
The Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins Protects Against Microglia-Mediated Neuronal Loss In Vitro


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Reagents and Treatments
2.3. Cell Culture Experiments
2.4. MTT Assay
2.5. Gene Expression Analysis
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Phagocytosis Assay
2.8. Atomic Force Microscopy (AFM)
2.9. Statistics
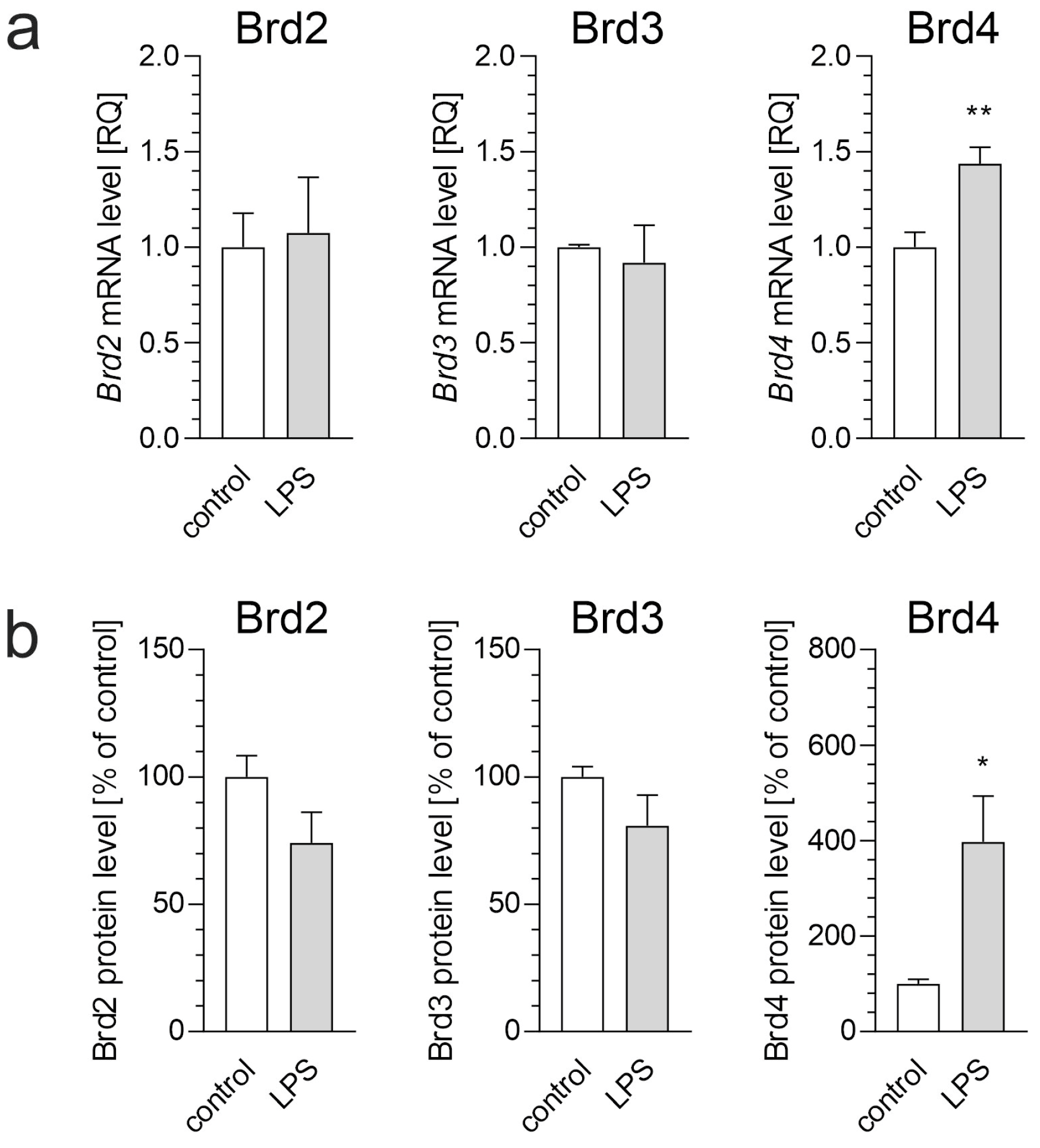
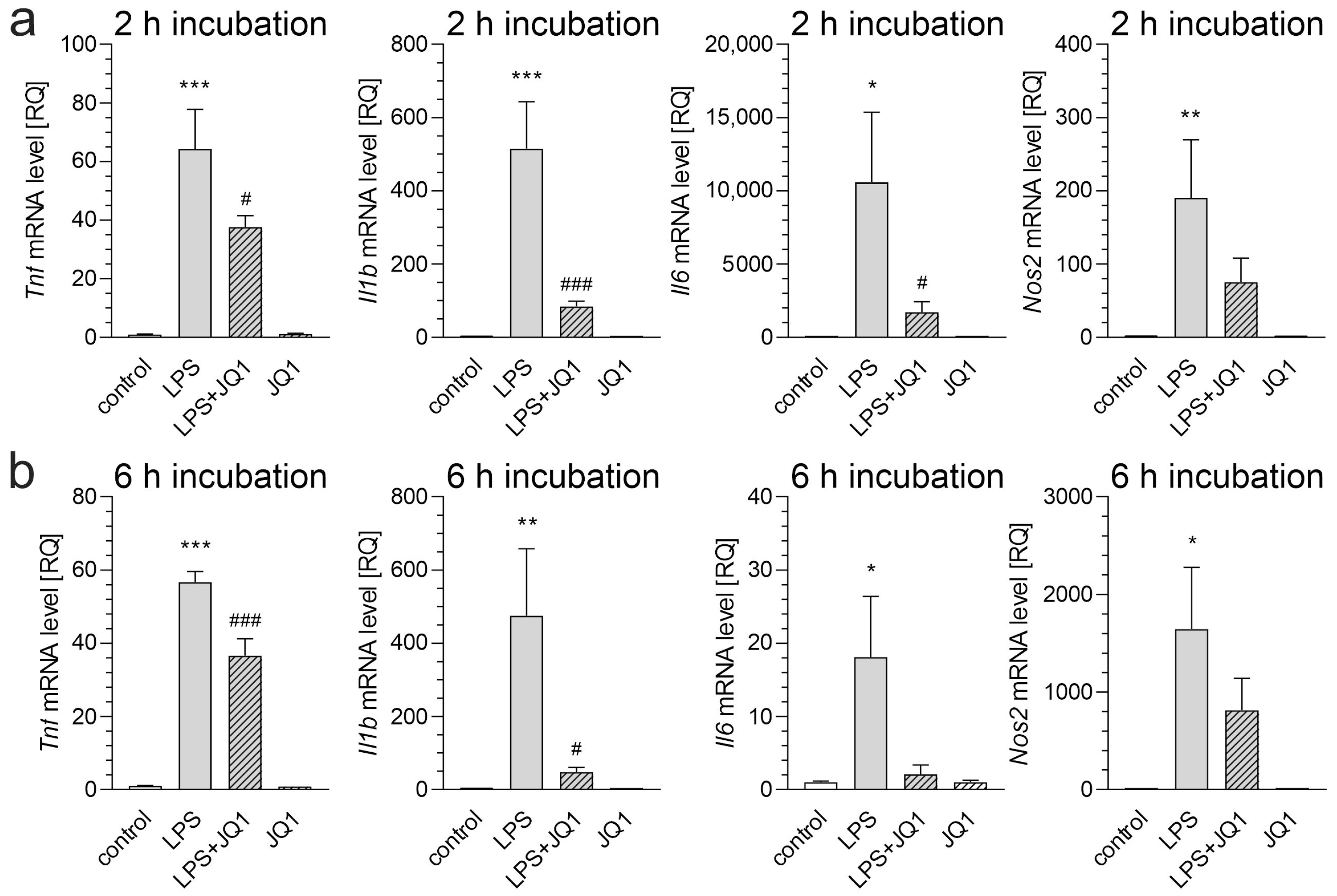
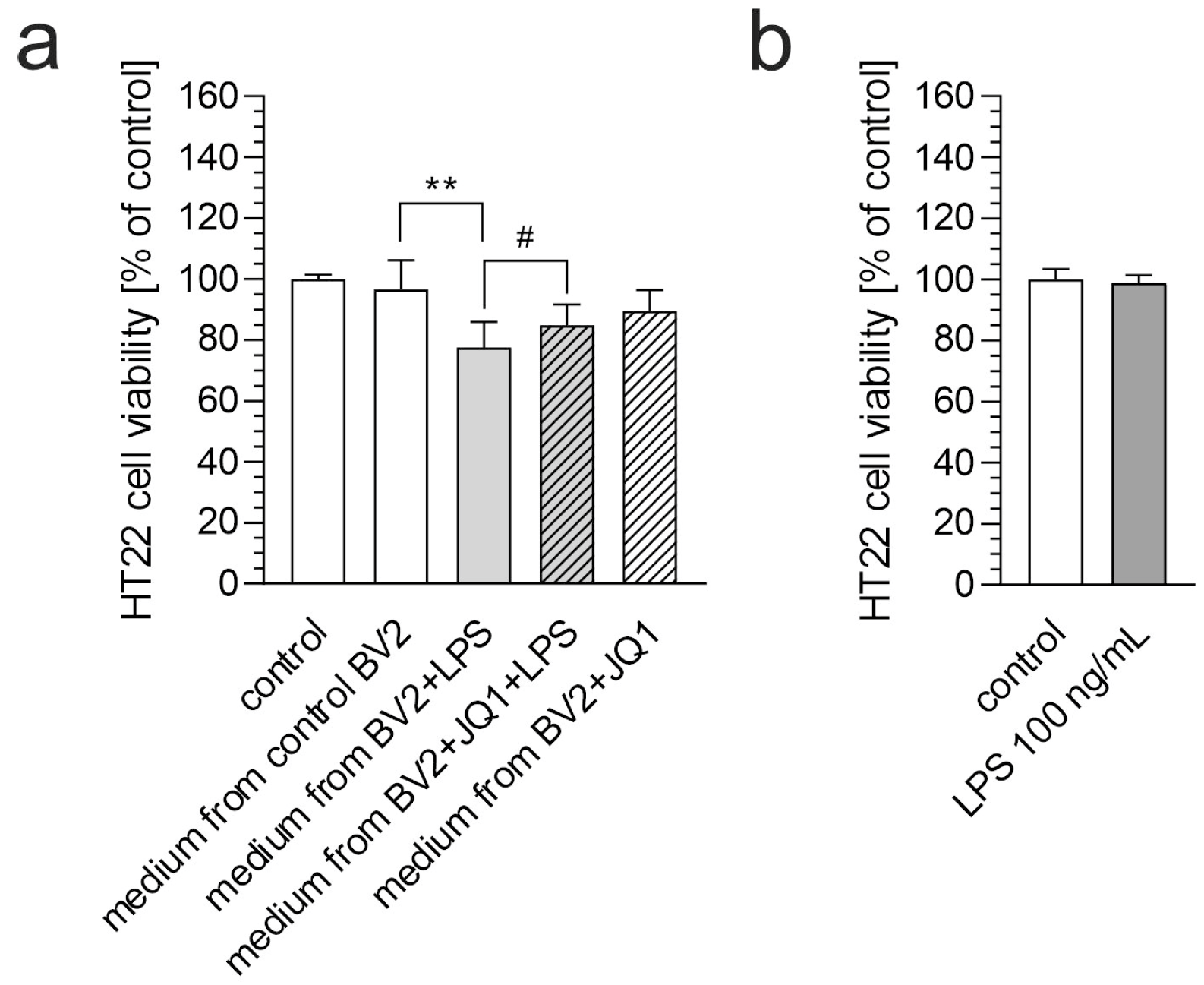
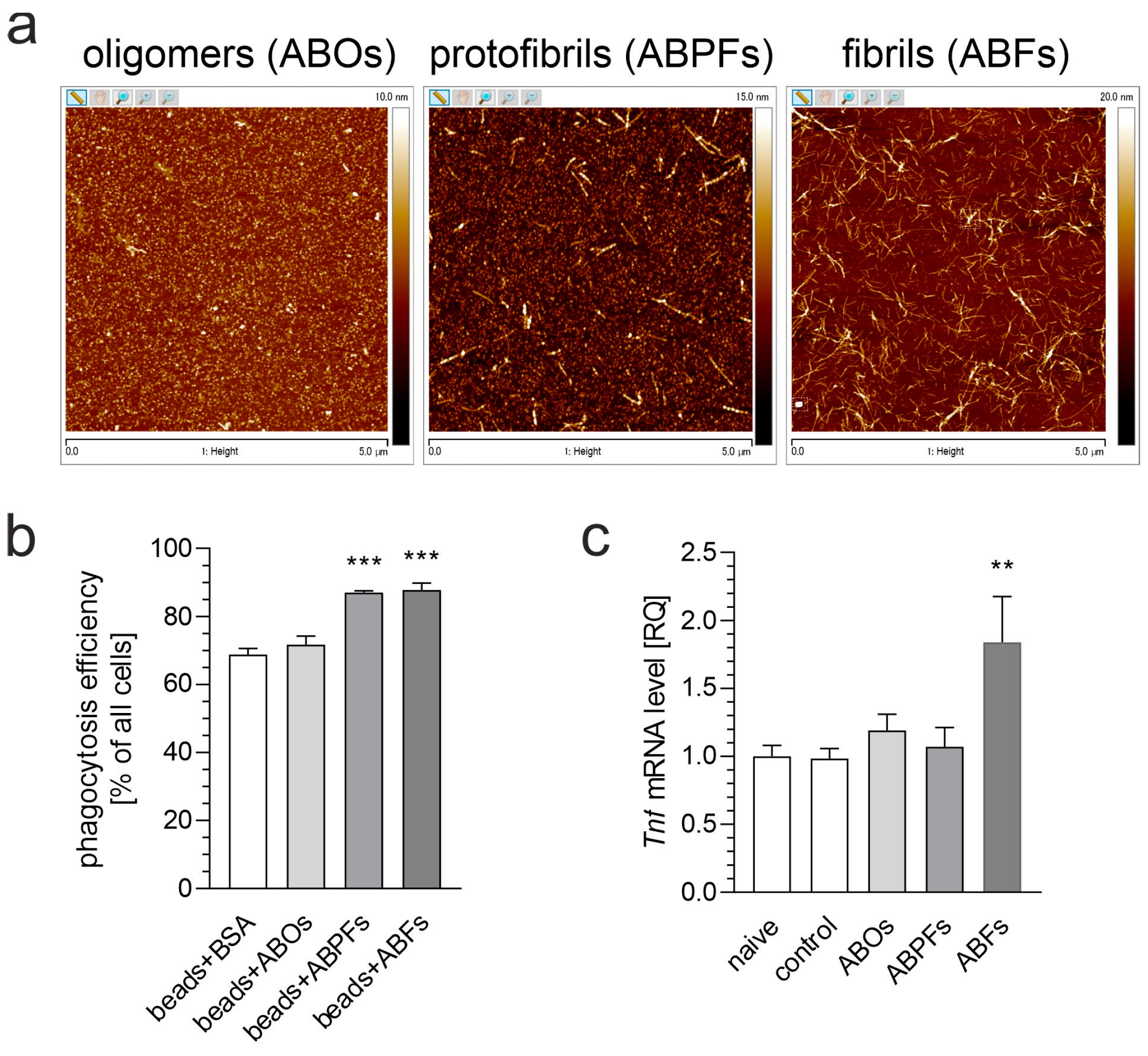
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| ABOs | amyloid-β oligomers |
| ABFs | amyloid-β fibrils |
| ABPFs | amyloid-β protofibrils |
| AD | Alzheimer’s disease |
| AFM | atomic force microscopy |
| BET | bromodomain and extraterminal domain |
| CNS | central nervous system |
| DMSO | dimethyl sulfoxide |
| FMS | fluorescent microspheres |
| HFIP | hexafluoroisopropanol |
| LPS | lipopolysaccharide |
| PD | Parkinson’s disease |
| RQ | relative quantity |
References
- Shah, S.; Jain, H. Microglia-Associated Neuroinflammation in Alzheimer’s Disease and Its Therapeutic Potential. Neuroglia 2024, 5, 452–466. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Sobue, A.; Komine, O.; Yamanaka, K. Neuroinflammation in Alzheimer’s disease: Microglial signature and their relevance to disease. Inflamm. Regen. 2023, 43, 26. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, C. Microglia activation linking amyloid-β drive tau spatial propagation in Alzheimer’s disease. Front. Neurosci. 2022, 16, 951128. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, Z.C.; Wu, Y.Y.; Pi, Y.N.; Lou, S.H.; Liu, T.B.; Lou, G.; Yang, C. Bromodomain and extraterminal (BET) proteins: Biological functions, diseases, and targeted therapy. Signal Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Jacques, M.T.; Saso, L.; Farina, M. LPS-Activated Microglial Cell-Derived Conditioned Medium Protects HT22 Neuronal Cells against Glutamate-Induced Ferroptosis. Int. J. Mol. Sci. 2023, 24, 2910. [Google Scholar] [CrossRef]
- Lajqi, T.; Lang, G.P.; Haas, F.; Williams, D.L.; Hudalla, H.; Bauer, M.; Groth, M.; Wetzker, R.; Bauer, R. Memory-Like Inflammatory Responses of Microglia to Rising Doses of LPS: Key Role of PI3Kγ. Front. Immunol. 2019, 10, 2492. [Google Scholar] [CrossRef]
- Dubnovitsky, A.; Sandberg, A.; Rahman, M.M.; Benilova, I.; Lendel, C.; Härd, T. Amyloid-β protofibrils: Size, morphology and synaptotoxicity of an engineered mimic. PLoS ONE 2013, 8, e66101. [Google Scholar] [CrossRef]
- Hughes, C.; Choi, M.L.; Yi, J.H.; Kim, S.C.; Drews, A.; George-Hyslop, P.S.; Bryant, C.; Gandhi, S.; Cho, K.; Klenerman, D. Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun. Biol. 2020, 3, 79. [Google Scholar] [CrossRef]
- Matuszewska, M.; Cieślik, M.; Wilkaniec, A.; Strawski, M.; Czapski, G.A. The Role of Bromodomain and Extraterminal (BET) Proteins in Controlling the Phagocytic Activity of Microglia In Vitro: Relevance to Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 13. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Morimoto, B.H.; Koshland, D.E., Jr. Induction and expression of long- and short-term neurosecretory potentiation in a neural cell line. Neuron 1990, 5, 875–880. [Google Scholar] [CrossRef]
- Das, A.; Kim, S.H.; Arifuzzaman, S.; Yoon, T.; Chai, J.C.; Lee, Y.S.; Park, K.S.; Jung, K.H.; Chai, Y.G. Transcriptome sequencing reveals that LPS-triggered transcriptional responses in established microglia BV2 cell lines are poorly representative of primary microglia. J. Neuroinflammation 2016, 13, 182. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Gassowska-Dobrowolska, M.; Strawski, M.; Adamczyk, A.; Czapski, G.A. Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity. J. Neuroinflamm. 2018, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.; Christensen, K.V.; Hedtjärn, M.; Mortensen, A.L.; Hagberg, H.; Falsig, J.; Hasseldam, H.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J. Neuroimmunol. 2006, 180, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Roqué, P.J.; Costa, L.G. Co-Culture of Neurons and Microglia. Curr. Protoc. Toxicol. 2017, 74, 11.24.11–11.24.17. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lambert, J.P.; Mitchell, L.; Rudner, A.; Baetz, K.; Figeys, D. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol. Cell. Proteom. 2009, 8, 870–882. [Google Scholar] [CrossRef]
- Moore, A.H.; O’Banion, M.K. Neuroinflammation and anti-inflammatory therapy for Alzheimer’s disease. Adv. Drug Deliv. Rev. 2002, 54, 1627–1656. [Google Scholar] [CrossRef]
- Gao, C.; Shen, X.; Tan, Y.; Chen, S. Pathogenesis, therapeutic strategies and biomarker development based on “omics” analysis related to microglia in Alzheimer’s disease. J. Neuroinflammation 2022, 19, 215. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Jin, H.; Lin, D.; Chen, Y.; Chen, X.; Wang, B.; Hu, S.; Wu, Y.; Wu, Y.; et al. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J. Cell. Mol. Med. 2019, 23, 3214–3223. [Google Scholar] [CrossRef]
- Huang, C.; Liang, Y.; Jiang, A.; Chen, L.; Sun, C.; Luo, D.; Xia, Z.; Li, L.; Jiang, Y. Dynamic proteome and phosphoproteome profiling reveals regulatory mechanisms in LPS-stimulated macrophage inflammatory responses. Biochem. Biophys. Res. Commun. 2025, 750, 151341. [Google Scholar] [CrossRef]
- Li, W.; Shen, X.; Feng, S.; Liu, Y.; Zhao, H.; Zhou, G.; Sang, M.; Sun, X.; Jiao, R.; Liu, F. BRD4 inhibition by JQ1 protects against LPS-induced cardiac dysfunction by inhibiting activation of NLRP3 inflammasomes. Mol. Biol. Rep. 2022, 49, 8197–8207. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Matuszewska, M.; Cieślik, M.; Strosznajder, J.S. Inhibitor of bromodomain and extraterminal domain proteins decreases transcription of Cd33 in the brain of mice subjected to systemic inflammation; a promising strategy for neuroprotection. Folia Neuropathol. 2024, 62, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fu, H.; Zhu, R.; Wu, X.; Ji, X.; Li, X.; Jiang, H.; Lin, Z.; Tang, X.; Sun, S.; et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 2020, 12, 9240–9259. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, C.; Candelario-Jalil, E. Role of BET Proteins in Inflammation and CNS Diseases. Front. Mol. Biosci. 2021, 8, 748449. [Google Scholar] [CrossRef]
- Baek, M.; Yoo, E.; Choi, H.I.; An, G.Y.; Chai, J.C.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. The BET inhibitor attenuates the inflammatory response and cell migration in human microglial HMC3 cell line. Sci. Rep. 2021, 11, 8828. [Google Scholar] [CrossRef]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflammation 2009, 6, 1. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Ferrera, D.; Mazzaro, N.; Canale, C.; Gasparini, L. Resting microglia react to Aβ42 fibrils but do not detect oligomers or oligomer-induced neuronal damage. Neurobiol. Aging 2014, 35, 2444–2457. [Google Scholar] [CrossRef]
- McFarland, K.N.; Ceballos, C.; Rosario, A.; Ladd, T.; Moore, B.; Golde, G.; Wang, X.; Allen, M.; Ertekin-Taner, N.; Funk, C.C.; et al. Microglia show differential transcriptomic response to Aβ peptide aggregates ex vivo and in vivo. Life Sci. Alliance 2021, 4, e202101108. [Google Scholar] [CrossRef]
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef]
- Parvathy, S.; Rajadas, J.; Ryan, H.; Vaziri, S.; Anderson, L.; Murphy, G.M., Jr. Abeta peptide conformation determines uptake and interleukin-1alpha expression by primary microglial cells. Neurobiol. Aging 2009, 30, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bai, P.; Lei, D.; Liang, Y.; Zhen, S.; Bakiasi, G.; Pang, H.; Choi, S.H.; Wang, C.; Tanzi, R.E.; et al. Degradation and inhibition of epigenetic regulatory protein BRD4 exacerbate Alzheimer’s disease-related neuropathology in cell models. J. Biol. Chem. 2022, 298, 101794. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Liang, M.; Shi, Y.; Zhang, C.; Li, Q.; Liu, M.; Shou, Y.; Yin, H.; Zhu, X.; et al. (+)-JQ1 attenuated LPS-induced microglial inflammation via MAPK/NFκB signaling. Cell Biosci. 2018, 8, 60. [Google Scholar] [CrossRef]
- Magistri, M.; Velmeshev, D.; Makhmutova, M.; Patel, P.; Sartor, G.C.; Volmar, C.H.; Wahlestedt, C.; Faghihi, M.A. The BET-Bromodomain Inhibitor JQ1 Reduces Inflammation and Tau Phosphorylation at Ser396 in the Brain of the 3xTg Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 985–995. [Google Scholar] [CrossRef]
- Badrikoohi, M.; Esmaeili-Bandboni, A.; Babaei, P. Simultaneous administration of bromodomain and histone deacetylase I inhibitors alleviates cognition deficit in Alzheimer’s model of rats. Brain Res. Bull. 2022, 179, 49–56. [Google Scholar] [CrossRef]
- Nikkar, R.; Esmaeili-Bandboni, A.; Badrikoohi, M.; Babaei, P. Effects of inhibiting astrocytes and BET/BRD4 chromatin reader on spatial memory and synaptic proteins in rats with Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 1119–1131. [Google Scholar] [CrossRef]
- Benito, E.; Ramachandran, B.; Schroeder, H.; Schmidt, G.; Urbanke, H.; Burkhardt, S.; Capece, V.; Dean, C.; Fischer, A. The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl. Psychiatry 2017, 7, e1239. [Google Scholar] [CrossRef]
- Quezada, E.; Cappelli, C.; Diaz, I.; Jury, N.; Wightman, N.; Brown, R.H., Jr.; Montecino, M.; van Zundert, B. BET bromodomain inhibitors PFI-1 and JQ1 are identified in an epigenetic compound screen to enhance C9ORF72 gene expression and shown to ameliorate C9ORF72-associated pathological and behavioral abnormalities in a C9ALS/FTD model. Clin. Epigenetics 2021, 13, 56. [Google Scholar] [CrossRef]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Darnell, R.B.; Allis, C.D. BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat. Neurosci. 2015, 18, 1464–1473. [Google Scholar] [CrossRef]
- Bilecki, W.; Wawrzczak-Bargieła, A.; Majcher-Maślanka, I.; Chmelova, M.; Maćkowiak, M. Inhibition of BET Proteins during Adolescence Affects Prefrontal Cortical Development: Relevance to Schizophrenia. Int. J. Mol. Sci. 2021, 22, 8710. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Han, D.; Wang, J.I.; Park, J.; Kim, H.; Kim, Y. Quantitative Proteomics Reveals Temporal Proteomic Changes in Signaling Pathways during BV2 Mouse Microglial Cell Activation. J. Proteome Res. 2017, 16, 3419–3432. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Martinez-Muriana, A.; Davis, E.; Daniels, M.J.D.; Hou, P.; Mancuso, R.; Brenes, A.J.; Sinclair, L.V.; Geric, I.; Snellinx, A.; et al. Deep proteomic analysis of microglia reveals fundamental biological differences between model systems. Cell Rep. 2024, 43, 114908. [Google Scholar] [CrossRef]
- Ozbey, G.; Gorczynski, R.; Erin, N.U.R.A.Y. Stability of cytokines in supernatants of stimulated mouse immune cells. Eur. Cytokine Netw. 2014, 25, 30–34. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Li, Y.; Xu, L.; Yu, X.; Ge, L.; Li, J.; Zhu, Y.; He, S. Necroptosis mediates TNF-induced toxicity of hippocampal neurons. BioMed Res. Int. 2014, 2014, 290182. [Google Scholar] [CrossRef]
- Gelibter, S.; Marostica, G.; Mandelli, A.; Siciliani, S.; Podini, P.; Finardi, A.; Furlan, R. The impact of storage on extracellular vesicles: A systematic study. J. Extracell. Vesicles 2022, 11, e12162. [Google Scholar] [CrossRef]
- Kwon, S.B.; Ernst, J. Learning a genome-wide score of human-mouse conservation at the functional genomics level. Nat. Commun. 2021, 12, 2495. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, Z.; Kim, B.H.; Wu, W.; Cayting, P.; Boyle, A.P.; Sundaram, V.; Xing, X.; Dogan, N.; Li, J.; et al. Principles of regulatory information conservation between mouse and human. Nature 2014, 515, 371–375. [Google Scholar] [CrossRef]
- Lin, S.; Lin, Y.; Nery, J.R.; Urich, M.A.; Breschi, A.; Davis, C.A.; Dobin, A.; Zaleski, C.; Beer, M.A.; Chapman, W.C.; et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 17224–17229. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuszewska, M.; Wilkaniec, A.; Cieślik, M.; Strawski, M.; Czapski, G.A. The Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins Protects Against Microglia-Mediated Neuronal Loss In Vitro. Biomolecules 2025, 15, 528. https://doi.org/10.3390/biom15040528
Matuszewska M, Wilkaniec A, Cieślik M, Strawski M, Czapski GA. The Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins Protects Against Microglia-Mediated Neuronal Loss In Vitro. Biomolecules. 2025; 15(4):528. https://doi.org/10.3390/biom15040528
Chicago/Turabian StyleMatuszewska, Marta, Anna Wilkaniec, Magdalena Cieślik, Marcin Strawski, and Grzegorz A. Czapski. 2025. "The Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins Protects Against Microglia-Mediated Neuronal Loss In Vitro" Biomolecules 15, no. 4: 528. https://doi.org/10.3390/biom15040528
APA StyleMatuszewska, M., Wilkaniec, A., Cieślik, M., Strawski, M., & Czapski, G. A. (2025). The Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins Protects Against Microglia-Mediated Neuronal Loss In Vitro. Biomolecules, 15(4), 528. https://doi.org/10.3390/biom15040528