
Cell-Based Small-Molecule Screening Identifying Proteostasis Regulators Enhancing Factor VIII Missense Mutant Secretion

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line, Bacterial Strains, Reagents, and Antibodies
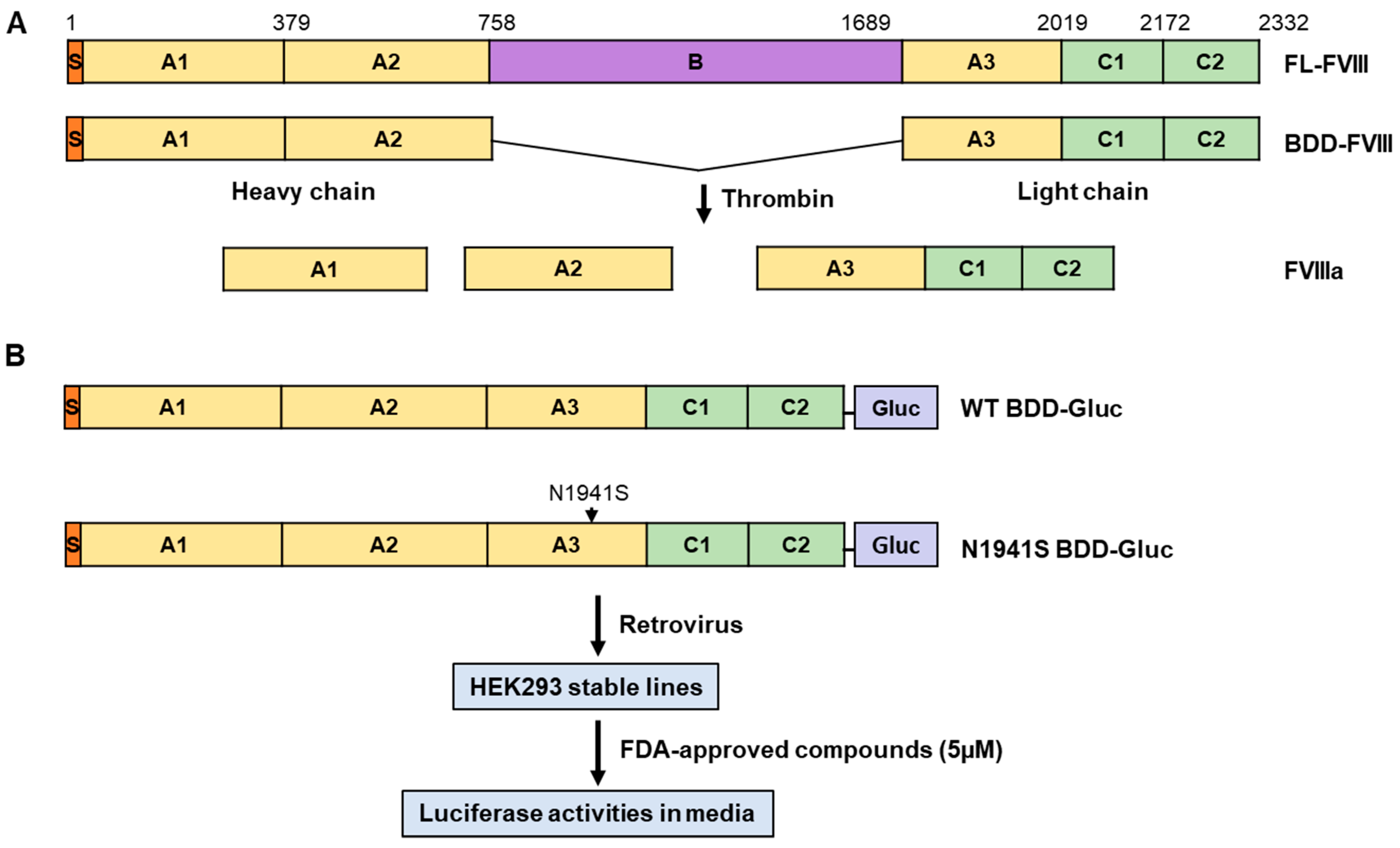
2.2. Establishment of Cell Lines Stably Expressing B-Domain-Deleted FVIII–Gaussia Luciferase Fusion
2.3. Construction of Plasmids Expressing Full-Length FVIII and Individual FVIII Domains with Missense Mutations
2.4. High-Throughput and Secondary Screening Using a Secreted Luciferase Assay
2.5. FVIII Antigen and Activity Assays
2.6. Immunoblotting and Immunoprecipitation
2.7. Animal Study
2.8. Statistical Analysis
3. Results
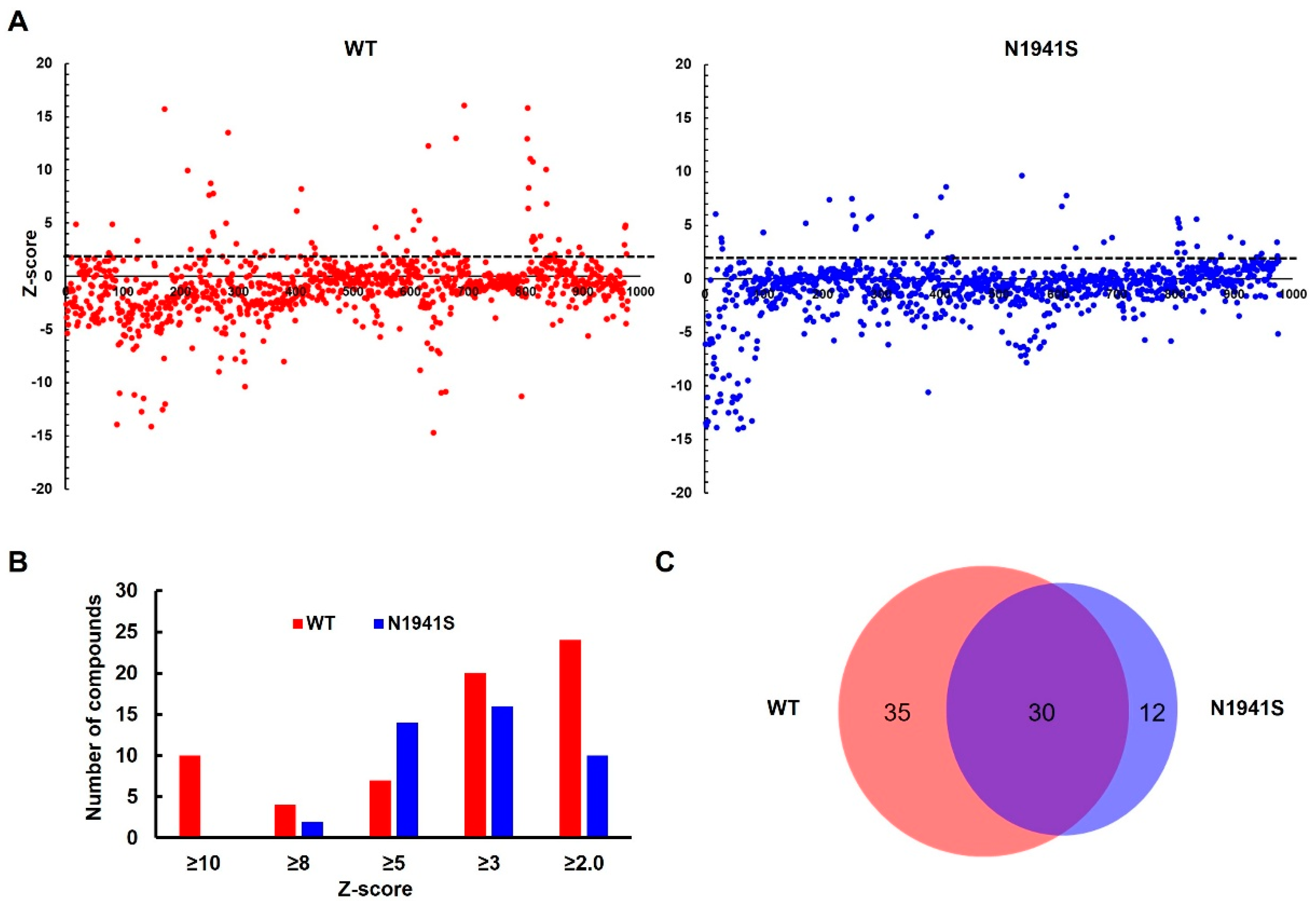
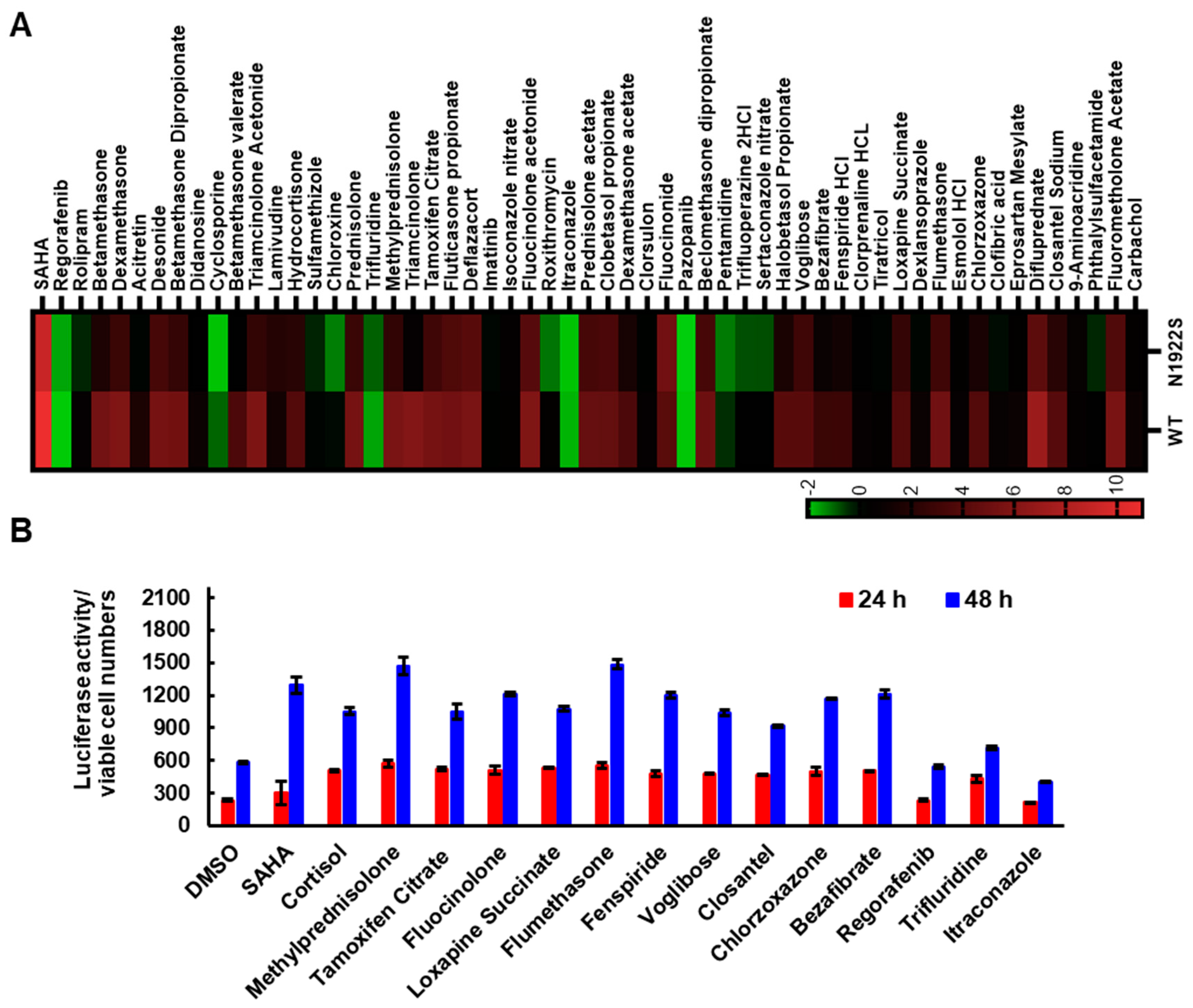
3.1. Screening of FDA-Approved Compound Library to Identify Compounds That Enhance FVIII Secretion
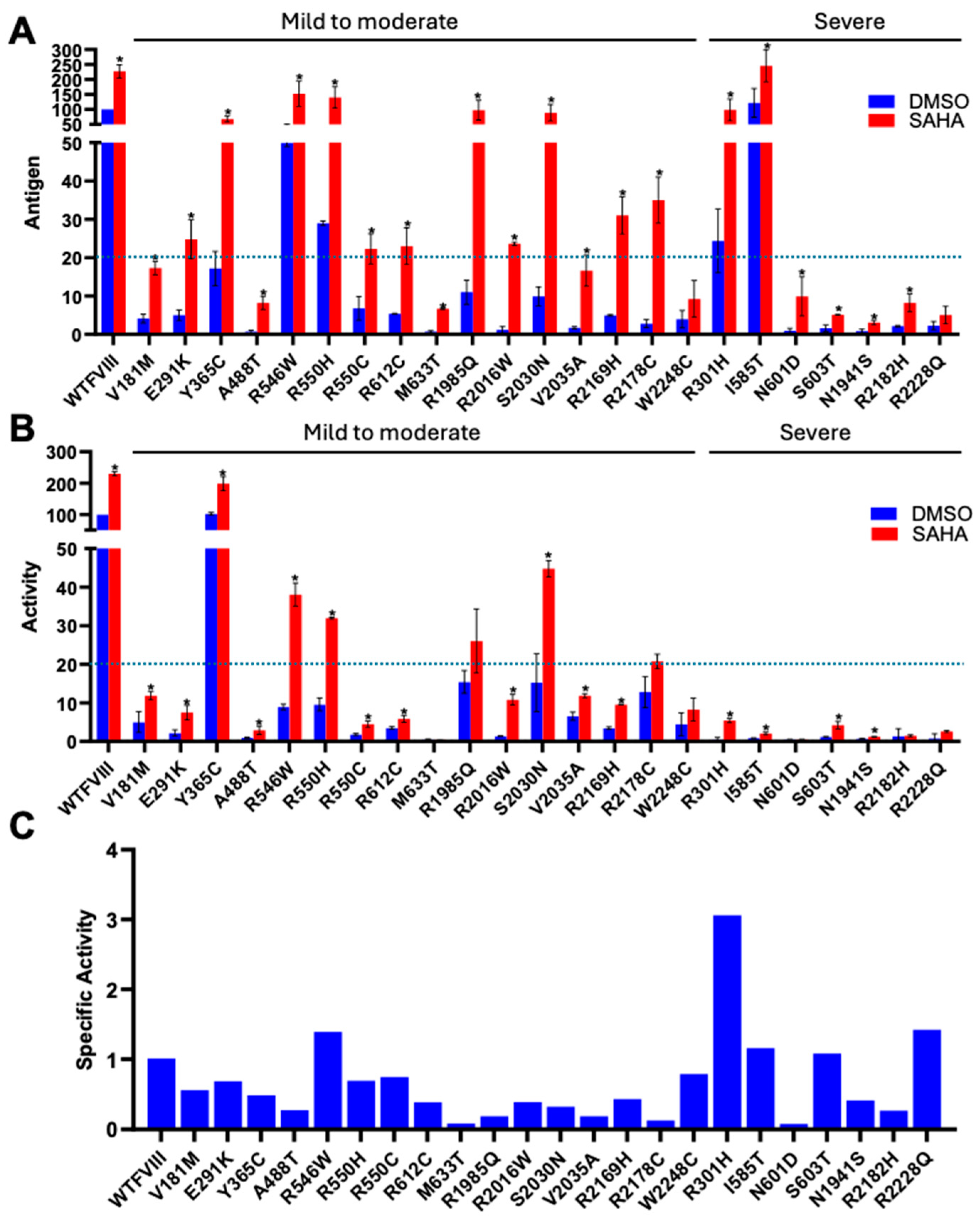
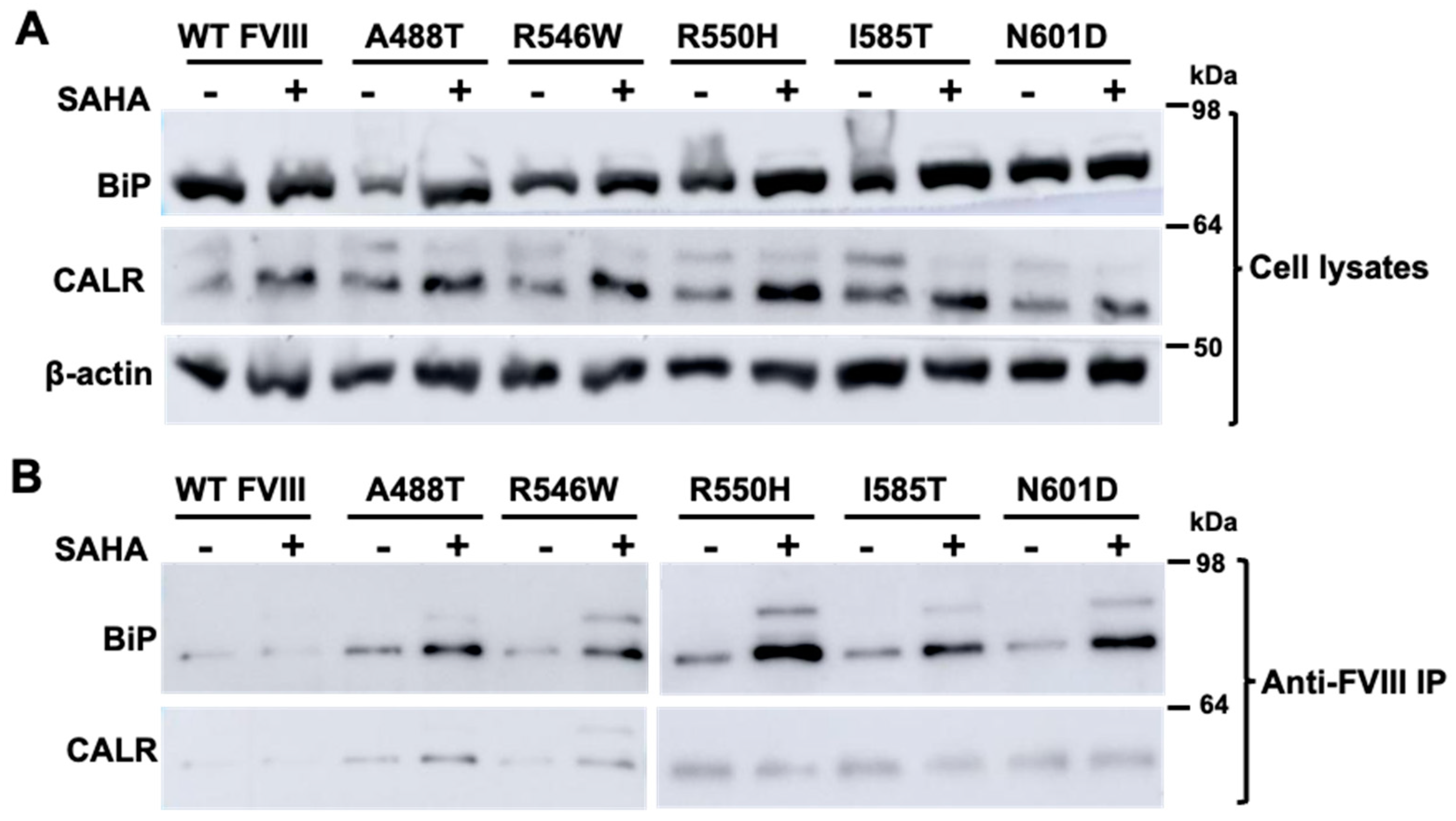
3.2. SAHA Significantly Enhanced the Secretion of FVIII Missense Mutants
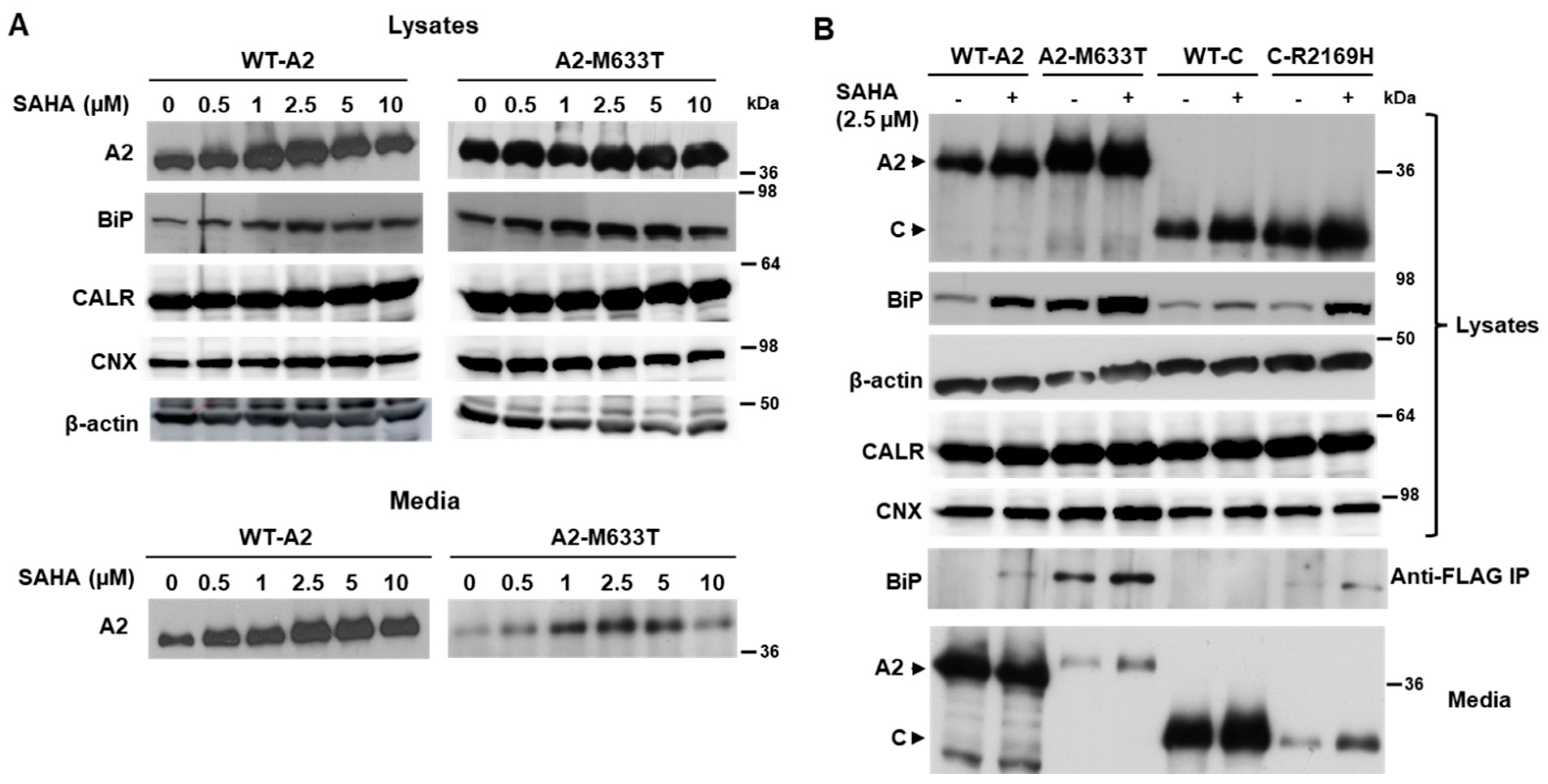
3.3. SAHA Increased the Interaction of FVIII with an ER Chaperone BiP
3.4. SAHA Increased the Secretion of Individual FVIII Subunits
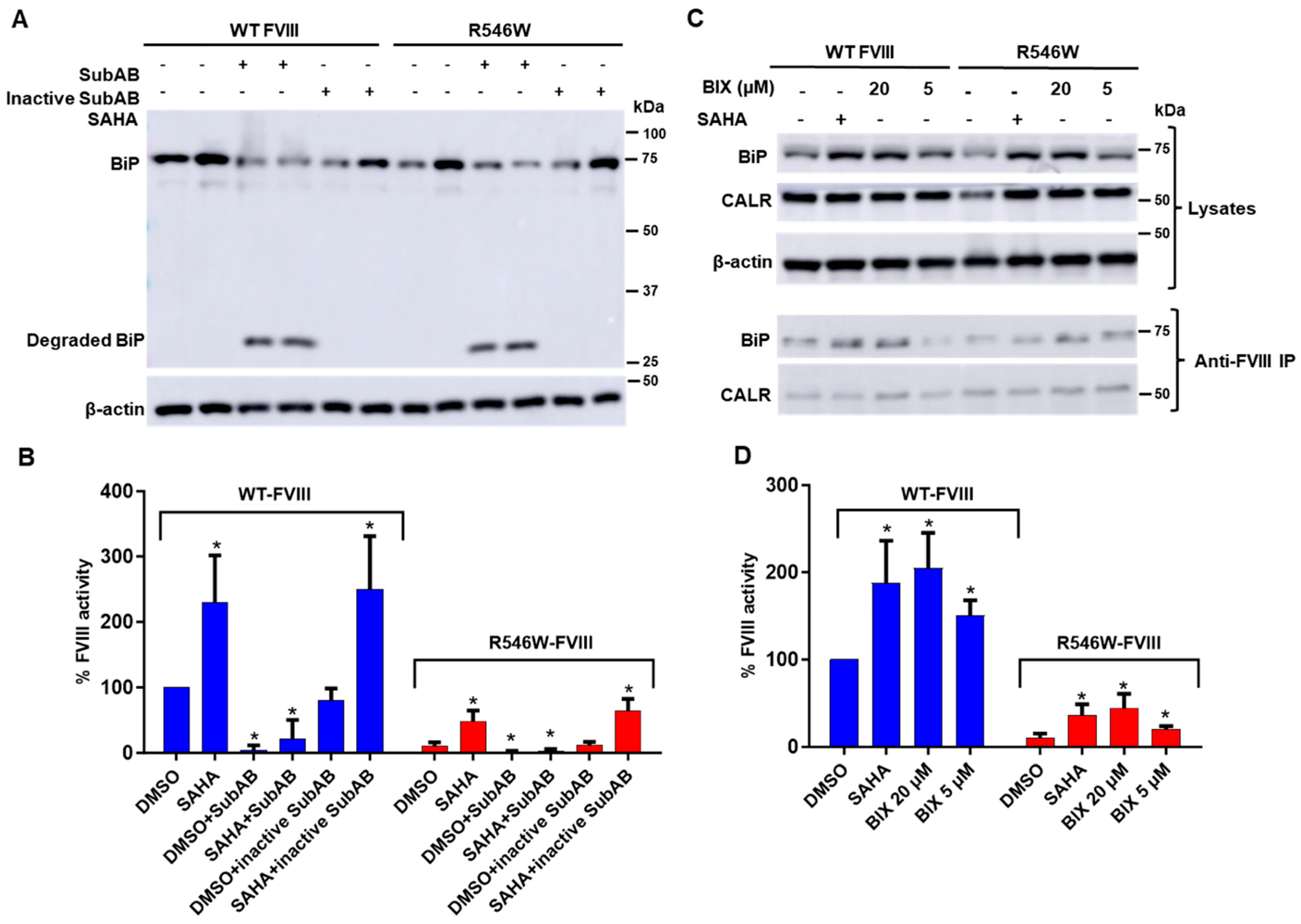
3.5. Decreasing BiP Levels Reversed SAHA-Mediated Stimulation of FVIII Secretion
3.6. A BiP Activator Enhanced FVIII Secretion
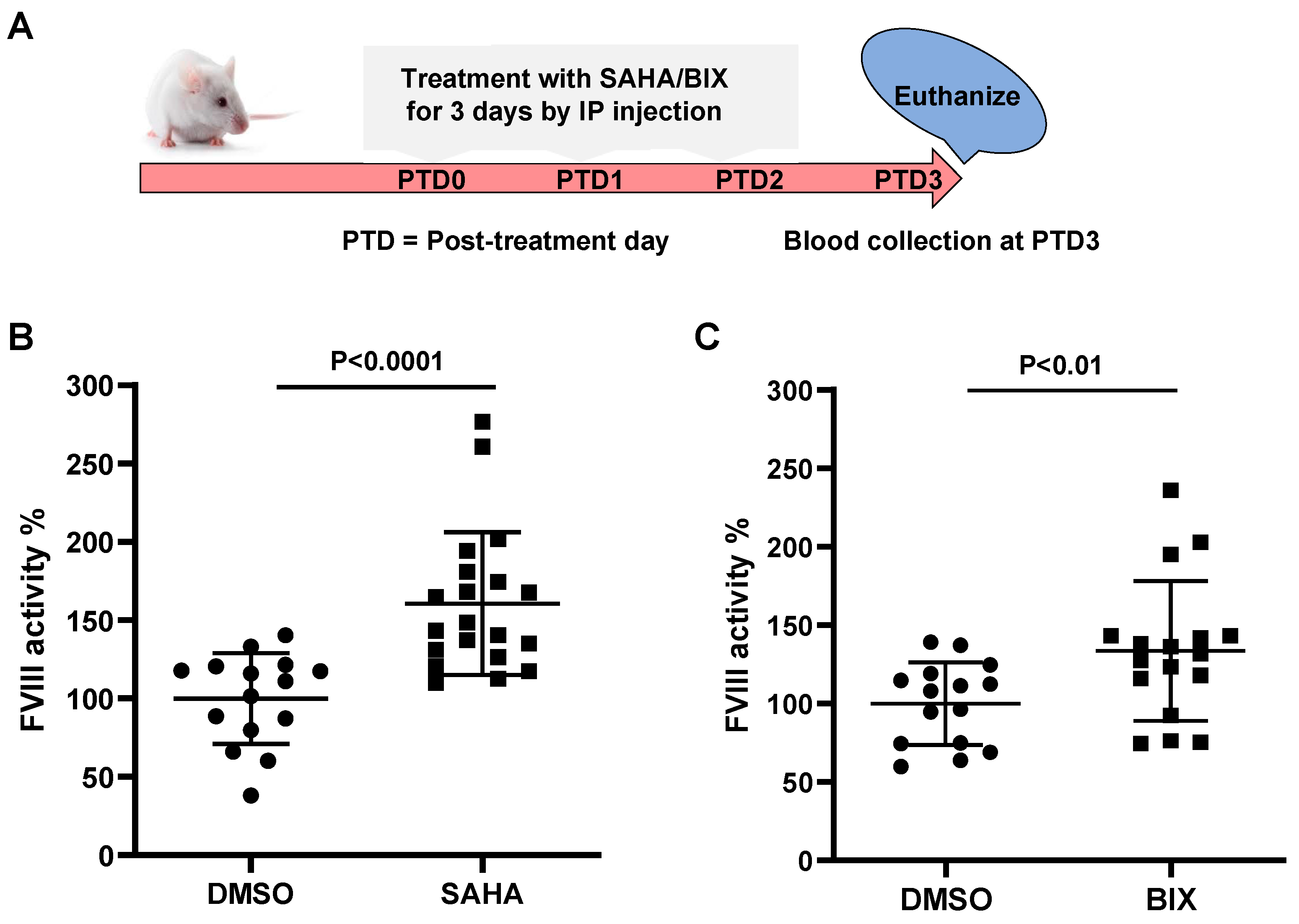
3.7. SAHA and BIX Treatments Increase Endogenous FVIII Levels in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HA | hemophilia A |
| FVIII | factor VIII |
| FIX | factor IX |
| FL | full-length |
| BDD | B-domain-deleted |
| SAHA | suberoylanilide hydroxamic acid |
| EC50 | half maximal effective concentration |
| ER | endoplasmic reticulum |
| BiP | immunoglobulin binding protein |
| CANX | calnexin |
| CALR | calreticulin |
| COPII | coat protein complex II |
| WT | wild type |
| UPR | unfolded protein response |
| ERAD | ER-associated degradation |
| PR | proteostasis regulator |
| FDA | Food and Drug Administration |
| DMSO | dimethyl sulfoxide |
| BIX | BiP Inducer X |
| FBS | Fetal Bovine Serum |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| Gluc | Gaussia luciferase |
| HTS | high-throughput screening |
| RLU | relative luciferase unit |
| HDACi | histone deacetylase inhibitor |
References
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Primers 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Iorio, A.; Stonebraker, J.S.; Chambost, H.; Makris, M.; Coffin, D.; Herr, C.; Germini, F.; Data and Demographics Committee of the World Federation of Hemophilia. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males: A Meta-analytic Approach Using National Registries. Ann. Intern. Med. 2019, 171, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J.; Fay, P.J.; Popolo, L.; Ortel, T.L. Factor V and Factor VIII. In Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 6th ed.; Marder, V.J., Aird, W.C., Bennet, J.S., Schulman, S., White, G.C., Eds.; Lippincott William & Wilkins: Philadelphia, PA, USA, 2013; pp. 179–196. [Google Scholar]
- Espanol, M.G.; Mistretta, J.N.; Tarantino, M.D.; Roberts, J.C. The Evolution of Hemophilia Therapeutics: An Illustrated Review. Res. Pract. Thromb. Haemost. 2024, 8, 102308. [Google Scholar] [CrossRef]
- Thornburg, C.D.; Adamski, K.; Cook, K.; Vembusubramanian, M.; Sendhil, S.R.; Hinds, D.; Chen, E.; Sammon, J.; Solari, P.; Garrison, L.P., Jr.; et al. Health care costs and resource utilization among commercially insured adult patients with hemophilia A managed with FVIII prophylaxis in the United States. J. Manag. Care Spec. Pharm. 2022, 28, 449–460. [Google Scholar] [CrossRef]
- Balestra, D.; Branchini, A. Molecular Mechanisms and Determinants of Innovative Correction Approaches in Coagulation Factor Deficiencies. Int. J. Mol. Sci. 2019, 20, 3036. [Google Scholar] [CrossRef] [PubMed]
- Loomans, J.I.; Kruip, M.; Carcao, M.; Jackson, S.; van Velzen, A.S.; Peters, M.; Santagostino, E.; Platokouki, H.; Beckers, E.; Voorberg, J.; et al. Desmopressin in moderate hemophilia A patients: A treatment worth considering. Haematologica 2018, 103, 550–557. [Google Scholar] [CrossRef]
- Candy, V.; Whitworth, H.; Grabell, J.; Thibeault, L.; Harpell, L.; Bowman, M.; Good, D.; Hopman, W.M.; Sidonio, R.F., Jr.; James, P.D. A decreased and less sustained desmopressin response in hemophilia A carriers contributes to bleeding. Blood Adv. 2018, 2, 2629–2636. [Google Scholar] [CrossRef]
- Chen, Z.; Herzog, R.W.; Kaufman, R.J. Cellular stress and coagulation factor production: When more is not necessarily better. J. Thromb. Haemost. 2023, 21, 3329–3341. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef]
- Yakovleva, E.; Zhang, B. Clinical, Laboratory, Molecular, and Reproductive Aspects of Combined Deficiency of Factors V and VIII. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers, Inc.: New York, NY, USA, 2024. [Google Scholar] [CrossRef]
- Zhang, Y.; Srivastava, V.; Zhang, B. Mammalian cargo receptors for endoplasmic reticulum-to-Golgi transport: Mechanisms and interactions. Biochem. Soc. Trans. 2023, 51, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef]
- Poothong, J.; Pottekat, A.; Siirin, M.; Campos, A.R.; Paton, A.W.; Paton, J.C.; Lagunas-Acosta, J.; Chen, Z.; Swift, M.; Volkmann, N.; et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. Blood 2020, 135, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916. [Google Scholar] [CrossRef]
- Hakeos, W.H.; Miao, H.; Sirachainan, N.; Kemball-Cook, G.; Saenko, E.L.; Kaufman, R.J.; Pipe, S.W. Hemophilia A mutations within the factor VIII A2-A3 subunit interface destabilize factor VIIIa and cause one-stage/two-stage activity discrepancy. Thromb. Haemost. 2002, 88, 781–787. [Google Scholar] [CrossRef]
- Pipe, S.W.; Kaufman, R.J. Factor VIII C2 domain missense mutations exhibit defective trafficking of biologically functional proteins. J. Biol. Chem. 1996, 271, 25671–25676. [Google Scholar] [CrossRef]
- Summers, R.J.; Meeks, S.L.; Healey, J.F.; Brown, H.C.; Parker, E.T.; Kempton, C.L.; Doering, C.B.; Lollar, P. Factor VIII A3 domain substitution N1922S results in hemophilia A due to domain-specific misfolding and hyposecretion of functional protein. Blood 2011, 117, 3190–3198. [Google Scholar] [CrossRef]
- Gilbert, G.E.; Novakovic, V.A.; Kaufman, R.J.; Miao, H.; Pipe, S.W. Conservative mutations in the C2 domains of factor VIII and factor V alter phospholipid binding and cofactor activity. Blood 2012, 120, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Misra, S.; Cannon, M.V.; Yang, R.; Zhu, X.; Gilmore, R.; Zhu, M.; Zhang, B. Molecular mechanisms of missense mutations that generate ectopic N-glycosylation sites in coagulation factor VIII. Biochem. J. 2018, 475, 873–886. [Google Scholar] [CrossRef]
- Wei, W.; Zheng, C.; Zhu, M.; Zhu, X.; Yang, R.; Misra, S.; Zhang, B. Missense mutations near the N-glycosylation site of the A2 domain lead to various intracellular trafficking defects in coagulation factor VIII. Sci. Rep. 2017, 7, 45033. [Google Scholar] [CrossRef]
- Grandjean, J.M.D.; Wiseman, R.L. Small molecule strategies to harness the unfolded protein response: Where do we go from here? J. Biol. Chem. 2020, 295, 15692–15711. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W. Pharmacologic Approaches for Adapting Proteostasis in the Secretory Pathway to Ameliorate Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2020, 12, a034108. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 2022, 21, 115–140. [Google Scholar] [CrossRef]
- Tannous, B.A.; Kim, D.E.; Fernandez, J.L.; Weissleder, R.; Breakefield, X.O. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 2005, 11, 435–443. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Zhu, M.; Zhang, B. Identification of candidate nonsense mutations of FVIII for ribosomal readthrough therapy. Haematologica 2019, 104, e573–e576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kaufman, R.J.; Ginsburg, D. LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. J. Biol. Chem. 2005, 280, 25881–25886. [Google Scholar] [CrossRef]
- Bouchecareilh, M.; Hutt, D.M.; Szajner, P.; Flotte, T.R.; Balch, W.E. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of alpha1-antitrypsin deficiency. J. Biol. Chem. 2012, 287, 38265–38278. [Google Scholar] [CrossRef]
- Di, X.J.; Wang, Y.J.; Cotter, E.; Wang, M.; Whittsette, A.L.; Han, D.Y.; Sangwung, P.; Brown, R.; Lynch, J.W.; Keramidas, A.; et al. Proteostasis Regulators Restore Function of Epilepsy-Associated GABA(A) Receptors. Cell Chem. Biol. 2021, 28, 46–59.e47. [Google Scholar] [CrossRef]
- Han, D.Y.; Di, X.J.; Fu, Y.L.; Mu, T.W. Combining valosin-containing protein (VCP) inhibition and suberanilohydroxamic acid (SAHA) treatment additively enhances the folding, trafficking, and function of epilepsy-associated gamma-aminobutyric acid, type A (GABAA) receptors. J. Biol. Chem. 2015, 290, 325–337. [Google Scholar] [CrossRef]
- Hutt, D.M.; Herman, D.; Rodrigues, A.P.; Noel, S.; Pilewski, J.M.; Matteson, J.; Hoch, B.; Kellner, W.; Kelly, J.W.; Schmidt, A.; et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010, 6, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Mumford, A.D.; Laffan, M.; O’Donnell, J.; McVey, J.H.; Johnson, D.J.; Manning, R.A.; Kemball-Cook, G. A Tyr346→Cys substitution in the interdomain acidic region a1 of factor VIII in an individual with factor VIII:C assay discrepancy. Br. J. Haematol. 2002, 118, 589–594. [Google Scholar] [CrossRef]
- Paton, A.W.; Beddoe, T.; Thorpe, C.M.; Whisstock, J.C.; Wilce, M.C.; Rossjohn, J.; Talbot, U.M.; Paton, J.C. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Miskiewicz, P.; Milczarek-Banach, J.; Rutkowska-Hinc, B.; Kondracka, A.; Bednarczuk, T. High-dose intravenous methylprednisolone therapy in patients with Graves’ orbitopathy is associated with the increased activity of factor VIII. J. Endocrinol. Investig. 2019, 42, 217–225. [Google Scholar] [CrossRef]
- Ozturk, G.; Ozsoylu, S.; Gursel, T. Effects of methylprednisolone on FVIII:C and vWF levels. Eur. J. Haematol. 1994, 53, 119–120. [Google Scholar] [CrossRef]
- Ozsoylu, S.; Strauss, H.S.; Diamond, L.K. Effects of corticosteroids on coagulation of the blood. Nature 1962, 195, 1214–1215. [Google Scholar] [CrossRef]
- Halsall, J.A.; Turner, B.M. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. BioEssays 2016, 38, 1102–1110. [Google Scholar] [CrossRef]
- DuBois, S.G.; Granger, M.M.; Groshen, S.; Tsao-Wei, D.; Ji, L.; Shamirian, A.; Czarnecki, S.; Goodarzian, F.; Berkovich, R.; Shimada, H.; et al. Randomized Phase II Trial of MIBG Versus MIBG, Vincristine, and Irinotecan Versus MIBG and Vorinostat for Patients With Relapsed or Refractory Neuroblastoma: A Report From NANT Consortium. J. Clin. Oncol. 2021, 39, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Saldanha, S.A.; Hodder, P.; Tait, B.D.; Garza, D.; et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2011, 8, 185–196. [Google Scholar] [CrossRef]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative strategies to treat protein misfolding in inborn errors of metabolism: Pharmacological chaperones and proteostasis regulators. J. Inherit. Metab. Dis. 2014, 37, 505–523. [Google Scholar] [CrossRef]
- Lu, J.; Yang, C.; Chen, M.; Ye, D.Y.; Lonser, R.R.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc. Natl. Acad. Sci. USA 2011, 108, 21200–21205. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Rahimpour, S.; Lu, J.; Pacak, K.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. USA 2013, 110, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Poothong, J.; Jang, I.; Kaufman, R.J. Defects in Protein Folding and/or Quality Control Cause Protein Aggregation in the Endoplasmic Reticulum. Cell. Biol. Endoplasmic Reticulum 2021, 59, 115–143. [Google Scholar] [CrossRef]
- Otero, J.H.; Lizak, B.; Hendershot, L.M. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 2010, 21, 472–478. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. The degradation pathway of a model misfolded protein is determined by aggregation propensity. Mol. Biol. Cell 2018, 29, 1422–1434. [Google Scholar] [CrossRef]
- Roth, S.D.; Schuttrumpf, J.; Milanov, P.; Abriss, D.; Ungerer, C.; Quade-Lyssy, P.; Simpson, J.C.; Pepperkok, R.; Seifried, E.; Tonn, T. Chemical chaperones improve protein secretion and rescue mutant factor VIII in mice with hemophilia A. PLoS ONE 2012, 7, e44505. [Google Scholar] [CrossRef]
- Andersen, E.; Chollet, M.E.; Baroni, M.; Pinotti, M.; Bernardi, F.; Skarpen, E.; Sandset, P.M.; Skretting, G. The effect of the chemical chaperone 4-phenylbutyrate on secretion and activity of the p.Q160R missense variant of coagulation factor FVII. Cell Biosci. 2019, 9, 69. [Google Scholar] [CrossRef]
- Pignani, S.; Todaro, A.; Ferrarese, M.; Marchi, S.; Lombardi, S.; Balestra, D.; Pinton, P.; Bernardi, F.; Pinotti, M.; Branchini, A. The chaperone-like sodium phenylbutyrate improves factor IX intracellular trafficking and activity impaired by the frequent p.R294Q mutation. J. Thromb. Haemost. 2018, 16, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.C.; Dougan, S.K.; Winter, S.V.; Paton, A.W.; Paton, J.C.; Ploegh, H.L. Subtilase cytotoxin cleaves newly synthesized BiP and blocks antibody secretion in B lymphocytes. J. Exp. Med. 2009, 206, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Codon | Protein | Domain | Exon | Patients | Severity | |
|---|---|---|---|---|---|---|---|
| 1 | c.1834C>T | CGC-TGC | p.R612C | A2 | 12 | 242 | Mild/Moderate |
| 2 | c.6506G>A | CGT-CAT | p.R2169H | C1 | 23 | 182 | Moderate/Mild |
| 3 | c.6046C>T | CGG-TGG | p.R2016W | A3 | 19 | 100 | Moderate/Severe |
| 4 | c.1094A>G | TAT-TGA | p.Y365C | A1 | 8 | 97 | Mild |
| 5 | c.6532C>T | CGC-TGC | p.R2178C | C1 | 23 | 93 | Mild |
| 6 | c.5954G>A | CGA-CAA | p.R1985Q | A3 | 18 | 89 | Mild |
| 7 | c.1648C>T | CGC-TGC | p.R550C | A2 | 11 | 89 | Mild/Moderate |
| 8 | c.1636C>T | CGG-TGG | p.R546W | A2 | 11 | 87 | Mild |
| 9 | c.6545G>A | CGC-CAC | p.R2182H | C1 | 23 | 67 | Severe/Moderate |
| 10 | c.5822A>G | AAT-AGT | p.N1941S | A3 | 18 | 67 | Severe/Moderate |
| 11 | c.1649G>A | CGC-CAC | p.R550H | A2 | 11 | 64 | Mild |
| 12 | c.6089G>A | AGC-AAC | p.S2030N | A3 | 19 | 63 | Mild |
| 13 | c.541G>A | GTG-ATG | p.R181M | A1 | 4 | 60 | Mild/Moderate |
| 14 | c.6683G>A | CGA-CAA | p.R2228Q | C2 | 24 | 57 | Severe/Moderate |
| 15 | c.6744G>T | TGG-TGT | p.R2248C | C2 | 25 | 43 | Mild/Moderate |
| 16 | c.902G>A | CGC-CAC | p.R301H | A1 | 7 | 43 | Severe |
| 17 | c.873G>A | GAA-AAA | p.E291K | A1 | 7 | 41 | Mild/Moderate |
| 18 | c.6104T>C | GTG-GCG | p.V2035A | A3 | 19 | 38 | Mild |
| 19 | c.1754T>C | ATA-ACA | p.I585T | A2 | 12 | 10 | Severe |
| 20 | c.1801A>G | AAC-GAC | p.N601D | A2 | 12 | 4 | Severe |
| 21 | c.1808G>C | AGC-ACC | p.S603T | A2 | 12 | 1 | Severe |
| 22 | c.1898T>C | ATG ACG | p.M633T | A2 | 12 | 1 | Mild |
| 23 | c.1462G>A | GCA-TCA | p.A488S | A2 | 10 | 1 | Not reported |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, V.; Liu, Z.; Wei, W.; Zhang, Y.; Paton, J.C.; Paton, A.W.; Mu, T.; Zhang, B. Cell-Based Small-Molecule Screening Identifying Proteostasis Regulators Enhancing Factor VIII Missense Mutant Secretion. Biomolecules 2025, 15, 458. https://doi.org/10.3390/biom15040458
Srivastava V, Liu Z, Wei W, Zhang Y, Paton JC, Paton AW, Mu T, Zhang B. Cell-Based Small-Molecule Screening Identifying Proteostasis Regulators Enhancing Factor VIII Missense Mutant Secretion. Biomolecules. 2025; 15(4):458. https://doi.org/10.3390/biom15040458
Chicago/Turabian StyleSrivastava, Vishal, Zhigang Liu, Wei Wei, Yuan Zhang, James C. Paton, Adrienne W. Paton, Tingwei Mu, and Bin Zhang. 2025. "Cell-Based Small-Molecule Screening Identifying Proteostasis Regulators Enhancing Factor VIII Missense Mutant Secretion" Biomolecules 15, no. 4: 458. https://doi.org/10.3390/biom15040458
APA StyleSrivastava, V., Liu, Z., Wei, W., Zhang, Y., Paton, J. C., Paton, A. W., Mu, T., & Zhang, B. (2025). Cell-Based Small-Molecule Screening Identifying Proteostasis Regulators Enhancing Factor VIII Missense Mutant Secretion. Biomolecules, 15(4), 458. https://doi.org/10.3390/biom15040458