
Cold-Active Starch-Degrading Enzymes from a Cold and Alkaline Greenland Environment: Role of Ca2+ Ions and Conformational Dynamics in Psychrophilicity

 , , , ,
, , , ,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of Samples
2.2. Identification of Amylase-Coding Genes and Homology Alignment
2.3. Cloning, Expression, and Purification
2.4. Determination of Carbohydrate Breakdown Products
2.5. Activity Assays
2.6. Biophysical Analysis
2.7. Molecular Dynamics Simulations
3. Results & Discussion
3.1. Isolation, Identification, and Recombinant Expression of Amylases
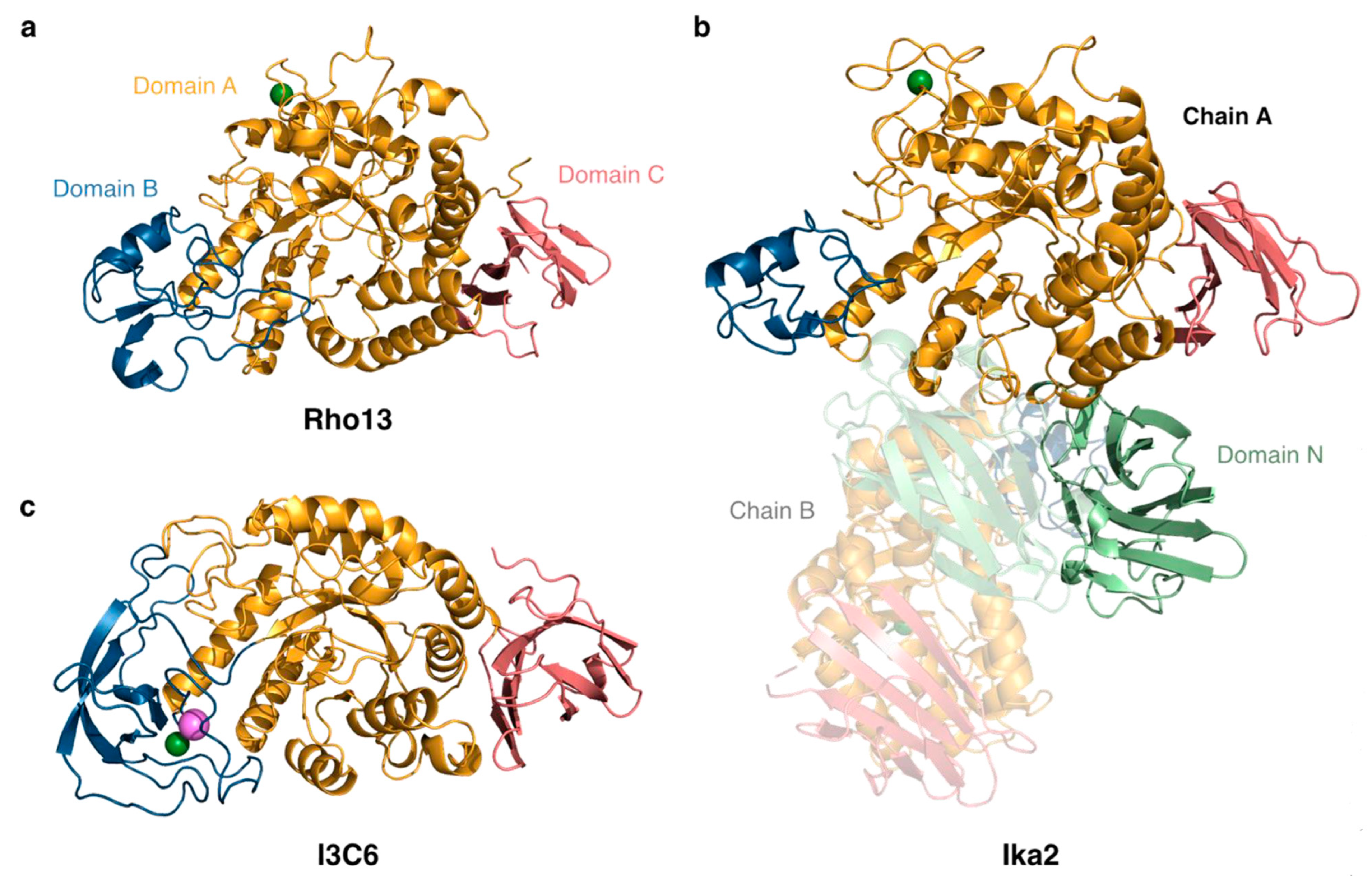
3.1.1. Rho13
3.1.2. Ika2
3.2. Structural and Biophysical Analysis of the Amylases
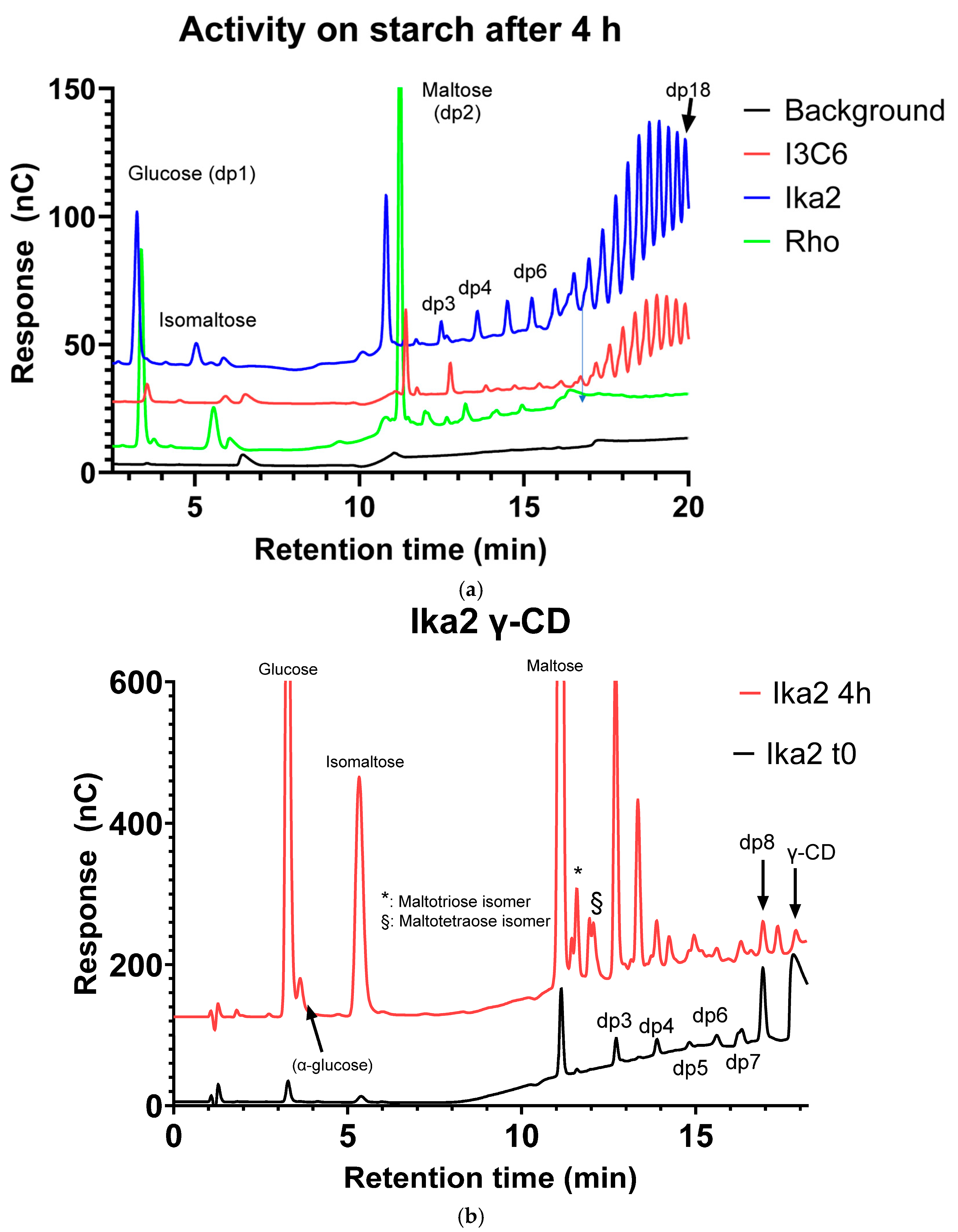
3.2.1. Substrate Specificity and Cleavage Products
Ika2
Rho13
I3C6
3.3. Ika2 Forms a Stable Dimer, While Rho13 and I3C6 Are Mainly Monomeric
3.4. Activity Profiles as a Function of Temperature, pH, and Ca2+
3.4.1. Temperature
3.4.2. pH Profile
3.4.3. Salt Profile
3.4.4. Analysis of Ca2+ Binding of Ika2, Rho13, and I3C6
Ca2+ Has Little Effect on Ika2 Activity, but Rho13 and I3C6 Follow a Bell-Shaped Dependence
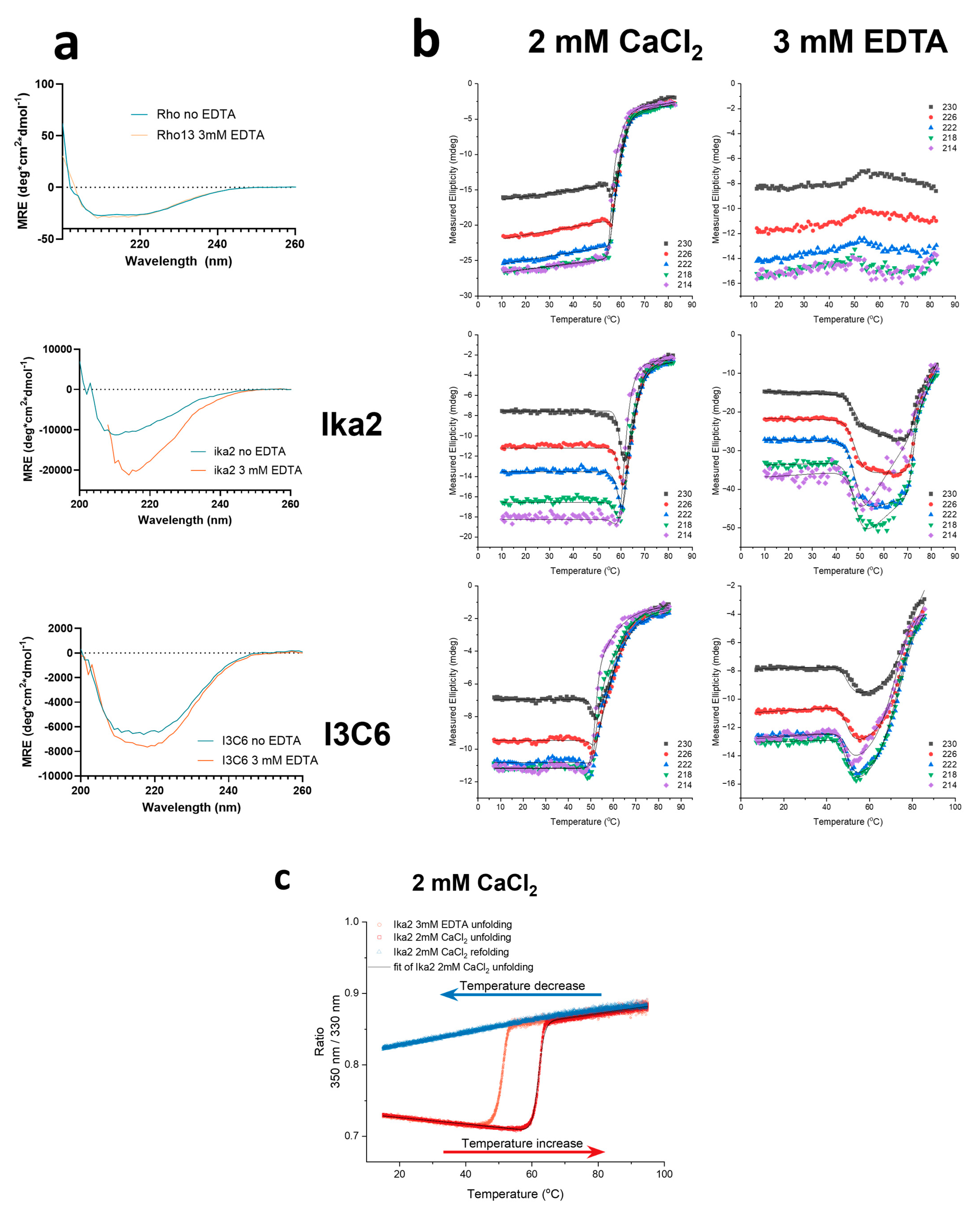
Removal of Ca2+ Affects Enzyme Structure and Reduces Stability
- Rho13
- Ika2
- I3C6
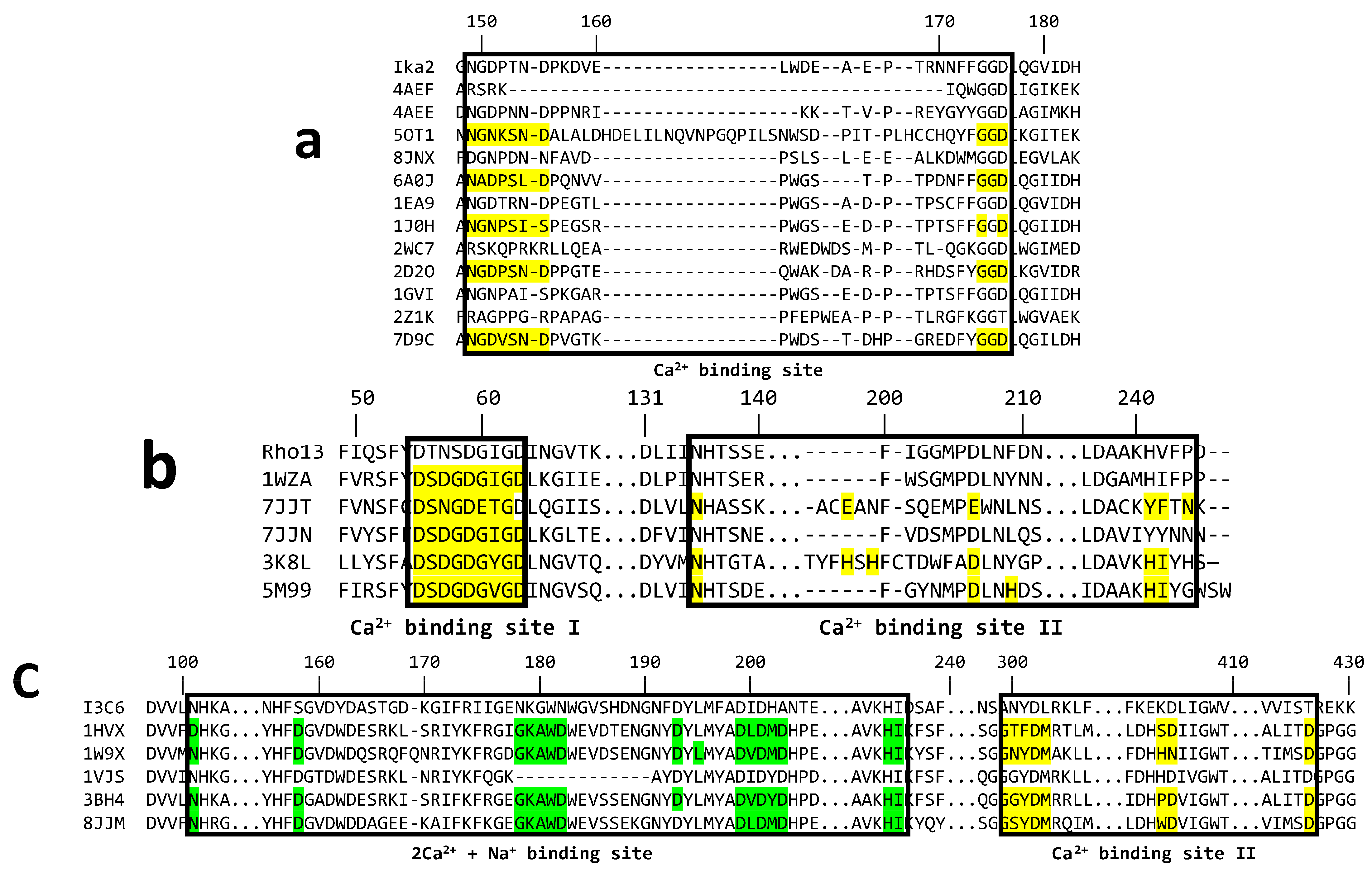
3.5. Computational Analysis of Ca2+ Binding to the Three Enzymes
3.5.1. Ika2 Shows No Evidence of Stable Ca2+ Binding
3.5.2. Rho13 Likely Has One Ca2+ Binding Site
3.5.3. I3C6 Only Maintains One Stably Bound Ca2+ Ion
3.6. Psychrophilic Features of the Three Amylases Analyzed by MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CD | cyclodextrin |
| CiD | circular dichroism |
| ΔH | melting enthalpy |
| dp | degree of polymerization (glucose units) |
| DSF | differential scanning fluorometry |
| G2 | maltose |
| GH | glycoside hydrolase |
| I3C6 | amylase isolated from metagenomic analysis of ikaite columns |
| Ika2 | amylase from Bacillaceae isolated from ikaite columns |
| MBTH | 3-methyl-2-benzothiazolinone hydrazone hydrochloride |
| MD | molecular dynamics |
| PCA | principal component analysis |
| PDB | Protein Data Bank |
| Rg | radius of gyration |
| Rho13 | amylase from R. psychrophilum |
| RMSF | root mean square fluctuation |
| SEC | size exclusion chromatography |
| SAXS | small-angle X-ray scattering |
| tm | melting temperature |
References
- Liu, Y.; Zhang, N.; Ma, J.; Zhou, Y.; Wei, Q.; Tian, C.; Fang, Y.; Zhong, R.; Chen, G.; Zhang, S. Advances in cold-adapted enzymes derived from microorganisms. Front. Microbiol. 2023, 14, 1152847. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.S.; Otzen, D.E. Helping proteins come in from the cold: 5 burning questions about cold-active enzymes. BBA Adv. 2024, 5, 100104. [Google Scholar] [CrossRef]
- Paul, J.S.; Gupta, N.; Beliya, E.; Tiwari, S.; Jadhav, S.K. Aspects and Recent Trends in Microbial α-Amylase: A Review. Appl. Biochem. Biotechnol. 2021, 193, 2649–2698. [Google Scholar] [CrossRef] [PubMed]
- Rani, K.; Rana, R.; Datt, S. Review on characteristics and application of amylases. Int. J. Microbiol. Bioinform. 2015, 5, 1–5. [Google Scholar]
- Sundarram, A.; Murthy, T.P.K. α-Amylase Production and Applications: A Review. J. Appl. Environ. Microbiol. 2014, 2, 166–175. [Google Scholar]
- Kuddus, M.; Roohi, A.J.; Ramteke, P.W. Structural adaptation and biocatalytic prospective of microbial cold-active α-amylase. Afr. J. Microbiol. Res. 2012, 6, 206–213. [Google Scholar]
- Sarian, F.D.; Janeček, Š.; Pijning, T.; Ihsanawati; Nurachman, Z.; Radjasa, O.K.; Dijkhuizen, L.; Natalia, D.; Van Der Maarel, M.J.E.C. A new group of glycoside hydrolase family 13 α-amylases with an aberrant catalytic triad. Sci. Rep. 2017, 7, srep44230. [Google Scholar] [CrossRef]
- Takata, H.; Kuriki, T.; Okada, S.; Takesada, Y.; Iizuka, M.; Minamiura, N.; Imanaka, T. Action of neopullulanase. Neopullulanase catalyzes both hydrolysis and transglycosylation at α-(1→4)- and α-(1→6)-glucosidic linkages. J. Biol. Chem. 1992, 267, 18447–18452. [Google Scholar] [CrossRef]
- Wang, X.; Nie, Y.; Xu, Y. Industrially produced pullulanases with thermostability: Discovery, engineering, and heterologous expression. Bioresour. Technol. 2019, 278, 360–371. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, S.-Y.; Luo, Z.-G.; Zong, M.-H.; Li, X.-X.; Lou, W.-Y. Biotechnology and bioengineering of pullulanase: State of the art and perspectives. World J. Microbiol. Biotechnol. 2021, 37, 43. [Google Scholar] [CrossRef]
- Park, K.H.; Kim, T.J.; Cheong, T.K.; Kim, J.W.; Oh, B.H.; Svensson, B. Structure, specificity and function of cyclomaltodextrinase, a multispecific enzyme of the α-amylase family. Biochim. Biophys. Acta—Protein Struct. Mol. Enzymol. 2000, 1478, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Tonozuka, T.; Ohtsuka, M.; Sakano, Y.; Mogi, S.i.; Sakai, H.; Ohta, T. A Neopullulanase-type α-Amylase Gene from Thermoactinomyces vulgaris R-47. Biosci. Biotechnol. Biochem. 1993, 57, 395–401. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, M.S.; Cho, H.S.; Kim, J.I.; Kim, T.J.; Choi, J.H.; Park, C.; Lee, H.S.; Oh, B.H.; Park, K.H. Cyclomaltodextrinase, neopullulanase, and maltogenic amylase are nearly indistinguishable from each other. J. Biol. Chem. 2002, 277, 21891–21897. [Google Scholar] [CrossRef] [PubMed]
- Arabaci, N.; Arikan, B. Isolation and characterization of a cold-active, alkaline, detergent stable α-amylase from a novel bacterium Bacillus subtilis N8. Prep. Biochem. Biotechnol. 2018, 48, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Roohi, R.; Kuddus, M.; Saima, S. Cold-active detergent-stable extracellular α-amylase from Bacillus cereus GA6: Biochemical characteristics and its perspectives in laundry detergent formulation. J. Biochem. Technol. 2013, 4, 636–644. [Google Scholar]
- Qin, Y.; Huang, Z.; Liu, Z. A novel cold-active and salt-tolerant α-amylase from marine bacterium Zunongwangia profunda: Molecular cloning, heterologous expression and biochemical characterization. Extremophiles 2014, 18, 271–281. [Google Scholar] [CrossRef]
- Aghajari, N.; Georges, F.; Gerday, C.; Haser, R. Crystal Structures of the Psychrophilic a-Amylase from Alteromonas haloplanctis in Its Native Form and Complexed with an Inhibitor; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Feller, G.; D’Amico, D.; Gerday, C. Thermodynamic stability of a cold-active α-amylase from the antarctic bacterium Alteromonas haloplanctis. Biochemistry 1999, 38, 4613–4619. [Google Scholar] [CrossRef]
- Feller, G.; Lonhienne, T.; Deroanne, C.; Libioulle, C.; Van Beeumen, J.; Gerday, C. Purification, characterization, and nucleotide sequence of the thermolabile α-amylase from the antarctic psychrotroph Alteromonas haloplanctis A23. J. Biol. Chem. 1992, 267, 5217–5221. [Google Scholar] [CrossRef]
- Feller, G.; Payan, F.; Theys, F.; Qian, M.; Haser, R.; Gerday, C. Stability and structural analysis of alpha-amylase from the antarctic psychrophile Alteromonas haloplanctis A23. Eur. J. Biochem. 1994, 222, 441–447. [Google Scholar] [CrossRef]
- Ito, S. Alkaline Enzymes in Current Detergency. In Extremophiles Handbook; Horikoshi, K., Ed.; Springer: Tokyo, Japan, 2011; pp. 229–251. [Google Scholar]
- Buchardt, B.; Seaman, P.; Stockmann, G.; Vous, M.; Wilken, U.; Düwel, L.; Kristiansen, A.; Jenner, C.; Whiticar, M.J.; Kristensen, R.M.; et al. Submarine columns of ikaite tufa. Nature 1997, 390, 129–130. [Google Scholar] [CrossRef]
- Schmidt, M.; Priemé, A.; Stougaard, P. Bacterial diversity in permanently cold and alkaline ikaite columns from Greenland. Extremophiles 2006, 10, 551–562. [Google Scholar] [CrossRef]
- Schmidt, M.; Priemé, A.; Stougaard, P. Arsukibacterium ikkense gen. nov., sp. nov, a novel alkaliphilic, enzyme-producing γ-Proteobacterium isolated from a cold and alkaline environment in Greenland. Syst. Appl. Microbiol. 2007, 30, 197–201. [Google Scholar] [CrossRef]
- Vester, J.K.; Glaring, M.A.; Stougaard, P. Discovery of novel enzymes with industrial potential from a cold and alkaline environment by a combination of functional metagenomics and culturing. Microb. Cell Factories 2014, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Lylloff, J.E.; Hansen, L.B.S.; Jepsen, M.; Sanggaard, K.W.; Vester, J.K.; Enghild, J.J.; Sørensen, S.J.; Stougaard, P.; Glaring, M.A. Genomic and exoproteomic analyses of cold- and alkaline-adapted bacteria reveal an abundance of secreted subtilisin-like proteases. Microb. Biotechnol. 2016, 9, 245–256. [Google Scholar] [CrossRef]
- Vester, J.K.; Glaring, M.A.; Stougaard, P. An exceptionally cold-adapted alpha-amylase from a metagenomic library of a cold and alkaline environment. Appl. Microbiol. Biotechnol. 2014, 99, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, A.L.; Glaring, M.A.; Hallin, P.F.; Priemé, A.; Stougaard, P. Draft genome sequence of the psychrophilic and alkaliphilic Rhodonellum psychrophilum strain GCM71T. Genome Announc. 2013, 1, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Priemé, A.; Stougaard, P. Rhodonellum psychrophilum gen. nov., sp. nov., a novel psychrophilic and alkaliphilic bacterium of the phylum Bacteroidetes isolated from Greenland. Int. J. Syst. Evol. Microbiol. 2006, 56, 2887–2892. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Zervas, A.; Stougaard, P.; Thøgersen, M.S. Complete genome sequence of “Bacillaceae sp. strain IKA-2”: A cold-active, amylase-producing bacterium from ikaite columns in SW Greenland. Microbiol. Resour. Announc. 2023, 13, e00887-23. [Google Scholar] [CrossRef]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting Biomolecular Knowledge Via a Protein Database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Winder, A.J.; Harris, H. New assays for the tyrosine hydroxylase and dopa oxidase activities of tyrosinase. Eur. J. Biochem. 1991, 198, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.E.; Sehgal, P.; Otzen, D.E. Activation, inhibition, and destabilization of Thermomyces lanuginosus lipase by detergents. Biochemistry 2005, 44, 1719–1730. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. biorxiv 2021, 2021-10. [Google Scholar] [CrossRef]
- Lyngso, J.; Pedersen, J.S. A high-flux automated laboratory small-angle X-ray scattering instrument optimized for solution scattering. J. Appl. Crystallogr. 2021, 54, 295–305. [Google Scholar] [CrossRef]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Crystall. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Hansen, S.; Bauer, R. The Aggregation Behavior of Zinc-free Insulin Studied by Small-angle Neutron Scattering. Eur. Biophys. J. 1994, 23, 379–389. [Google Scholar] [CrossRef]
- Steiner, E.M.; Lyngsø, J.; Guy, J.E.; Bourenkov, G.; Lindqvist, Y.; Schneider, T.R.; Pedersen, J.S.; Schneider, G.; Schnell, R. The structure of the N-terminal module of the cell wall hydrolase RipA and its role in regulating catalytic activity. Proteins Struct. Funct. Bioinform. 2018, 86, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.L.; Lyngso, J.; Zarantonello, A.; Kjoge, K.; Nielsen, P.K.; Andersen, G.R.; Pedersen, J.S.; Enghild, J.J. Structural Investigations of Human A2M Identify a Hollow Native Conformation That Underlies Its Distinctive Protease-Trapping Mechanism. Mol Cell Proteom. 2021, 20, 100090. [Google Scholar] [CrossRef]
- Duvjir, S.; Fujimoto, Z.; Takase, K.; Matsumura, M.; Mizuno, H. Crystal Structure of Bacillus stearothermophilus a-Amylase: Possible Factors Determining the Thermostability. J. Biochem. 2001, 129, 461–468. [Google Scholar] [CrossRef]
- Hondoh, H.; Kuriki, T.; Matsuura, Y. Three-dimensional Structure and Substrate Binding of Bacillus stearothermophilus Neopullulanase. J. Mol. Biol. 2003, 326, 177–188. [Google Scholar] [CrossRef]
- Sivakumar, N.; Li, N.; Tang, J.W.; Patel, B.K.C.; Swaminathan, K. Crystal structure of AmyA lacks acidic surface and provide insights into protein stability at poly-extreme condition. FEBS Lett. 2006, 580, 2646–2652. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.D.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. AmberTools. J. Chem. Inf. Model. 2023, 63, 6183–6191. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Cauchy, A.-L. Méthode générale pour la résolution des systèmes d’équations simultanées. C. R. Acad. Sci. Paris 1847, 25, 536–538. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1987, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Zhang, Z.-B.; Xia, Y.-L.; Dong, G.-H.; Fu, Y.-X.; Liu, S.-Q. Exploring the Cold-Adaptation Mechanism of Serine Hydroxymethyltransferase by Comparative Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 1781. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, T.; Chen, K.; Shen, S.; Ruan, J.; Kurgan, L. On the relation between residue flexibility and local solvent accessibility in proteins. Proteins Struct. Funct. Bioinform. 2009, 76, 617–636. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.5.0.4. Available online: https://www.pymol.org/support.html (accessed on 15 February 2025).
- Kim, J.S.; Cha, S.S.; Kim, H.J.; Kim, T.J.; Ha, N.C.; Oh, S.T.; Cho, H.S.; Cho, M.J.; Kim, M.J.; Lee, H.S.; et al. Crystal structure of a maltogenic amylase provides insights into a catalytic versatility. J. Biol. Chem. 1999, 274, 26279–26286. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Tonozuka, T.; Yokota, T.; Shimura, Y.; Sakano, Y. Analysis of Catalytic Residues of Thermoactinomyces vulgaris R-47 α-Amylase II (TVA II) by Site-directed Mutagenesis. Biosci. Biotechnol. Biochem. 2000, 64, 2692–2695. [Google Scholar] [CrossRef] [PubMed]
- Kamitori, S.; Kondo, S.; Okuyama, K.; Yokota, T.; Shimura, Y.; Tonozuka, T.; Sakano, Y. Crystal structure of thermoactinomyces vulgaris R-47 α-Amylase II (TVAII) hydrolyzing cyclodextrins and pullulan at 2.6 Å resolution. J. Mol. Biol. 1999, 287, 907–921. [Google Scholar] [CrossRef]
- Mizuno, M.; Tonozuka, T.; Uechi, A.; Ohtaki, A.; Ichikawa, K.; Kamitori, S.; Nishikawa, A.; Sakano, Y. The crystal structure of Thermoactinomyces vulgaris R-47 α-amylase II (TVA II) complexed with transglycosylated product. Eur. J. Biochem. 2004, 271, 2530–2538. [Google Scholar] [CrossRef]
- Møller, M.S.; Svensson, B. Structural biology of starch-degrading enzymes and their regulation. Curr. Opin. Struct. Biol. 2016, 40, 33–42. [Google Scholar] [CrossRef]
- Kim, T.J.; Kim, M.J.; Kim, B.C.; Kim, J.C.; Cheong, T.K.; Kim, J.W.; Park, K.H. Modes of action of acarbose hydrolysis and transglycosylation catalyzed by a thermostable maltogenic amylase, the gene for which was cloned from a Thermus strain. Appl. Environ. Microbiol. 1999, 65, 1644–1651. [Google Scholar] [CrossRef]
- Pico, J.; Martinez, M.M.; Martin, M.T.; Gomez, M. Quantification of sugars in wheat flours with an HPAEC-PAD method. Food Chem 2015, 173, 674–681. [Google Scholar] [CrossRef]
- Carabetta, S.; Di Sanzo, R.; Campone, L.; Fuda, S.; Rastrelli, L.; Russo, M. High-Performance Anion Exchange Chromatography with Pulsed Amperometric Detection (HPAEC–PAD) and Chemometrics for Geographical and Floral Authentication of Honeys from Southern Italy (Calabria region). Foods 2020, 9, 1625. [Google Scholar] [CrossRef]
- Duedahl-Olesen, L.; Haastrup Pedersen, L.; Lambertsen Larsen, K. Suitability and limitations of methods for characterisation of activity of malto-oligosaccharide-forming amylases. Carbohydr. Res. 2000, 329, 109–119. [Google Scholar] [CrossRef]
- Anthon, G.E.; Barrett, D.M. Determination of reducing sugars with 3-methyl-2-benzothiazolinonehydrazone. Anal. Biochem. 2002, 305, 287–289. [Google Scholar] [CrossRef]
- Feller, G. Psychrophilic enzymes: From folding to function and biotechnology. Scientifica 2013, 2013, 512840. [Google Scholar] [CrossRef] [PubMed]
- Lonhienne, T.; Gerday, C.; Feller, G. Psychrophilic enzymes: Revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 2000, 1543, 1–10. [Google Scholar] [CrossRef]
- Sočan, J.; Isaksen, G.V.; Brandsdal, B.O.; Åqvist, J. Towards Rational Computational Engineering of Psychrophilic Enzymes. Sci. Rep. 2019, 9, 19147. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.S.; Cavicchioli, R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef]
- Papaleo, E.; Tiberti, M.; Invernizzi, G. Molecular Dynamics Simulations to Study Structure-Function Relationship in Psychrophilic Enzymes. In Biotechnology of Extremophiles: Advances and Challenges; Rampelotto, P.H., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 675–698. [Google Scholar]
- Papaleo, E.; Riccardi, L.; Villa, C.; Fantucci, P.; De Gioia, L. Flexibility and enzymatic cold-adaptation: A comparative molecular dynamics investigation of the elastase family. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2006, 1764, 1397–1406. [Google Scholar] [CrossRef]
- Isaksen, G.V.; Aqvist, J.; Brandsdal, B.O. Enzyme surface rigidity tunes the temperature dependence of catalytic rates. Proc. Natl. Acad. Sci. USA 2016, 113, 7822–7827. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | ConcentrationAbs280 (mg/mL) | Rg (Å) a | Model | χ2 b | ConcentrationSAXS (mg/mL) | n-mers a (SAXS b) |
|---|---|---|---|---|---|---|
| Ika2 | 1.99 | 33.8 ± 0.1 | AF dimer | 4.3 | 1.69 | 1.67 (1.97) |
| 1.00 | 33.8 ± 0.1 | AF dimer | 1.9 | 0.84 | 1.67 (1.99) | |
| 0.51 | 34 ± 0.2 | AF dimer | 1.4 | 0.42 | 1.63 (1.96) | |
| Rho | 0.94 | 49.8 ± 0.5 | Monomer0.03 mg/mL + dimer0.69 mg/mL | 2.9 | 0.72 | 1.58 (1.96) |
| 0.46 | 42.6 ± 0.7 | Monomer0.06 mg/mL + dimer0.29 mg/mL | 1.4 | 0.35 | 1.38 (1.83) | |
| I3C6 | 1.76 | 64.5 ± 1 | - | - | - | 1.6 |
| 0.87 | 94 ± 0.8 | - | - | - | 3.42 | |
| 0.46 | 111.4 ± 0.4 | - | - | - | 7.4 |
| Activity Optimum (°C) | tm (DSF) (°C) b | tm (CiD) (°C) c | |||||
|---|---|---|---|---|---|---|---|
| 2 mM CaCl2 | 2 mM CaCl2 + 3 mM EDTA | 2 mM CaCl2 | 2 mM CaCl2 + 3 mM EDTA | ||||
| tm | tm | tmN-I | tmI-D | tmN-I | tmI-D | ||
| Ika2 d | 35 | 61.97 ± 0.01 | 50.82 ± 0.01 | 57.16 ± 1.77 | 62.10 ± 0.99 | 47.80 ± 0.23 | 72.16 ± 0.20 |
| Rho13 e | 30 | 55.21 ± 0.01 | 53.38 ± 0.01 | 55.98 ± 0.04 | 58.32 ± 0.14 | N/A g | N/A g |
| I3C6 f | 30 | 55.40 ± 0.01 | 52.28 ± 0.01 | 52.47 ± 0.05 | 53.85 ± 0.52 | 55.67 ± 0.25 | 75.42 ± 0.70 |
| Average RMSD (Backbone)/Å | Average RMSF (Backbone)/Å | Average No. H-Bonds | Average Rg (Backbone)/Å | Average SASA/nm2 | |
|---|---|---|---|---|---|
| Rho13 | 2.763 ± 0.006 | 1.02 ± 0.02 | 413.81 ± 0.09 | 23.46 ± 0.02 | 216.56 ± 0.03 |
| 1WZA (homolog) | 2.600 ± 0.004 | 0.88 ± 0.02 | 417.01 ± 0.09 | 22.88 ± 0.02 | 208.68 ± 0.03 |
| Ika2 | 2.837 ± 0.005 | 1.295 ± 0.008 | 1000.6 ± 0.1 | 33.400 ± 0.002 | 462.69 ± 0.06 |
| 1J0H (homolog) | 2.123 ± 0.004 | 1.075 ± 0.008 | 999.8 ± 0.1 | 33.467 ± 0.002 | 458.63 ± 0.06 |
| I3C6 | 1.670 ± 0.003 | 0.92 ± 0.02 | 410.79 ± 0.09 | 24.11 ± 0.02 | 205.81 ± 0.03 |
| 1HVX (homolog) | 1.073 ± 0.002 | 0.72 ± 0.01 | 424.97 ± 0.09 | 23.73 ± 0.02 | 186.45 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bendtsen, M.K.; Nowak, J.S.; Paiva, P.; López Hernández, M.; Ferreira, P.; Pedersen, J.S.; Bekker, N.S.; Viezzi, E.; Bisiak, F.; Brodersen, D.E.; et al. Cold-Active Starch-Degrading Enzymes from a Cold and Alkaline Greenland Environment: Role of Ca2+ Ions and Conformational Dynamics in Psychrophilicity. Biomolecules 2025, 15, 415. https://doi.org/10.3390/biom15030415
Bendtsen MK, Nowak JS, Paiva P, López Hernández M, Ferreira P, Pedersen JS, Bekker NS, Viezzi E, Bisiak F, Brodersen DE, et al. Cold-Active Starch-Degrading Enzymes from a Cold and Alkaline Greenland Environment: Role of Ca2+ Ions and Conformational Dynamics in Psychrophilicity. Biomolecules. 2025; 15(3):415. https://doi.org/10.3390/biom15030415
Chicago/Turabian StyleBendtsen, Malthe Kjær, Jan Stanislaw Nowak, Pedro Paiva, Marcos López Hernández, Pedro Ferreira, Jan Skov Pedersen, Nicolai Sundgaard Bekker, Elia Viezzi, Francesco Bisiak, Ditlev E. Brodersen, and et al. 2025. "Cold-Active Starch-Degrading Enzymes from a Cold and Alkaline Greenland Environment: Role of Ca2+ Ions and Conformational Dynamics in Psychrophilicity" Biomolecules 15, no. 3: 415. https://doi.org/10.3390/biom15030415
APA StyleBendtsen, M. K., Nowak, J. S., Paiva, P., López Hernández, M., Ferreira, P., Pedersen, J. S., Bekker, N. S., Viezzi, E., Bisiak, F., Brodersen, D. E., Pedersen, L. H., Zervas, A., Fernandes, P. A., Ramos, M. J., Stougaard, P., Thøgersen, M. S., & Otzen, D. E. (2025). Cold-Active Starch-Degrading Enzymes from a Cold and Alkaline Greenland Environment: Role of Ca2+ Ions and Conformational Dynamics in Psychrophilicity. Biomolecules, 15(3), 415. https://doi.org/10.3390/biom15030415