1. Introduction
Gravity is ubiquitously affecting life on Earth. All organisms must adapt to handle this force, which is constant in strength and orientation. Man is the first species in life history that has been able to live for a prolonged time in microgravity. Near-zero gravity is a challenging situation for both the psyche and physique and is also a decisive parameter on the cellular level [
1]. However, despite the fact that gravity force is constant in strength and orientation, mobile organisms change their orientation versus gravity due to movement and, therefore, vary the toehold of the vector. A reduction in this movement is a non-physiological situation, and in humans, the prevention of movement (“bed rest”) can be utilized as an analogue to mimic the effects of zero gravity in space [
2]. Many body functions, e.g., the cardiovascular system, are highly susceptible to this challenge. Just recently, it has been recognized that these complex systems are not only influenced as a whole by the omission of gravity or the frequent change in orientation, but effects are also found at the cellular and subcellular levels [
3].
The simulation of microgravity through continuous changes in orientation in samples versus the gravity vector is an accepted ground-based method in space research [
4]. Sophisticated motion sequences generated by so-called random-positioning machines (RPMs) result in the average gravity over time being near zero. In contrast to real microgravity, the cellular structures on the RPM are exposed to permanent load cycles, a situation which is, due to gut peristaltic, similar for mammal intestinal enterocytes in vivo.
On the other hand, the question arises of whether the dynamic change in gravity vector orientation is necessary for correct function on the cellular level. The early development of vertebrates under complete unloading conditions in space often resulted in malformations and developmental disorders [
5].
This means that the inclusion of gravity is especially important in the context of in vitro cell culture systems that are widely used in research and development. Cell culture methods are essential in vitro tools in biomedical research, enabling the analysis of cellular processes under simplified controlled conditions. The validity of the obtained results is essentially dependent on the capacity of the model system to mimic the in vitro system. In contrast to biochemical inputs, the role of physical factors is addressed far less frequently.
A preeminent example of biological structures in a dynamic environment is epithelial cells or enterocytes in the intestinal tract. Due to the functional requirements necessary for transport, the mixing and digestion of irregularly shaped bolus and liquid nutrition, the intestine is a highly dynamic system, and the cellular components must adapt to this environment. The multifarious mechanical stimuli influencing the epithelial and non-epithelial cellular components of the intestine play an essential role in proliferation and migration [
6]. Moreover, the peristaltic activity, as reflected by the neuronal spikeburst periodicity, triggers contractions in 10 to 20 min intervals. Together with other classes of contractions, the mucosa is rarely completely static in living organisms [
7]. Consequently, intestinal epithelial cells are constantly exposed to various orientations of the gravity vector caused by the gut dynamics and movement of the whole organism [
6]. As an interface between the internal body milieu and the outside world, enterocytes of the mucosa play a central role in nutrient uptake, immune response, food allergies, microbiome interaction, and toxin defence [
8]. Here, we introduce this dynamic aspect into the cultivation of intestinal epithelial cell culture mimicking the in vivo motility of the intestine. In the present investigation, we asked the question of whether a constant change in cellular orientation versus the Earth’s gravity is a relevant factor influencing cellular morphology, growth, and energy metabolism in enterocytes.
2. Materials and Methods
2.1. Cell Culture Mode
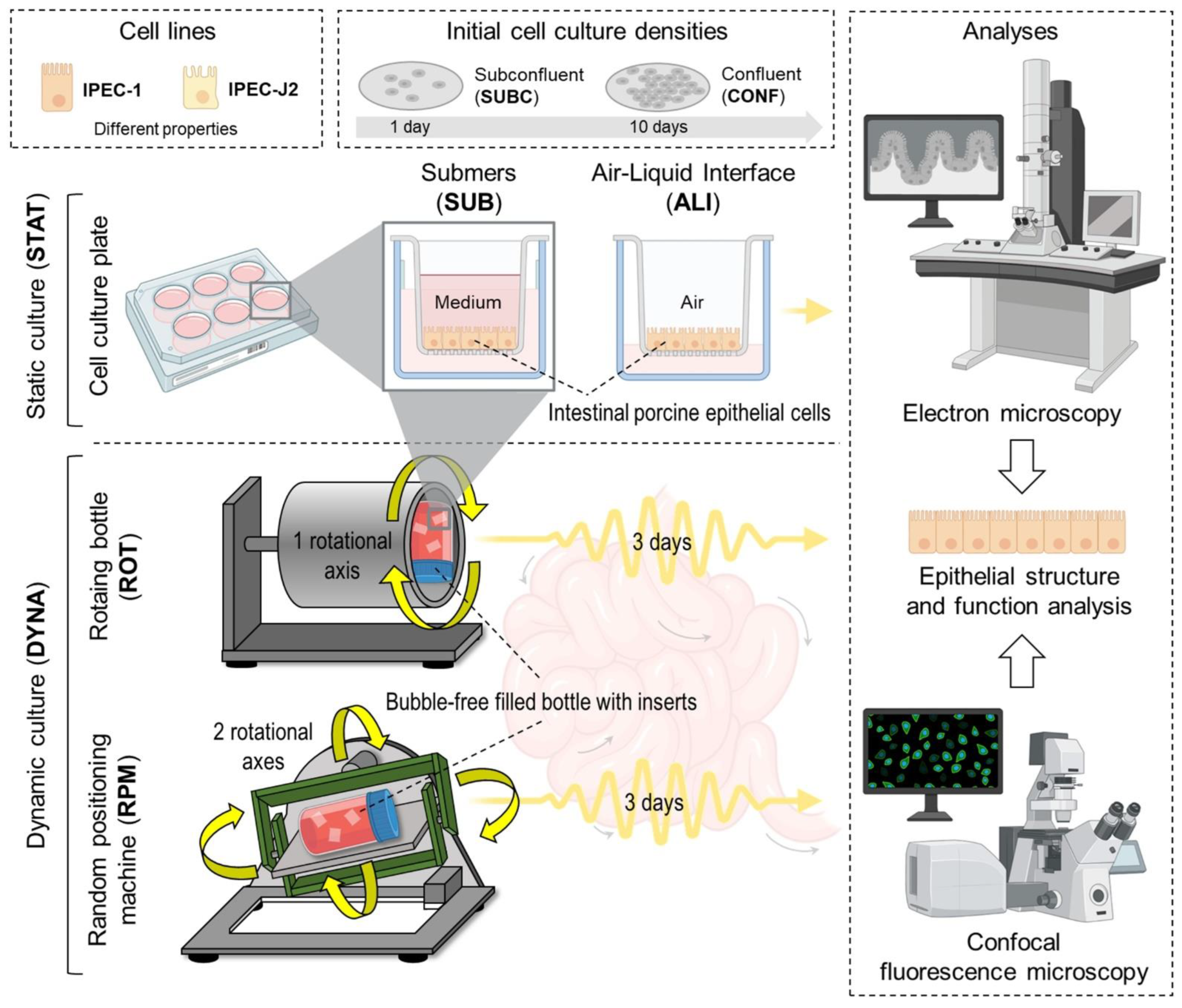
We developed and applied a set of six different culture conditions mimicking various in vivo situations in the intestinal epithelium, as summarized in
Figure 1. A. Mode of motion. B. Initial cell density.
2.1.1. Static Submers/Subconfluent (STAT-SUB/SUBC)
The cells were grown submerged without movement for 4 days (1 day +3 days) before analysis. Cells were still subconfluent after 1 day.
2.1.2. Static Submerse/Confluent (STAT-SUB/CONF)
This condition is the standard situation in conventional cell culture and is utilized as a reference for comparison. Here, the cells were grown for 13 days (10 days +3 days) on membranes without any movement besides handling for medium exchange.
2.1.3. Dynamic Random-Positioning Machine/Confluent (DYNA-RPM/CONF)
The cells were grown for 10 days to reach confluence and were then exposed to microgravity (>0.01 µg) in RPM for 3 days. This condition is a model for zero gravity in space; however, the cells were still exposed to gravitational acceleration.
2.1.4. Dynamic Rotating Vessel/Subconfluent (DYNA-ROT/SUBC)
Here, the subconfluent membrane cultures (1 day) were placed in a rotating vessel (ROT) and cultured for another 3 days. In contrast to the above (dynamic/confluent), growing cells were exposed to the dynamic situation, mimicking the epithelial constellation on the crypt base (cell division).
2.1.5. Dynamic Rotating Vessel/Confluent (DYNA-ROT/CONF)
Confluent membrane cultures (10 days) were placed in a rotating vessel (ROT) after 10 days of preincubation and cultured for another 3 days. Similarly to the RPM condition, the cells on the membranes were exposed to gravity in a random orientation; however, the integral of the gravitational acceleration was not near zero (microgravity). This culture condition is an approximation for the confluent epithelial cell layer distal of the crypt base.
2.1.6. Static Air–Liquid Interface/Subconfluent (STAT-ALI/SUBC)
The cells were cultured under static conditions; the apical medium was reduced to a thin film. This maximized the oxygen supply to the cells by reducing the diffusion gradients.
2.2. Cell Culture
In all experiments, cells were seeded with a density of 1 × 10
5 cells/cm
2 on a permeable support (ThinCerts; pore size: 1 µm; polyester, Greiner bio-one, Frickenhausen, Germany). DMEM/HAMs F12 supplemented with 5% fetal bovine serum (FBS), ITS (insulin–transferrin–selenium), 5 mL/500 mL of cell culture medium, 16 mM of 4-(2-hydroxyethyl)-1-piperazineethansulfonic acid (HEPES) (all: PAN-Biotech, Aidenbach, Germany) and 5 ng/mL of the epidermal growth factor (EGF, Biochrome, Berlin, Germany) were used as the cell culture medium. The cells grew at 39 °C in an atmosphere of 5% CO
2 and 95% relative humidity. The transepithelial electrical resistance (TEER) was measured using a Millicell-TERS electrode (Millipore, Berlin, Germany). Electrodes in the basal and apical compartment of the membrane insert could perform sensitive detection to determine the intactness of the epithelial layer. Intestinal porcine epithelial cells (IPEC-1, ACC 705; IPEC J2, ACC701; [
9,
10]) were regularly tested and found to be free of mycoplasma contamination (Venor GeM Mycoplasma Detection Kit; Minerva Biolabs, Berlin, Germany).
2.3. Cultivation
The IPEC-1 and IPEC-J2 epithelial cell lines were cultured in a static and dynamic environment. Static conditions include a conventional cell culture environment with cells on the bottom of a liquid column and air–liquid cultures, where the apical liquid column is reduced to a thin film (approximately 20 µL/cm
2 [
11]). Dynamic conditions were realized by a rotating vessel system (a 100 mL bottle fixed in a plastic tube with overhead rotation at 60 rpm; DYNA-ROT) and a microgravity-simulating system (DYNA-RPM) [
12]. In dynamic conditions, membrane inserts were allowed to move freely in the container bottle. See
Figure 1 for more detail.
2.4. Immunofluorescence Staining
The cells were fixed with Histofix (Roth, 3% PFA) or with ethanol (30 min, 4 °C) and acetone (90 s, −20 °C precooled). Phosphate buffer (PB) at 0.1 M was used as the wash buffer, and every step was performed three times for 5 min (RT). After washing the cells, they were permeabilized with 0.3% Triton/0.1 M PB for 30 min (RT), blocked with 1% normal goat serum (Biozol, Eching, Germany) in 0.1 M PB supplemented with 10% bovine serum albumin (30 min, RT), and washed.
2.5. F-Actin Was Labelled with Phalloidin-A488 (1:50) in 0.1 M PB for 2 h
Primary antibodies were incubated at room temperature (rabbit anti-ZO-1, Invitrogen, ThermoFisher Scientific, Darmstadt, Germany, 40–2200; 1:100 in 0.1 M PB) for 2 h. Cells were washed and supplemented with a secondary antibody (goat anti-rabbit IgG2b Alexa 555, Life Technology, Darmstadt, Germany; 1:400) for 1 h at room temperature. After undergoing washing, the cells were counterstained with DAPI (1:10 in 0.1 M PB) and washed again. Membranes were embedded in a Vectashield-filled micro-chamber consisting of spacers and the top coverslip.
2.6. RNA Isolation of IPEC-1 and qPCR
After the withdrawal of the apical and basolateral medium, the cells were covered with the TRIzol reagent (Invitrogen), as described by the manufacturer’s protocol, and the membrane was scraped off. After adding chloroform to the cell lysate, the supernatant was extracted, and RNA was precipitated using isopropanol alcohol. Using 75% ethanol, the RNA was purified and stored in RNA-free water peqGOLD (peqlab, VWR, Darmstadt, Germany) at −80 °C until further processing.
RNA obtained from five independently repeated experiments was used as the template for qPCR. Each 1 µg of the template RNA was subjected to reverse transcription with First Aid Reverse Transcription Reagents (Fermentas, ThermoFisher Scientific, Darmstadt, Germany), as described by the manufacturer with the supplied random hexamer primers in ThermalCycler TC1 (Biometra, Göttingen, Germany). The resulting cDNA samples were used for the qPCR amplification of the seven chosen genes.
Quantitative PCR amplification was performed for all genes under the following conditions on an iCycler (BioRad, Feldkirchen, Germany): 5 min at 95 °C followed by 40 cycles of 30 s at 95 °C and 60 s at an optimal primer annealing temperature (
Table 1). The melting curve analysis (50–95 °C) was used to assess amplification specificity. The reaction volume of 20 µL contained 10 µL Maxima Mastermix (2×, Fermentas, Germany) with SYBR
® Green and Fluorescein used as the internal standard, 3 µL of the respective primers (2 pmol/µL), 2 µL nuclease-free water, and 2 µL cDNA (60 ng/µL).
The analysis comprised five independent experiments with each sample in triplicate. The results are given as Ct to avoid uncertainties of potentially regulated housekeeper genes.
2.7. Western Blot Experiments and Quantitative Analysis
The fractionation of cell lysate was performed with NE-PER Nuclear and Cytoplasmic Extraction reagents (Thermo Fisher Scientific, Darmstadt, Germany, 88332) according to the manufacturer’s instructions. Separation efficiency was monitored by the presence or absence of Histone H3 (Cell Signalling; Danvers, MA, USA; #9715; 1:1000) in the cytosolic and nuclear fraction. The protein fractions were treated as described below. Samples for SDS-PAGE and Western blot analysis protein fractions were solubilized with the 4×SDS sample buffer (250 mM Tris-HCl, pH 6.8, 1% SDS, 40% glycerol, 20% β-mercaptoethanol, and 0.004% bromophenol blue), boiled for five minutes (95 °C) and separated on 5–20% SDS–Polyacrylamide gradient gels, subsequently followed by transfer onto nitro-cellulose membranes. Transfer efficiency was routinely monitored by Ponceau staining (Ponceau S solution; Cell Signalling; #59803; 5 min staining and distaining with H2O and 0.1% Tween 20 in 1×TBS). After blotting, membranes were blocked with a blocking solution (5% dry milk, 0.1% Tween 20 in 1×TBS) for 1 h. Incubation with primary antibodies (mouse monoclonal anti-β-Actin, Sigma-Aldrich, Merck, Darmstadt, Germany; A1978 clone AC-15; 1:10,000) was performed at 4 °C overnight in a blocking solution. After intensive washing, the blots were incubated for 90 min at room temperature with HRP-conjugated secondary antibodies (1:10,000) in the blocking solution, and these were finally developed with the ECL reagent (Thermo Fisher Scientific) using the Odyssey® Fc luminescence detector (LI-COR, Bad Homburg, Germany).
For normalization, identical samples were separated by SDS-PAGE and stained for 1 h with 0.05% Coomassie brilliant blue dissolved in 50% methanol and 10% acetic acid, followed by distaining using a solution consisting of 5% methanol and 7% acetic acid. The determination and quantification of the Western blot signals and Coomassie-stained gels were performed using the Image Studio Lite version 5.0 software from LI-COR.
2.8. Confocal Microscopy
The slides were examined using a ZEISS LSM 800 confocal laser scanning microscope (Carl Zeiss, Göttingen, Germany). The system was equipped with a laser module (405,488,561,640 nm) and an Airyscan detector module (Carl Zeiss, Göttingen, Germany). The standard objective was Plan-Apochromat 63×/1.4 oil. The standard z-step size was 170 nm. To ensure comparability for the intensity quantification, all the images were acquired with the same settings using the ZEISS Airyscan detector and ZEN 3.4 software (Carl Zeiss, Göttingen, Germany). The Airyscan processing settings were optimized for each antibody–wavelength combination and manually applied to the corresponding samples. To check for the non-specific binding of the secondary antibody and, thus, a false positive signal, the secondary antibodies were applied to separate the samples of the same condition without the primary antibody. The epithelial height was estimated in the orthogonal view of 3D reconstruction above the centre of the nucleus.
2.9. Electron Microscopy
Samples were treated with a fixation solution containing 0.5% paraformaldehyde and 0.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) overnight. After fixation, the dishes, membranes, and tissue were washed with phosphate buffer, cut out, and kept in a phosphate buffer. Subsequently, samples were treated with 1% osmium tetroxide (Science Services, Munich, Germany) in 0.1 M phosphate buffer.
Samples were rinsed in a buffer, dehydrated, and bloc-contrasted for 1 h with 1% uranyl acetate in 70% ethanol. Finally, the dishes, membranes, and tissue were infiltrated with Durcupan ACM (Fluka, Buchs, Switzerland) and embedded flat between plastic foil. The resin was polymerized in an oven at 70 °C for 2 days. Regions of interest were cut out and glued vertically onto a blank block of resin with a small groove. With an Ultracut S ultramicrotome (Reichert, Leica AG, Vienna, Austria), semi-thin sections (1 µm) were cut and counterstained with toluidine blue for light microscopic measurements on the slides. Ultrathin sections (50–70 nm) were collected on Formvar-coated slot grids of copper. They were further contrasted with 2% uranyl acetate and 0.1% lead citrate and examined in a LEO 906 E transmission electron microscope of Zeiss (Oberkochen, Germany) equipped with a digital 1K BioScan camera (Gatan Inc., Pleasanton, CA, USA).
2.10. Analysis of Mitochondrial Function
After dynamic cultivation, a mitostress test was performed using a Seahorse Analyzer (Agilent Technologies, Waldbronn, Germany). The day before, a sensor cartridge had to be hydrated with 200 µL/well ampuwa® and at the same time, 20 mL of the calibrant (Agilent, Santa Clara, CA, USA) was stored overnight in the same incubator (37 °C; non-CO2). On the day of the experiment, ampuwa was aspirated, and the calibrant was added (200 µL/well; pre-warmed, overnight). Furthermore, a sensor cartridge was incubated for a further 45 min (37 °C; non-CO2) before starting the experiment. In the next step, an assay medium was prepared with the supplementation of 17.5 mM glucose, 1 mM pyruvate and 2.5 mM glutamine to the XF base medium (Agilent Technologies). The pH was adjusted to 7.40 at 37 °C with 0.1 M NaOH, and the medium was filtered in a sterile way. Stock solutions were produced according to the manufacturer’s protocol, which was used to prepare the operating concentrations (oligomycin: 15 µM; FCCP: 20 µM; rotenone/antimycin A: 5 µM). Membrane-cultured IPEC-1 cells were washed with the assay medium and circular (3.6 mm diameter) membrane sections were transferred into a Seahorse XF24 Islet Capture plate (Agilent) and fixed with a plastic grid. The cell layer was orientated versus the detector compartment. The cell plate was washed with the assay medium, and 500 µL of the assay medium was added to the wells, and the plate was incubated for 45 min at 37 °C/non-CO2. During incubation, ports of the sensor cartridge were filled (port A: 56 µL; port B: 62 µL; port: C: 69 µL) with operating concentrations of oligomycin, FCCP, and rotenone/antimycin A. The measurement was performed as follows: 3 × 3 cycles (15 min: 3 min mix; 2 min wait) and 1 × 9 cycles for rotenone/antimycin A.
2.11. Senescence-Associated Beta-Galactosidase (SA-ßgal)
Membrane-based cells were washed in PBS for 15 min and fixed in a fixation solution (2% formaldehyde (
v/
v), 0.2% glutaraldehyde (
v/
v)) in PBS. After 2 washing steps, the inserts were immersed (250 µL apical and basal) with a staining solution (40 mM citric acid/Na phosphate buffer, 5 mM K
4[Fe(CN)
6] 3H2O, 5 mM K
3[Fe(CN)
6], 150 mM sodium chloride, 2 mM magnesium chloride, and 1 mg/mL X-gal in distilled water, freshly prepared) and incubated at 37 °C overnight. The absorbance of staining was measured in the TECAN reader (Tecan, Crailsheim, Germany) at 690 nm, and membranes were subsequently counterstained with DAPI and inspected in a bright field and DAPI fluorescence channel. The protocol was used according to Debacq-Chainiaux et al. [
13]
2.12. Statistical Analysis
Data are expressed as boxplots. Data were tested on normal distribution using the Shapiro–Wilk test. In the case of normal distribution and two experimental groups, a paired t-test was applied (two-tailed). If not otherwise indicated, multi-group comparisons were performed with ANOVA and Dunnett’s post hoc test. In the case of non-normal distribution, Wilcoxon’s matched-pairs signed-rank test was used to compare two groups and the Kruskal–Wallis test was used for more than two experimental groups. (p ≤ 0.05 *; p ≤ 0.01 **; n.s. = not significant; GraphPad Prism 10, GraphPad Software, Boston, MA, USA).
4. Discussion
Microgravity simulated on Earth created by a continuous change in orientation versus the gravity vector influences cellular mechanisms at the subcellular level. The mechanical impact on cellular systems has long been underestimated. Parameters such as shear stress induced by streaming fluids, the stiffness and rigidity of matrix [
14] or gravity forces [
3] are key players in the processes defining the morphological and functional appearance of cellular structures and organs, e.g., in the digestive system [
15].
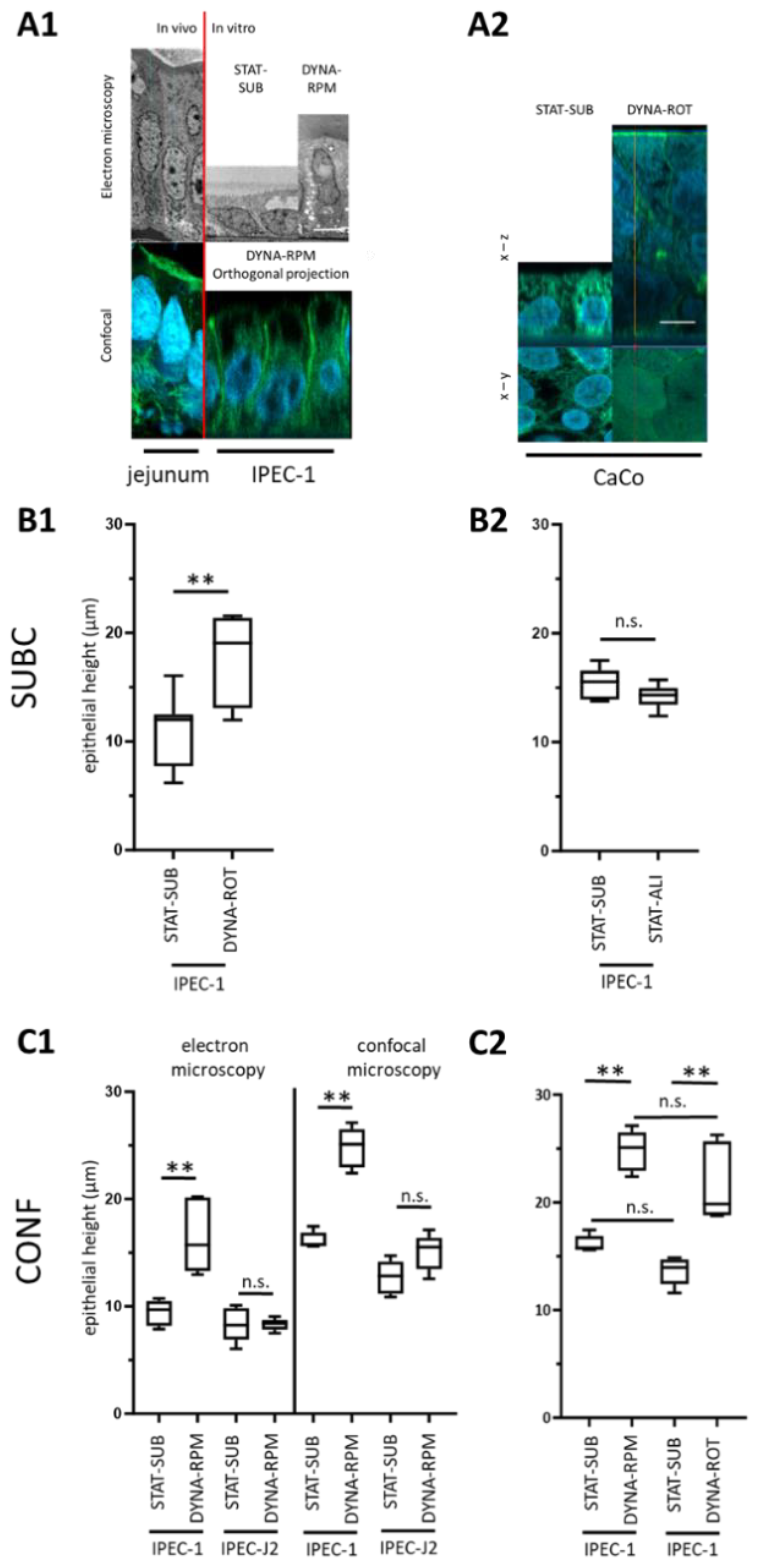
In vivo, the principal enterocyte morphology shows a columnar and luminal border as well as microvilli structures. The enterocyte cell height in vertebrates in vivo was found in a broad range. This range was reported to be between 30 and 45 µm in the phyton, depending on the nutrition composition [
16]. In mice, 37 µm from the microvillus tip to the basal membrane has been measured [
17]. A measurement of 30 µm was reported for the ileum of post-hatch chicks. In the duodenum and ileum, the height further increased to 50 to 60 µm without a strong change in width [
18]. The increase in cell height, as observed here in the dynamic approaches, is an indication of the role of the load cycle in the development of the epithelial cell line IPEC-1. As expected, the load cycle is not an isolated factor in development. We have shown in previous investigations that optimized oxygen supply (the air–liquid interface culture, ALI) can also trigger morphology modifications [
11]. ALI culture techniques have been shown to be valuable tools not only in respiratory cell culture models but also in intestinal cell culture approaches [
19]. In IPEC-1, the application of ALI changes the cellular morphology to a columnar shape. However, in those experiments, the cells were initially cultured for 10 days under conventional, static conditions and then transferred for 21 days to an air–liquid culture. The effects of dynamic cultivation were seen after only 3 days. As illustrated in
Figure 2(B2) the morphological effect after 3 days of ALI was absent. Besides the cell height, the formation of apical microvilli is an enterocyte-typical feature in vivo [
20]. About 2000 microvilli structures per cell were reported [
21]. In our simplified dynamic model, we found a significant but moderate increase in villin1 (
Figure 5D). However, in the EM and confocal approaches, we found no indications of an enhanced occurrence of microvilli (
Figure 3B,D).
The analysis of actin comprised the transcription level in the form of quantitative mRNA (defined by specific primers), the translation level of the monomer in the form of Western blot (defined by a specific antibody and molecular weight) and the structural appearance of polymerized actin (F-actin) defined by the binding of phalloidin [
22] and detection with confocal microscopy.
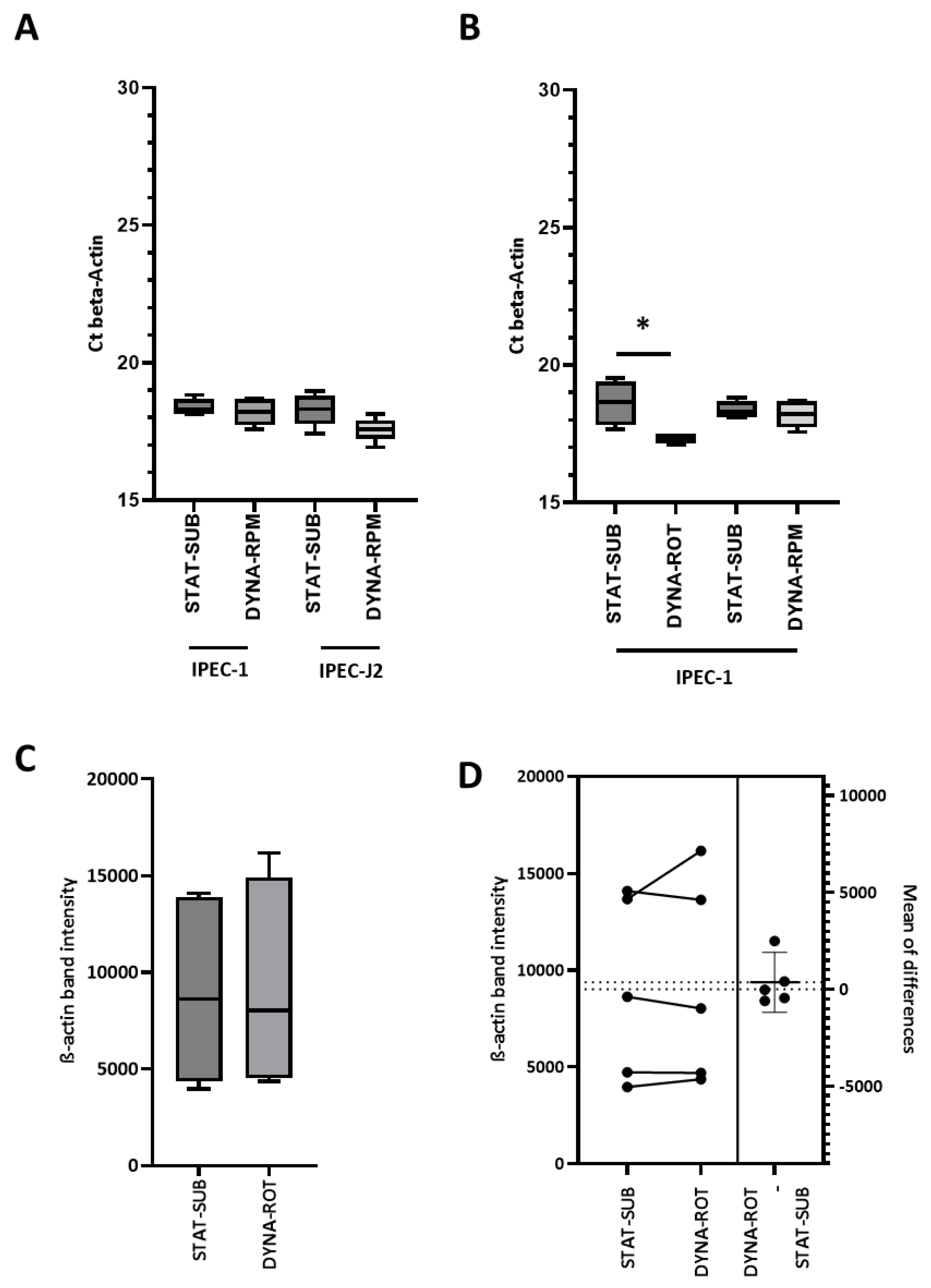
In
Figure 3A,B, we analyzed the mRNA levels of actin at static and dynamic levels. A significant increase in specific mRNA was only found with IPEC-1 cells, which were transferred into a rotating vessel (DYNA-ROT) in a subconfluent growth state. However, this increased mRNA level was not reflected in the overall actin protein level (
Figure 3C,D). We conclude that actin protein biosynthesis is not an essential part of the response of IPEC-1 cells to dynamic cell-culture conditions within the experimental time frame
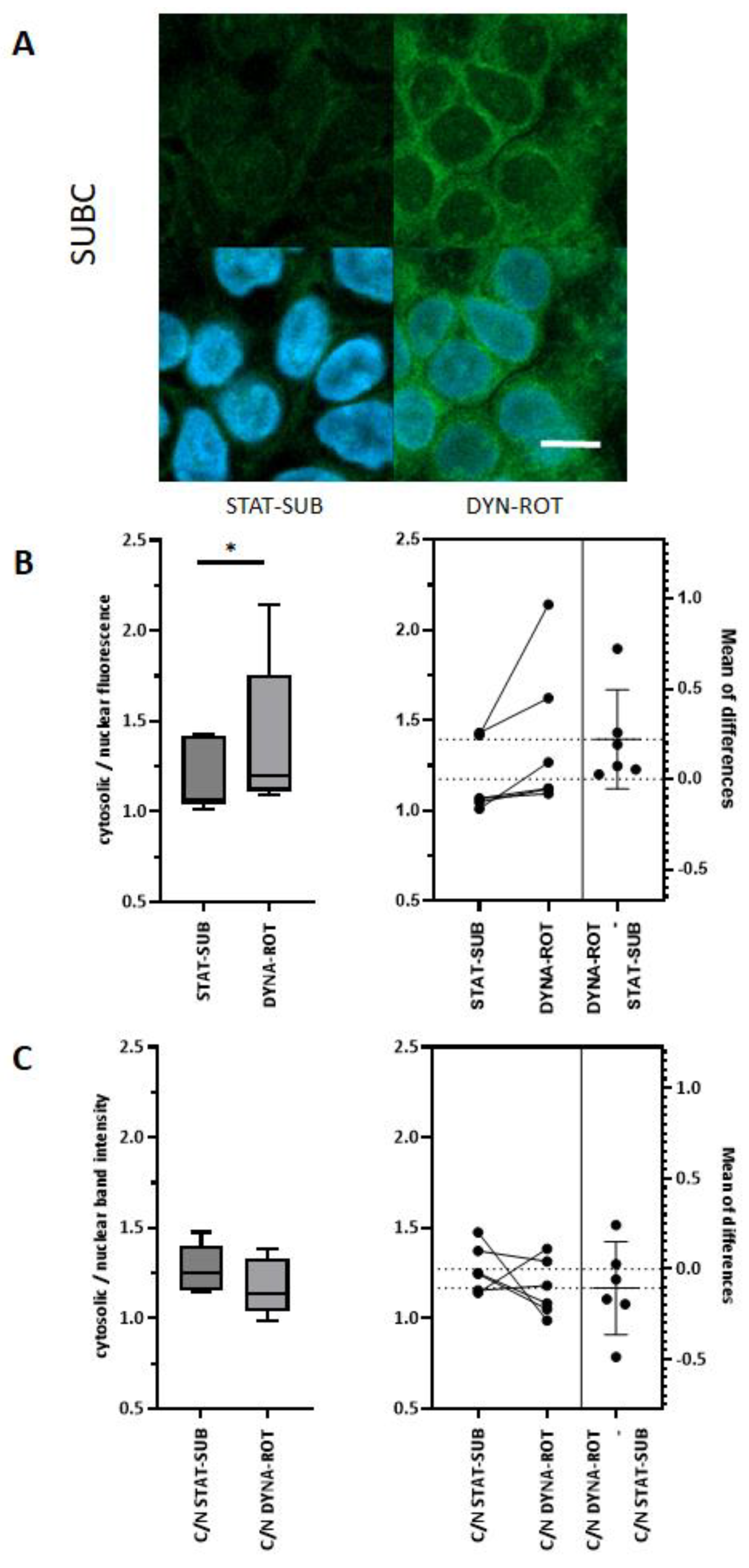
In the confocal microscopic approach, we found an enrichment of phalloidin-detectable F-actin in the perinuclear-cytosolic compartment. Shao et al. [
23] reported a similar observation in response to a mechanical stimulus in NIH 3T3 fibroblasts. In general, the effect of simulated microgravity on F-actin seems to be cell-type-dependent. In primary rat mesenchymal stem cells (BMSC), for example, a polymerized and thicker F-actin network was found after 24 h of clinorotation. In human umbilical vein endothelial cells (HUVEC), fewer microfilaments were detected after 24 h clinostat-exposure [
24]. Cell-type specificity was also found on the
ACTB expression level in mammary adenocarcinoma cells. Whereas minimally invasive MCF-7 showed a reduced amount of
ACTB mRNA, in corresponding highly invasive MDA-MB-231, the amount was increased after random positioning. Further differences were seen between 2D growth and 3D spheroid formation [
25]. The detection of G-actin in whole-cell homogenate did not indicate an influence on dynamic cultivation [
24].
A further clue to the role of motion in intestinal epithelial structures is the significant increase in villin mRNA, as shown in
Figure 8. It has been shown that villin is a trigger of the polymerization/depolymerization cycle in intestinal cells [
26].
It has been previously shown that the gene expression of components of the intestinal barrier is reduced in humans in space [
27]. The tight junction component ZO-1 did not exhibit prominent morphological characteristics when IPEC-1 was cultured in dynamic conditions. In the colorectal adenocarcinoma cell line HT-29, the distribution and localization of ZO-1 were rather modified compared to the protein expression.
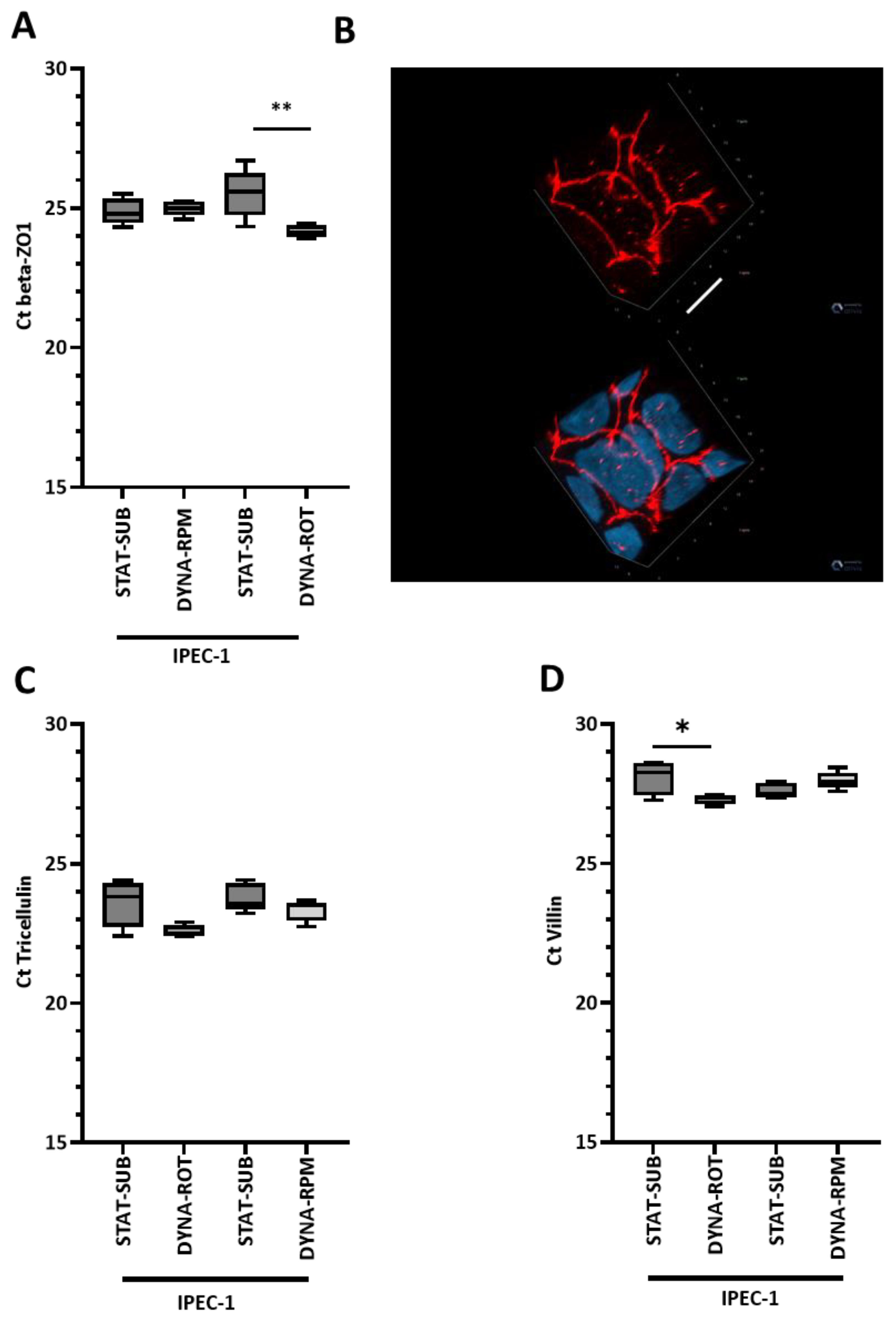
In IPEC-1, we also did not see an effect on the protein level, but a gentle significant increase in the TJP1 mRNA level was detected in the simplified dynamic system.
A previous study using a desktop RPM showed that after 3 days of rotation, there was a significant upregulation of ZO-1 in thyroid carcinoma cells but not in thyroid epithelial cells [
28]. Similarly, tricellulin, as another important component of the tight junction system [
29], was not significantly modified by dynamic conditions in either the structure or mRNA level.
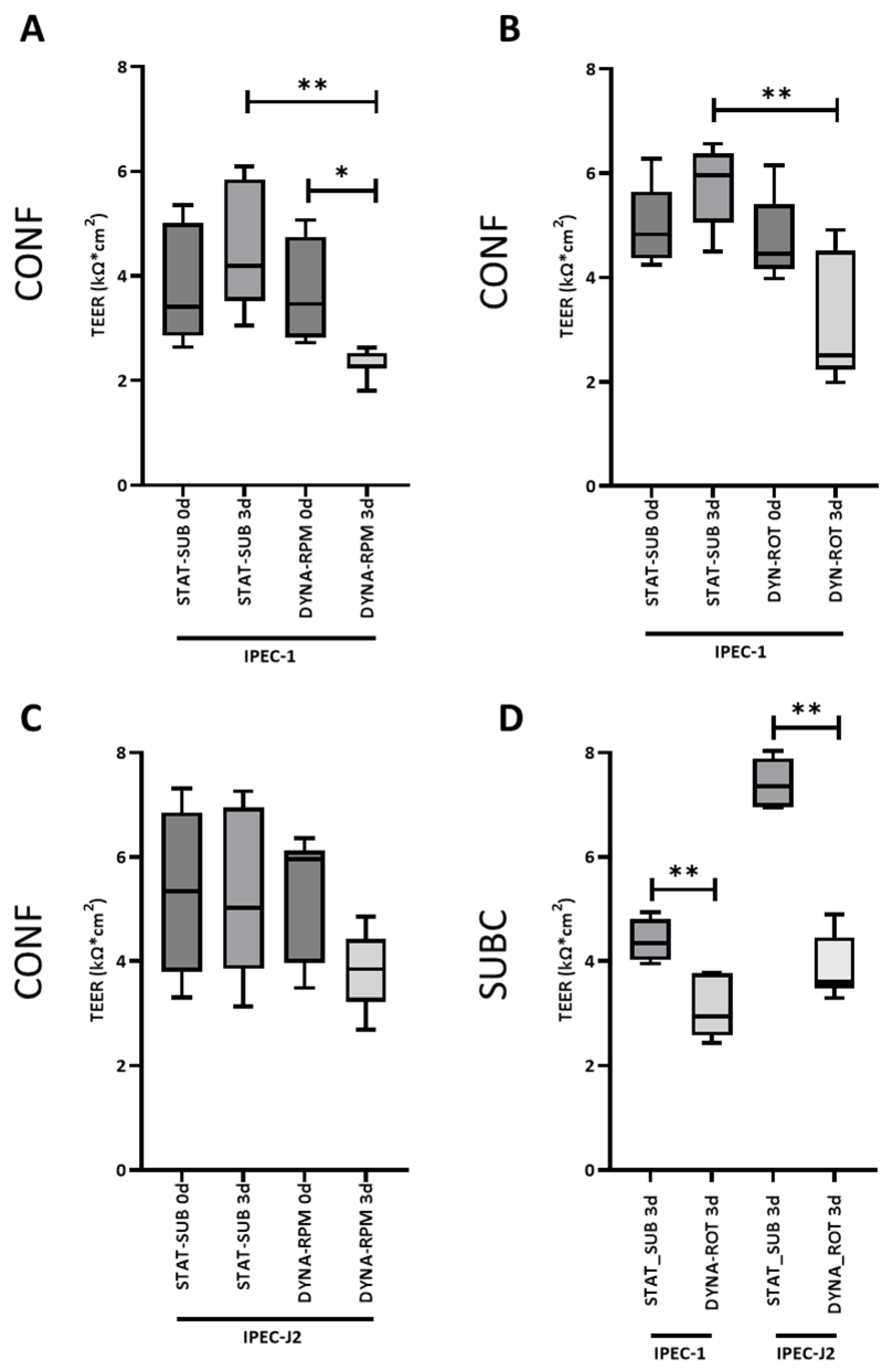
A direct, measurable effect of tight junctional complexes of an intact epithelial layer is electrical resistance (transepithelial electrical resistance; TEER). TEER is accessible in in vivo and in vitro culture systems. In vivo TEER values were reported from 50 to 400 Ω·cm
2. In many cell culture systems, electrical resistance is on a non-physiologic level, indicating a modified tight junction configuration [
30]. In our approach, the dynamic cultivation significantly reduced the TEER; however, it was still comparably high. This means, on the other hand, that the cell layer was still intact after RPM or ROT culture. A comparable but moderate effect was found in the HT-29 cell line cultured in a rotating wall vessel [
31].
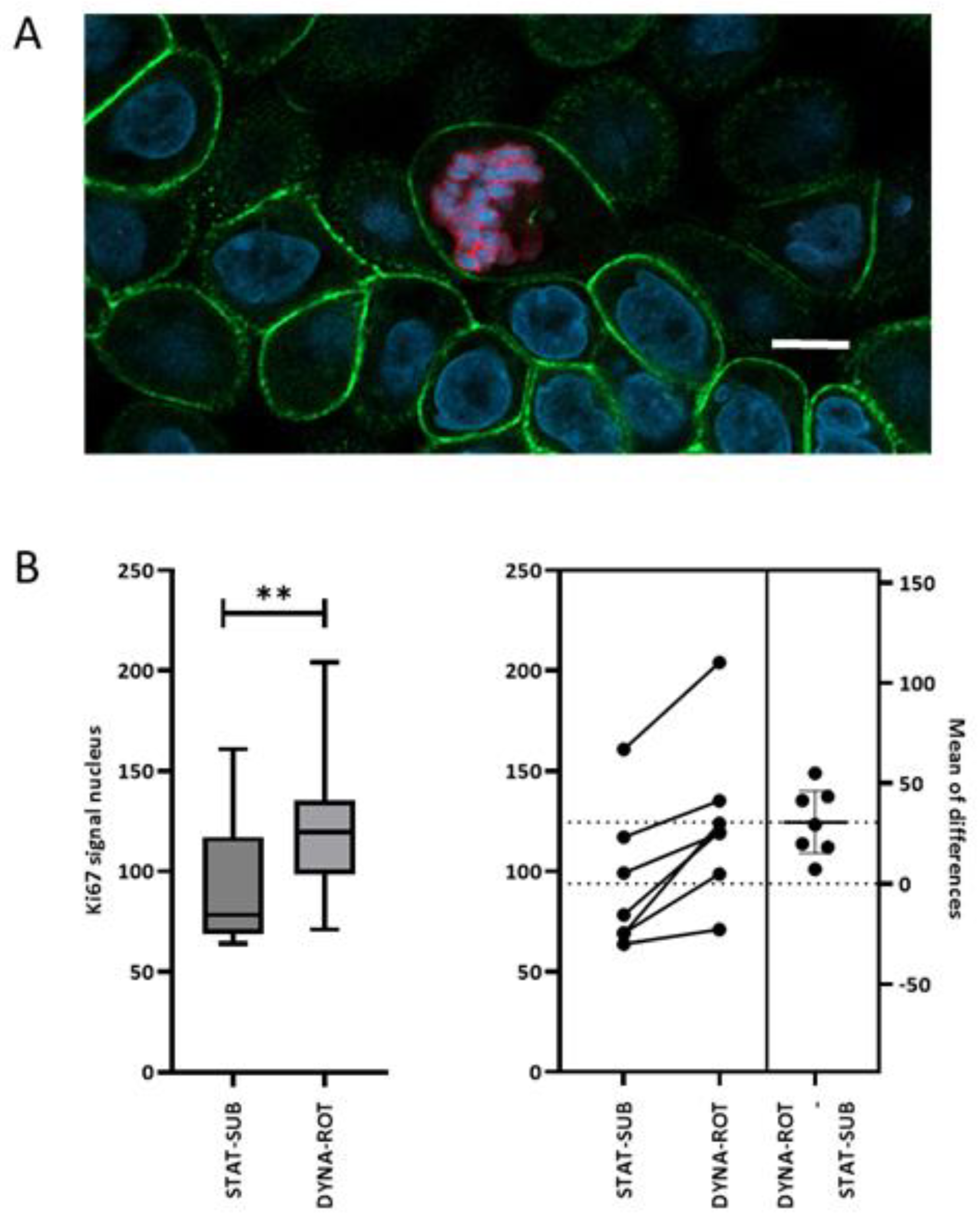
It is known that in weaning piglets, Ki-67-positive cells increase with crypt depth [
32]. The hindlimb unloading mouse model, a rodent analogy for microgravity, however, leads to reduced numbers of Ki67-positive cells in the crypts of the colon but not along the length of the epithelial border. The authors reported a generally impaired barrier function in the colon [
33]. Contradictory effects were also found in other cell types. The growth of bone marrow mesenchymal stem cells was inhibited by 1 d to 3 d exposure to simulated microgravity [
34].
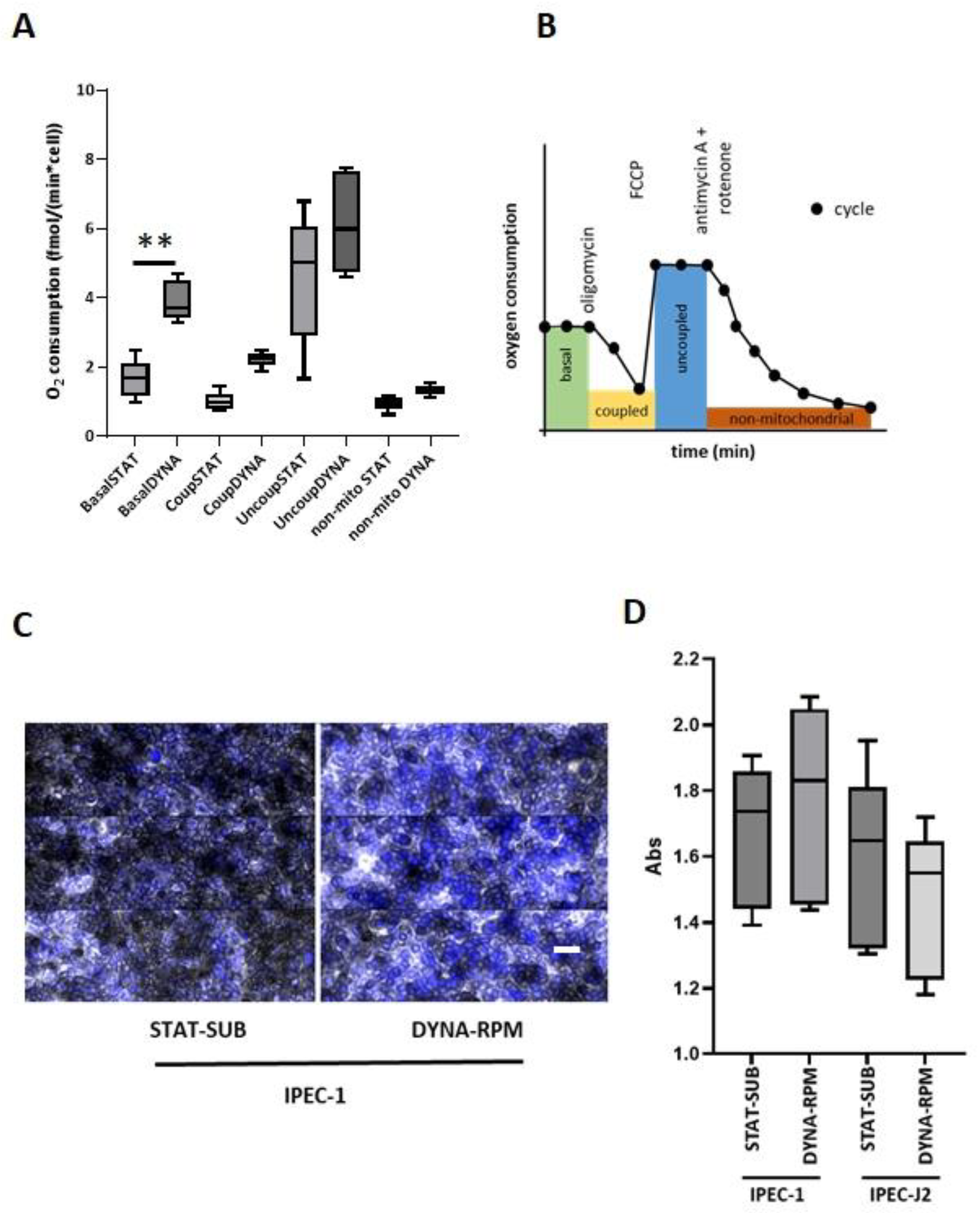
The influence of dynamic cultivation, when used for the simulation of microgravity on mitochondrial respiration, was again shown to be heterogeneous. In most investigations, mitochondrial components were detected at the mRNA or protein level [
35], whereas functional mitochondrial respiration was seldom tested [
36].
In neonatal rat cardiomyocytes, mitochondrial protein synthesis was decreased after 5 days (rotating wall vessel), while apoptosis, cell viability, and protein degradation were largely unaffected [
37]. The exposure of human Hodgkin lymphoma cells to simulated microgravity for 2 days increased their reactive oxygen species (ROS) production and NADPH oxidase family gene expression, while mitochondrial mass, ATPase, ATP synthase, and intracellular ATP levels decreased [
38]. Similarly, the elevation in ROS generation and NADPH oxidase activity was also reported from human lung bronchial epithelial cells (BEAS-2B) after a 2-day exposure to a 3D clinostat [
39]. Interestingly, in real microgravity (ISS), the mRNA of human-induced pluripotent stem cell-derived cardiomyocytes showed an enrichment of modifications in mitochondrial and respiratory processes [
40].
In the final step, we asked the question of whether dynamic cultivation conditions may trigger signs of senescence. It is known that real microgravity occurring over longer exposure times in spaceflights can evoke signs which are similar to those seen in ageing processes [
41]. An experimental parameter which allows the detection of ageing and senescence on the cellular level is the expression of senescence-associated beta-galactosidase (SA-ßgal) [
13]. In the present investigation, we did not see a significant influence of RPM treatment on the senescence marker in IPEC-1 or IPEC-J2 cells.
Taken together, the data illuminate the influence of motion on the cellular level of the intestinal epithelial border cells. The simple change in cultivation conditions obtained by ROT within three days is sufficient to trigger significant morphological and cell physiological modifications, which improve the expressiveness of the IPEC cell culture model.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}