Treatment. Cancer Gene Ther. 2024, 31, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gullett, J.M.; Tweedell, R.E.; Kanneganti, T. Innate Immune Inflammatory Cell Death: PANoptosis and PANoptosomes in Host Defense and Disease. Eur. J. Immunol. 2023, 53, 2250235. [Google Scholar] [CrossRef]
- Pandian, N.; Kanneganti, T.-D. PANoptosis: A Unique Innate Immune Inflammatory Cell Death Modality. J. Immunol. 2022, 209, 1625–1633. [Google Scholar] [CrossRef]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef]
- Zhu, P.; Ke, Z.-R.; Chen, J.-X.; Li, S.-J.; Ma, T.-L.; Fan, X.-L. Advances in Mechanism and Regulation of PANoptosis: Prospects in Disease Treatment. Front. Immunol. 2023, 14, 1120034. [Google Scholar] [CrossRef]
- Nadella, V.; Kanneganti, T.-D. Inflammasomes and Their Role in PANoptosomes. Curr. Opin. Immunol. 2024, 91, 102489. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Liu, Z.; Busscher, B.M.; Storl-Desmond, M.; Xiao, T.S. Mechanisms of Gasdermin Recognition by Proteases. J. Mol. Biol. 2022, 434, 167274. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, Q.; Zhong, X.; Zeng, M.; Zeng, H.; Shi, X.; Li, Z.; Wang, Y.; Zhao, Q.; Shao, F.; et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 2020, 180, 941–955.e20. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Chai, Q.; Yu, S.; Zhong, Y.; Lu, Z.; Qiu, C.; Yu, Y.; Zhang, X.; Zhang, Y.; Lei, Z.; Qiang, L.; et al. A Bacterial Phospholipid Phosphatase Inhibits Host Pyroptosis by Hijacking Ubiquitin. Science 2022, 378, eabq0132. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ruan, J. Mechanistic Insights into Gasdermin Pore Formation and Regulation in Pyroptosis. J. Mol. Biol. 2022, 434, 167297. [Google Scholar] [CrossRef]
- Chen, X.; He, W.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and Its Morphology Is Different from MLKL Channel-Mediated Necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Du, G.; Healy, L.B.; David, L.; Walker, C.; Fontana, P.; Dong, Y.; Devant, P.; Puthenveetil, R.; Ficarro, S.B.; Banerjee, A.; et al. ROS-dependent palmitoylation is an obligate licensing modification for GSDMD pore formation. Nature 2024, 630, 437–446. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Ghimire, L.; Hsu, A.Y.; Kambara, H.; Liu, X.; Hasegawa, T.; Xu, R.; Tahir, M.; Yu, H.; Lieberman, J.; et al. Palmitoylation of gasdermin D directs its membrane translocation and pore formation in pyroptosis. Sci. Immunol. 2024, 9, eadn1452. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Borges, J.P.; Sætra, R.S.; Volchuk, A.; Bugge, M.; Devant, P.; Sporsheim, B.; Kilburn, B.R.; Evavold, C.L.; Kagan, J.C.; Goldenberg, N.M.; et al. Glycine Inhibits NINJ1 Membrane Clustering to Suppress Plasma Membrane Rupture in Cell Death. eLife 2022, 11, e78609. [Google Scholar] [CrossRef]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 Mediates Plasma Membrane Rupture during Lytic Cell Death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef]
- Degen, M.; Santos, J.C.; Pluhackova, K.; Cebrero, G.; Ramos, S.; Jankevicius, G.; Hartenian, E.; Guillerm, U.; Mari, S.A.; Kohl, B.; et al. Structural Basis of NINJ1-Mediated Plasma Membrane Rupture in Cell Death. Nature 2023, 618, 1065–1071. [Google Scholar] [CrossRef]
- Martens, S.; Bridelance, J.; Roelandt, R.; Vandenabeele, P.; Takahashi, N. MLKL in Cancer: More than a Necroptosis Regulator. Cell Death Differ. 2021, 28, 1757–1772. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Meng, Y.; Garnish, S.E.; Davies, K.A.; Black, K.A.; Leis, A.P.; Horne, C.R.; Hildebrand, J.M.; Hoblos, H.; Fitzgibbon, C.; Young, S.N.; et al. Phosphorylation-Dependent Pseudokinase Domain Dimerization Drives Full-Length MLKL Oligomerization. Nat. Commun 2023, 14, 6804. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.; Liu, Z.-G. Plasma Membrane Translocation of Trimerized MLKL Protein Is Required for TNF-Induced Necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Huang, D.; Zheng, X.; Wang, Z.; Chen, X.; He, W.; Zhang, Y.; Xu, J.-G.; Zhao, H.; Shi, W.; Wang, X.; et al. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell. Biol. 2017, 37, e00497-16. [Google Scholar] [CrossRef]
- Davies, K.A.; Tanzer, M.C.; Griffin, M.D.W.; Mok, Y.F.; Young, S.N.; Qin, R.; Petrie, E.J.; Czabotar, P.E.; Silke, J.; Murphy, J.M. The Brace Helices of MLKL Mediate Interdomain Communication and Oligomerisation to Regulate Cell Death by Necroptosis. Cell Death Differ. 2018, 25, 1567–1580. [Google Scholar] [CrossRef]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL Trafficking and Accumulation at the Plasma Membrane Control the Kinetics and Threshold for Necroptosis. Nat. Commun 2020, 11, 3151. [Google Scholar] [CrossRef]
- Ramirez, R.X.; Campbell, O.; Pradhan, A.J.; Atilla-Gokcumen, G.E.; Monje-Galvan, V. Modeling the Molecular Fingerprint of Protein-Lipid Interactions of MLKL on Complex Bilayers. Front. Chem. 2023, 10, 1088058. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Ros, U.; Garcia-Saez, A.J. Pore Formation in Regulated Cell Death. EMBO J. 2020, 39, e105753. [Google Scholar] [CrossRef]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL Forms Disulfide Bond-Dependent Amyloid-like Polymers to Induce Necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM Structure of the Gasdermin A3 Membrane Pore. Nature 2018, 557, 62–67. [Google Scholar] [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein That Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e7. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Riquet, F.; Cappe, B. Necroptosis: (Last) Message in a Bubble. Immunity 2017, 47, 1–3. [Google Scholar] [CrossRef]
- Fan, W.; Guo, J.; Gao, B.; Zhang, W.; Ling, L.; Xu, T.; Pan, C.; Li, L.; Chen, S.; Wang, H.; et al. Flotillin-Mediated Endocytosis and ALIX–Syntenin-1–Mediated Exocytosis Protect the Cell Membrane from Damage Caused by Necroptosis. Sci. Signal. 2019, 12, eaaw3423. [Google Scholar] [CrossRef]
- Gong, Y.-N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300.e16. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL Triggers the NLRP3 Inflammasome in a Cell-Intrinsic Manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef]
- Dai, J.; Zhang, C.; Guo, L.; He, H.; Jiang, K.; Huang, Y.; Zhang, X.; Zhang, H.; Wei, W.; Zhang, Y.; et al. A Necroptotic-Independent Function of MLKL in Regulating Endothelial Cell Adhesion Molecule Expression. Cell Death Dis. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Bogdanov, K.; Kovalenko, A.; Wallach, D. Necroptosis Is Preceded by Nuclear Translocation of the Signaling Proteins That Induce It. Cell Death Differ. 2016, 23, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Murphy, J.M.; Vince, J.E. Gasdermin and MLKL Necrotic Cell Death Effectors: Signaling and Diseases. Immunity 2024, 57, 429–445. [Google Scholar] [CrossRef]
- Madeo, F.; Herker, E.; Maldener, C.; Wissing, S.; Lächelt, S.; Herlan, M.; Fehr, M.; Lauber, K.; Sigrist, S.J.; Wesselborg, S.; et al. A Caspase-Related Protease Regulates Apoptosis in Yeast. Mol. Cell 2002, 9, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.K.; Sherlock, G. Yca1 Metacaspase: Diverse Functions Determine How Yeast Live and Let Die. FEMS Yeast Res. 2023, 23, foad022. [Google Scholar] [CrossRef]
- Wilkinson, D.; Ramsdale, M. Proteases and Caspase-like Activity in the Yeast Saccharomyces cerevisiae. Biochem. Soc. Trans. 2011, 39, 1502–1508. [Google Scholar] [CrossRef]
- Meitzler, J.L.; Gray, J.J.; Hendrickson, T.L. Truncation of the Caspase-Related Subunit (Gpi8p) of Saccharomyces cerevisiae GPI Transamidase: Dimerization Revealed. Arch. Biochem. Biophys. 2007, 462, 83–93. [Google Scholar] [CrossRef]
- Yang, H.; Ren, Q.; Zhang, Z. Cleavage of Mcd1 by Caspase-like Protease Esp1 Promotes Apoptosis in Budding Yeast. Mol. Biol. Cell 2008, 19, 2127–2134. [Google Scholar] [CrossRef]
- Hawkins, C.J.; Wang, S.L.; Hay, B.A. Monitoring Activity of Caspases and Their Regulators in Yeast Saccharomyces cerevisiae. In Methods in Enzymology; Elsevier: Amsterdam, The Netherland, 2000; Volume 322, pp. 162–174. ISBN 978-0-12-182223-1. [Google Scholar]
- Puryer, M.A.; Hawkins, C.J. Human, Insect and Nematode Caspases Kill Saccharomyces cerevisiae Independently of YCA1 and Aif1p. Apoptosis 2006, 11, 509–517. [Google Scholar] [CrossRef]
- Kang, J.J.; Schaber, M.D.; Srinivasula, S.M.; Alnemri, E.S.; Litwack, G.; Hall, D.J.; Bjornsti, M.A. Cascades of Mammalian Caspase Activation in the Yeast Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 3189–3198. [Google Scholar] [CrossRef]
- Hawkins, C.J.; Wang, S.L.; Hay, B.A. A Cloning Method to Identify Caspases and Their Regulators in Yeast: Identification of Drosophila IAP1 as an Inhibitor of the Drosophila Caspase DCP-1. Proc. Natl. Acad. Sci. USA. 1999, 96, 2885–2890. [Google Scholar] [CrossRef]
- Wright, M.E.; Han, D.K.; Carter, L.; Fields, S.; Schwartz, S.M.; Hockenbery, D.M. Caspase-3 Inhibits Growth in Saccharomyces Cerevisiae without Causing Cell Death. FEBS Lett. 1999, 446, 9–14. [Google Scholar] [CrossRef]
- Ho, P.; Jabbour, A.M.; Ekert, P.G.; Hawkins, C.J. Caspase-2 Is Resistant to Inhibition by Inhibitor of Apoptosis Proteins (IAPs) and Can Activate Caspase-7. FEBS J. 2005, 272, 1401–1414. [Google Scholar] [CrossRef]
- Hawkins, C.J.; Silke, J.; Verhagen, A.M.; Foster, R.; Ekert, P.G.; Ashley, D.M. Analysis of Candidate Antagonists of IAP-Mediated Caspase Inhibition Using Yeast Reconstituted with the Mammalian Apaf-1-Activated Apoptosis Mechanism. Apoptosis 2001, 6, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Lopes-Rodrigues, V.; Coutinho, I.; Neves, M.P.; Lima, R.T.; Pinto, M.; Cidade, H.; Vasconcelos, M.H.; Saraiva, L. Potential Small-Molecule Activators of Caspase-7 Identified Using Yeast-Based Caspase-3 and -7 Screening Assays. Eur. J. Pharm. Sci. 2014, 54, 8–16. [Google Scholar] [CrossRef]
- Kitevska, T.; Roberts, S.J.; Pantaki-Eimany, D.; Boyd, S.E.; Scott, F.L.; Hawkins, C.J. Analysis of the Minimal Specificity of Caspase-2 and Identification of Ac-VDTTD-AFC as a Caspase-2-Selective Peptide Substrate. Biosci. Rep. 2014, 34, e00100. [Google Scholar] [CrossRef]
- Jabbour, A.M.; Ekert, P.G.; Coulson, E.J.; Knight, M.J.; Ashley, D.M.; Hawkins, C.J. The P35 Relative, P49, Inhibits Mammalian and Drosophila Caspases Including DRONC and Protects against Apoptosis. Cell Death Differ. 2002, 9, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.J.; Yoo, S.J.; Peterson, E.P.; Wang, S.L.; Vernooy, S.Y.; Hay, B.A. The Drosophila Caspase DRONC Cleaves Following Glutamate or Aspartate and Is Regulated by DIAP1, HID, and GRIM. J. Biol. Chem. 2000, 275, 27084–27093. [Google Scholar] [CrossRef]
- Jabbour, A.M.; Ho, P.K.; Puryer, M.A.; Ashley, D.M.; Ekert, P.G.; Hawkins, C.J. The Caenorhabditis Elegans CED-9 Protein Does Not Directly Inhibit the Caspase CED-3, in Vitro nor in Yeast. Cell Death Differ. 2004, 11, 1309–1316. [Google Scholar] [CrossRef]
- Beaumont, T.E.; Shekhar, T.M.; Kaur, L.; Pantaki-Eimany, D.; Kvansakul, M.; Hawkins, C.J. Yeast Techniques for Modeling Drugs Targeting Bcl-2 and Caspase Family Members. Cell Death Dis. 2013, 4, e619. [Google Scholar] [CrossRef]
- Ekert, P.G.; Silke, J.; Hawkins, C.J.; Verhagen, A.M.; Vaux, D.L. DIABLO Promotes Apoptosis by Removing MIHA/XIAP from Processed Caspase 9. J. Cell. Biol. 2001, 152, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.E.; Han, D.K.; Hockenbery, D.M. Caspase-3 and Inhibitor of Apoptosis Protein (s) Interactions in Saccharomyces cerevisiae and Mammalian Cells. FEBS Lett. 2000, 481, 13–18. [Google Scholar] [CrossRef]
- Silke, J.; Ekert, P.G.; Day, C.L.; Hawkins, C.J.; Baca, M.; Chew, J.; Pakusch, M.; Verhagen, A.M.; Vaux, D.L. Direct Inhibition of Caspase 3 Is Dispensable for the Anti-Apoptotic Activity of XIAP. EMBO J. 2001, 20, 3114–3123. [Google Scholar] [CrossRef] [PubMed]
- Glória, P.M.C.; Coutinho, I.; Gonçalves, L.M.; Baptista, C.; Soares, J.; Newton, A.S.; Moreira, R.; Saraiva, L.; Santos, M.M.M. Aspartic Vinyl Sulfones: Inhibitors of a Caspase-3-Dependent Pathway. Eur. J. Med. Chem. 2011, 46, 2141–2146. [Google Scholar] [CrossRef] [PubMed]
- Lisa-Santamaría, P.; Neiman, A.M.; Cuesta-Marbán, Á.; Mollinedo, F.; Revuelta, J.L.; Jiménez, A. Human Initiator Caspases Trigger Apoptotic and Autophagic Phenotypes in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2009, 1793, 561–571. [Google Scholar] [CrossRef]
- Brunette, S.; Sharma, A.; Bell, R.; Puente, L.; Megeney, L.A. Caspase 3 Exhibits a Yeast Metacaspase Proteostasis Function That Protects Mitochondria from Toxic TDP43 Aggregates. Microb. Cell 2023, 10, 157–169. [Google Scholar] [CrossRef]
- Lisa-Santamaría, P.; Jiménez, A.; Revuelta, J.L. The Protein Factor-Arrest 11 (Far11) Is Essential for the Toxicity of Human Caspase-10 in Yeast and Participates in the Regulation of Autophagy and the DNA Damage Signaling. J. Biol. Chem. 2012, 287, 29636–29647. [Google Scholar] [CrossRef]
- Valenti, M.; Molina, M.; Cid, V.J. Heterologous Expression and Auto-Activation of Human Pro-Inflammatory Caspase-1 in Saccharomyces cerevisiae and Comparison to Caspase-8. Front. Immunol. 2021, 12, 668602. [Google Scholar] [CrossRef]
- Manon, S.; Chaudhuri, B.; Guérin, M. Release of Cytochrome c and Decrease of Cytochrome c Oxidase in Bax-expressing Yeast Cells, and Prevention of These Effects by Coexpression of Bcl-x L. FEBS Lett. 1997, 415, 29–32. [Google Scholar] [CrossRef]
- Ligr, M.; Madeo, F.; Fröhlich, E.; Hilt, W.; Fröhlich, K.-U.; Wolf, D.H. Mammalian Bax Triggers Apoptotic Changes in Yeast. FEBS Lett. 1998, 438, 61–65. [Google Scholar] [CrossRef]
- Zha, H.; Fisk, H.A.; Yaffe, M.P.; Mahajan, N.; Herman, B.; Reed, J.C. Structure-Function Comparisons of the Proapoptotic Protein Bax in Yeast and Mammalian Cells. Mol. Cell. Biol. 1996, 16, 6494–6508. [Google Scholar] [CrossRef] [PubMed]
- Polčic, P.; Jaká, P.; Mentel, M. Yeast as a Tool for Studying Proteins of the Bcl-2 Family. Microb. Cell 2015, 2, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Ahmad, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Autoactivation of Procaspase-9 by Apaf-1-Mediated Oligomerization. Mol. Cell 1998, 1, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Cuddy, M.; Shu, V.C.-W.; Yip, K.W.; Madiraju, C.; Diaz, P.; Matsuyama, T.; Kaibara, M.; Taniyama, K.; Vasile, S.; et al. Versatile Assays for High Throughput Screening for Activators or Inhibitors of Intracellular Proteases and Their Cellular Regulators. PLoS ONE 2009, 4, e7655. [Google Scholar] [CrossRef]
- Valenti, M.; Molina, M.; Cid, V.J. Human Gasdermin D and MLKL Disrupt Mitochondria, Endocytic Traffic and TORC1 Signalling in Budding Yeast. Open Biol. 2023, 13, 220366. [Google Scholar] [CrossRef]
- Ji, Y.; Hawkins, C.J. Reconstitution of Human Pyroptotic Cell Death in Saccharomyces cerevisiae. Sci. Rep. 2023, 13, 3095. [Google Scholar] [CrossRef]
- Fontana, P.; Du, G.; Zhang, Y.; Zhang, H.; Vora, S.M.; Hu, J.J.; Shi, M.; Tufan, A.B.; Healy, L.B.; Xia, S.; et al. Small-Molecule GSDMD Agonism in Tumors Stimulates Antitumor Immunity without Toxicity. Cell 2024, 187, 6165–6181.e22. [Google Scholar] [CrossRef]
- Ji, Y.; Ward, L.A.; Hawkins, C.J. Reconstitution of Human Necrosome Interactions in Saccharomyces cerevisiae. Biomolecules 2021, 11, 153. [Google Scholar] [CrossRef]
- Liao, Y.; Chen, X.; Miller-Little, W.; Wang, H.; Willard, B.; Bulek, K.; Zhao, J.; Li, X. The Ras GTPase -activating-like Protein IQGAP1 Bridges Gasdermin D to the ESCRT System to Promote IL -1β Release via Exosomes. EMBO J. 2023, 42, e110780. [Google Scholar] [CrossRef]
- Evavold, C.L.; Hafner-Bratkovič, I.; Devant, P.; D’Andrea, J.M.; Ngwa, E.M.; Boršić, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; et al. Control of Gasdermin D Oligomerization and Pyroptosis by the Ragulator-Rag-mTORC1 Pathway. Cell 2021, 184, 4495–4511.e19. [Google Scholar] [CrossRef]
- Coronas-Serna, J.M.; Del Val, E.; Kagan, J.C.; Molina, M.; Cid, V.J. Heterologous Expression and Assembly of Human TLR Signaling Components in Saccharomyces cerevisiae. Biomolecules 2021, 11, 1737. [Google Scholar] [CrossRef]
- Agrawal, I.; Jha, S. Comprehensive Review of ASC Structure and Function in Immune Homeostasis and Disease. Mol. Biol. Rep. 2020, 47, 3077–3096. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Jucker, M. Neurodegenerative Diseases: Expanding the Prion Concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.-X.; Halfmann, R.; Chen, Z.J. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Poyet, J.-L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD Protein ASC Is an Activating Adaptor for Caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.-M.; Jiang, Z. MAVS Activates TBK1 and IKKε through TRAFs in NEMO Dependent and Independent Manner. PLoS. Pathog. 2017, 13, e1006720. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; Lindquist, S. Biochemical, Cell Biological, and Genetic Assays to Analyze Amyloid and Prion Aggregation in Yeast. In Methods in Enzymology; Elsevier: Amsterdam, The Netherland, 2010; Volume 470, pp. 709–734. ISBN 978-0-12-375172-0. [Google Scholar]
- Riek, R.; Saupe, S.J. The HET-S/s Prion Motif in the Control of Programmed Cell Death. Cold Spring Harb. Perspect. Biol. 2016, 8, a023515. [Google Scholar] [CrossRef]
- Zhao, L.; Zhu, Y.; Jia, H.; Han, Y.; Zheng, X.; Wang, M.; Feng, W. From Plant to Yeast—Advances in Biosynthesis of Artemisinin. Molecules 2022, 27, 6888. [Google Scholar] [CrossRef]
- Kjeldsen, T.; Balschmidt, P.; Diers, I.; Hach, M.; Kaarsholm, N.C.; Ludvigsen, S. Expression of Insulin in Yeast: The Importance of Molecular Adaptation for Secretion and Conversion. Biotechnol. Genet. Eng. Rev. 2001, 18, 89–121. [Google Scholar] [CrossRef]
- Mager, W.H.; Winderickx, J. Yeast as a Model for Medical and Medicinal Research. Trends Pharmacol. Sci. 2005, 26, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, J.; Yang, Y.; Shan, H.; Hou, S.; Fang, H.; Ma, M.; Chen, Z.; Tan, L.; Xu, D. A Palmitoylation–Depalmitoylation Relay Spatiotemporally Controls GSDMD Activation in Pyroptosis. Nat. Cell Biol. 2024, 26, 757–769. [Google Scholar] [CrossRef]
- Barry, R.; John, S.W.; Liccardi, G.; Tenev, T.; Jaco, I.; Chen, C.-H.; Choi, J.; Kasperkiewicz, P.; Fernandes-Alnemri, T.; Alnemri, E.; et al. SUMO-Mediated Regulation of NLRP3 Modulates Inflammasome Activity. Nat. Commun. 2018, 9, 3001. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Du, Y.; Fei, X.; Yang, H.; Li, X.; Yang, X.; Ma, J.; Huang, S.; Ma, Z.; Zheng, J.; et al. Inflammasome Activity Is Controlled by ZBTB16-Dependent SUMOylation of ASC. Nat. Commun. 2023, 14, 8465. [Google Scholar] [CrossRef]
- Madiraju, C.; Novack, J.P.; Reed, J.C.; Matsuzawa, S. K63 Ubiquitination in Immune Signaling. Trends Immunol. 2022, 43, 148–162. [Google Scholar] [CrossRef]
- Vanhelmont, T.; Vandebroek, T.; De Vos, A.; Terwel, D.; Lemaire, K.; Anandhakumar, J.; Franssens, V.; Swinnen, E.; Van Leuven, F.; Winderickx, J. Serine-409 Phosphorylation and Oxidative Damage Define Aggregation of Human Protein Tau in Yeast: Determinants of Tau Aggregation in Yeast. FEMS Yeast Res. 2010, 10, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, J.J.; Brandt, R. Signaling Pathways and Posttranslational Modifications of Tau in Alzheimer’s Disease: The Humanization of Yeast Cells. MIC 2016, 3, 135–146. [Google Scholar] [CrossRef]
- Lehle, L.; Strahl, S.; Tanner, W. Protein Glycosylation, Conserved from Yeast to Man: A Model Organism Helps Elucidate Congenital Human Diseases. Angew. Chem. Int. Ed. 2006, 45, 6802–6818. [Google Scholar] [CrossRef]
- Arico, C.; Bonnet, C.; Javaud, C. N-Glycosylation Humanization for Production of Therapeutic Recombinant Glycoproteins in Saccharomyces cerevisiae. In Glycosylation Engineering of Biopharmaceuticals; Beck, A., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 988, pp. 45–57. ISBN 978-1-62703-326-8. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caspase 1 | Inhibitor | Reference |
|---|---|---|
| Dm DCP-1 | DIAP1 | [82] |
| Baculovirus p35 protein | [88] | |
| Baculovirus p49 protein | [88] | |
| Dm DRONC | DIAP1 | [89] |
| Baculovirus p49 protein | [88] | |
| Dm drICE | Baculovirus p35 protein | [88] |
| Baculovirus p49 protein | [88] | |
| DIAP1 | [88] | |
| Ce CED-4 | Ce CED-9 | [90] |
| Hs CASP-8 | Baculovirus p35 protein | [81,88] |
| Cowpox virus CrmA | [91] | |
| Hs CASP-9 | XIAP | [92] |
| Hs CASP-3 | Cowpox virus CrmA-mut | [83] |
| XIAP, c-IAP1, c-IAP2 | [93,94] | |
| Baculovirus p35 protein | [88] | |
| Baculovirus p49 protein | [88] | |
| ZVAD-fluoromethyl ketone | [83] | |
| Q-VD-OPh | [91] | |
| Ac-DEVD-chloromethyl ketone | [95] | |
| Aspartic vinyl sulphones | [95] | |
| Hs CASP-7 | Baculovirus p35 protein | [88] |
| Baculovirus p49 protein | [88] | |
| XIAP | [88] | |
| Hs CASP-2 | Baculovirus p35 protein | [88] |
| Baculovirus p49 protein | [88] | |
| Hs CASP-4 | Baculovirus p35 protein | [88] |
| Hs CASP-5 | Baculovirus p35 protein | [88] |
| Hs CASP-1 | Q-VD-OPh | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbero-Úriz, Ó.; Valenti, M.; Molina, M.; Fernández-Acero, T.; Cid, V.J. Modeling Necroptotic and Pyroptotic Signaling in Saccharomyces cerevisiae. Biomolecules 2025, 15, 530. https://doi.org/10.3390/biom15040530
Barbero-Úriz Ó, Valenti M, Molina M, Fernández-Acero T, Cid VJ. Modeling Necroptotic and Pyroptotic Signaling in Saccharomyces cerevisiae. Biomolecules. 2025; 15(4):530. https://doi.org/10.3390/biom15040530
Chicago/Turabian StyleBarbero-Úriz, Óscar, Marta Valenti, María Molina, Teresa Fernández-Acero, and Víctor J. Cid. 2025. "Modeling Necroptotic and Pyroptotic Signaling in Saccharomyces cerevisiae" Biomolecules 15, no. 4: 530. https://doi.org/10.3390/biom15040530
APA StyleBarbero-Úriz, Ó., Valenti, M., Molina, M., Fernández-Acero, T., & Cid, V. J. (2025). Modeling Necroptotic and Pyroptotic Signaling in Saccharomyces cerevisiae. Biomolecules, 15(4), 530. https://doi.org/10.3390/biom15040530