

Distinct Roles of Common Genetic Variants and Their Contributions to Diabetes: MODY and Uncontrolled T2DM

Abstract
1. Introduction
2. Definition of Uncontrolled T2DM
3. Insight into the Monogenic and Polygenic Causes of Uncontrolled T2DM
3.1. The Complex Relationship Between MODY, T2DM, and Genomic Instability
3.2. Genetic Contributions to Diabetes: MODY and Uncontrolled T2DM
3.3. The Candidate Genes Associated with the Development of MODY
3.3.1. Genes Harboring Rare Variants That Cause GCA-MODY
3.3.2. An Update on HNF1A
3.3.3. HNF4A-MODY Overview
3.3.4. KCNJ11 Variants That Are Associated with an Increased Risk of T2DM in Adults
3.4. The Candidate Genes Associated with the Development of Late-Onset T2DM
3.4.1. The Transcription Factor 7-like 2 (TCF7L2) and Its Association with Beta-Cell Dysfunction in T2DM
3.4.2. GLIS3 Is Considered a Contributing Factor to Polygenic Nature of T2DM
3.4.3. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) in Diabetes, Obesity, and Atherosclerosis
3.4.4. The LEP Variant and Its Potential Contribution to Insulin Resistance and Metabolic Syndrome
3.4.5. The UCP1 rs45539933 Gene Variant Is Associated with Obesity and Diabetes
3.4.6. The OPG Variant and Its Potential Contribution to Hypertension, and Diabetes
4. Diabetes Inheritance and Personalized Treatment Approaches
4.1. Current Treatment Guidelines for Diabetes
4.2. Genetic Variability and Ethnic Differences in Drug Response
5. Obesity and Its Association with Insulin Resistance
5.1. Gene-Environment Interactions in Obesity
5.2. Implications for Personalized Medicine and Obesity Management
5.3. Treatment Options for MODY and Prediabetic Patients
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADA | American Diabetes Association |
| BMI | Body mass index |
| CV | Cardiovascular |
| CNS | Central nervous system |
| DPP-4 | dipeptidyl peptidase-4 |
| FTO | Fat mass and obesity-associated |
| FDA | Food and Drug Administration |
| GLIS3 | Gli-similar 3 |
| GLP-1 | Glucagon-like peptide-1 |
| GIP | Glucose-dependent insulinotropic polypeptide |
| GWAS | Genome-wide association studies |
| HNF4α | hepatocyte nuclear factor 4-alpha |
| HbA1C | Plasma glycosylated hemoglobin A1C |
| MC4R | Melanortin-4 receptor |
| MeSH | Medical subject headings |
| MODY | Maturity-onset diabetes of the young |
| NF-κB | Nuclear factor-κB |
| OPG | Osteoprotegerin |
| PPARγ | Peroxisome proliferator-activated receptor Gamma |
| SNPs | Single nucleotide polymorphisms |
| SGLT2 | Sodium-glucose co-transporter-2 |
| T2DM | Type 2 diabetes mellitus |
| TGFβ | Transforming growth factor beta |
| WHO | World Health Organization |
References
- Hollstein, T.; Basolo, A.; Ando, T.; Votruba, S.B.; Walter, M.; Krakoff, J.; Piaggi, P. Recharacterizing the Metabolic State of Energy Balance in Thrifty and Spendthrift Phenotypes. J. Clin. Endocrinol. Metab. 2020, 105, 1375–1392. [Google Scholar] [CrossRef]
- Heilbronn, L.K.; de Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-Month Calorie Restriction on Biomarkers of Longevity, Metabolic Adaptation, and Oxidative Stress in Overweight IndividualsA Randomized Controlled Trial. JAMA 2006, 295, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Chia, C.W.; Egan, J.M.; Ferrucci, L. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fang, R.; Wang, H.; Xu, D.-X.; Yang, J.; Huang, X.; Cozzolino, D.; Fang, M.; Huang, Y. A review of environmental metabolism disrupting chemicals and effect biomarkers associating disease risks: Where exposomics meets metabolomics. Environ. Int. 2022, 158, 106941. [Google Scholar] [CrossRef] [PubMed]
- Virolainen, S.J.; VonHandorf, A.; Viel, K.C.M.F.; Weirauch, M.T.; Kottyan, L.C. Gene–environment interactions and their impact on human health. Genes Immun. 2023, 24, 1–11. [Google Scholar] [CrossRef]
- Snykers, S.; Henkens, T.; De Rop, E.; Vinken, M.; Fraczek, J.; De Kock, J.; De Prins, E.; Geerts, A.; Rogiers, V.; Vanhaecke, T. Role of epigenetics in liver-specific gene transcription, hepatocyte differentiation and stem cell reprogrammation. J. Hepatol. 2009, 51, 187–211. [Google Scholar] [CrossRef]
- Trexler, E.T.; Smith-Ryan, A.E.; Norton, L.E. Metabolic adaptation to weight loss: Implications for the athlete. J. Int. Soc. Sports Nutr. 2014, 11, 7. [Google Scholar] [CrossRef]
- Hyde, P.N.; Sapper, T.N.; Crabtree, C.D.; LaFountain, R.A.; Bowling, M.L.; Buga, A.; Fell, B.; McSwiney, F.T.; Dickerson, R.M.; Miller, V.J.; et al. Dietary carbohydrate restriction improves metabolic syndrome independent of weight loss. JCI Insight 2019, 4, e128308. [Google Scholar] [CrossRef]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Park, C.; Pagnini, F.; Langer, E. Glucose metabolism responds to perceived sugar intake more than actual sugar intake. Sci. Rep. 2020, 10, 15633. [Google Scholar] [CrossRef]
- Mooradian, A.D. Cardiovascular Disease in Type 2 Diabetes Mellitus: Current Management Guidelines. Arch. Intern. Med. 2003, 163, 33–40. [Google Scholar] [CrossRef]
- Raz, I. Guideline approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care 2013, 36 (Suppl. S2), S139–S144. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.L.; Vellanki, P.; Blonde, L.; Christofides, E.A.; Galindo, R.J.; Hirsch, I.B.; Isaacs, S.D.; Izuora, K.E.; Low Wang, C.C.; Twining, C.L.; et al. American Association of Clinical Endocrinology Consensus Statement: Comprehensive Type 2 Diabetes Management Algorithm—2023 Update. Endocr. Pract. 2023, 29, 305–340. [Google Scholar] [CrossRef] [PubMed]
- Haw, J.S.; Shah, M.; Turbow, S.; Egeolu, M.; Umpierrez, G. Diabetes Complications in Racial and Ethnic Minority Populations in the USA. Curr. Diabetes Rep. 2021, 21, 2. [Google Scholar] [CrossRef]
- InterAct Consortium. The link between family history and risk of type 2 diabetes is not explained by anthropometric, lifestyle or genetic risk factors: The EPIC-InterAct study. Diabetologia 2013, 56, 60–69. [Google Scholar] [CrossRef]
- Kim, M.-J.; Lim, N.-K.; Choi, S.-J.; Park, H.-Y. Hypertension is an independent risk factor for type 2 diabetes: The Korean genome and epidemiology study. Hypertens. Res. 2015, 38, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Tomic, D.; Shaw, J.E.; Magliano, D.J. The burden and risks of emerging complications of diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 525–539. [Google Scholar] [CrossRef]
- Poulsen, P.; Kyvik, K.O.; Vaag, A.; Beck-Nielsen, H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—A population-based twin study. Diabetologia 1999, 42, 139–145. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef]
- Singh, S.; Usman, K.; Banerjee, M. Pharmacogenetic studies update in type 2 diabetes mellitus. World J. Diabetes 2016, 7, 302–315. [Google Scholar] [CrossRef]
- Jmel, H.; Sarno, S.; Giuliani, C.; Boukhalfa, W.; Abdelhak, S.; Luiselli, D.; Kefi, R. Genetic diversity of variants involved in drug response among Tunisian and Italian populations toward personalized medicine. Sci. Rep. 2024, 14, 5842. [Google Scholar] [CrossRef]
- Sivadas, A.; Sahana, S.; Jolly, B.; Bhoyar, R.C.; Jain, A.; Sharma, D.; Imran, M.; Senthivel, V.; Divakar, M.K.; Mishra, A.; et al. Landscape of pharmacogenetic variants associated with non-insulin antidiabetic drugs in the Indian population. BMJ Open Diabetes Res. Care 2024, 12, e003769. [Google Scholar] [CrossRef] [PubMed]
- Nicolaisen, S.K.; Thomsen, R.W.; Lau, C.J.; Sorensen, H.T.; Pedersen, L. Development of a 5-year risk prediction model for type 2 diabetes in individuals with incident HbA1c-defined pre-diabetes in Denmark. BMJ Open Diabetes Res. Care 2022, 10, e002946. [Google Scholar] [CrossRef]
- Lee, S.; Liu, T.; Zhou, J.; Zhang, Q.; Wong, W.T.; Tse, G. Predictions of diabetes complications and mortality using hba1c variability: A 10-year observational cohort study. Acta Diabetol. 2020, 58, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.M.; Haggerty, S.; Herman, W.H. HbA1c for the Diagnosis of Diabetes and Prediabetes: Is It Time for a Mid-Course Correction? J. Clin. Endocrinol. Metab. 2010, 95, 5203–5206. [Google Scholar] [CrossRef]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 6. Glycemic Targets: Standards of Care in Diabetes—2023. Diabetes Care 2022, 46 (Suppl. S1), S97–S110. [Google Scholar] [CrossRef]
- Yun, J.-S.; Jung, S.-H.; Shivakumar, M.; Xiao, B.; Khera, A.V.; Won, H.-H.; Kim, D. Polygenic risk for type 2 diabetes, lifestyle, metabolic health, and cardiovascular disease: A prospective UK Biobank study. Cardiovasc. Diabetol. 2022, 21, 131. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.; Fernandes Silva, L. Genetics of Type 2 Diabetes: Past, Present, and Future. Nutrients 2022, 14, 3201. [Google Scholar] [CrossRef]
- Yajnik, C.S.; Wagh, R.; Kunte, P.; Asplund, O.; Ahlqvist, E.; Bhat, D.; Shukla, S.R.; Prasad, R.B. Polygenic scores of diabetes-related traits in subgroups of type 2 diabetes in India: A cohort study. Lancet Reg. Health-Southeast Asia 2023, 14, 100182. [Google Scholar] [CrossRef]
- Prasad, R.B.; Groop, L. Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes 2015, 6, 87–123. [Google Scholar] [CrossRef]
- Praetorius, C.; Grill, C.; Stacey, S.N.; Metcalf, A.M.; Gorkin, D.U.; Robinson, K.C.; Van Otterloo, E.; Kim, R.S.; Bergsteinsdottir, K.; Ogmundsdottir, M.H.; et al. A Polymorphism in IRF4 Affects Human Pigmentation through a Tyrosinase-Dependent MITF/TFAP2A Pathway. Cell 2013, 155, 1022–1033. [Google Scholar] [CrossRef]
- Poon, K.-S.; Tan, K.M.-L.; Koay, E.S.-C.; Sng, A. Genetic testing of GCK-MODY identifies a novel pathogenic variant in a Chinese boy with early onset hyperglycemia. Hum. Genome Var. 2020, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Zhang, P.; Pang, W.; Ma, Y.; Gong, S.; Ba, T.; Liu, W.; Zhang, F.; Zhang, X.; Zhang, R.; et al. A Comparison of Daily Glucose Fluctuation Between GCK-MODY and Type 2 Diabetes Using Continuous Glucose Monitoring Technology. Diabetes 2023, 72, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Thuesen, A.C.B.; Jensen, R.T.; Maagensen, H.; Kristiansen, M.R.; Sørensen, H.T.; Vaag, A.; Beck-Nielsen, H.; Pedersen, O.B.; Grarup, N.; Nielsen, J.S.; et al. Identification of pathogenic GCK variants in patients with common type 2 diabetes can lead to discontinuation of pharmacological treatment. Mol. Genet. Metab. Rep. 2023, 35, 100972. [Google Scholar] [CrossRef]
- González, B.J.; Zhao, H.; Niu, J.; Williams, D.J.; Lee, J.; Goulbourne, C.N.; Xing, Y.; Wang, Y.; Oberholzer, J.; Blumenkrantz, M.H.; et al. Reduced calcium levels and accumulation of abnormal insulin granules in stem cell models of HNF1A deficiency. Commun. Biol. 2022, 5, 779. [Google Scholar] [CrossRef] [PubMed]
- Toaima, D.; Näke, A.; Wendenburg, J.; Praedicow, K.; Rohayem, J.; Engel, K.; Galler, A.; Gahr, M.; Lee-Kirsch, M.A. Identification of novel GCK and HNF1A/TCF1 mutations and polymorphisms in German families with maturity-onset diabetes of the young (MODY). Hum. Mutat. 2005, 25, 503–504. [Google Scholar] [CrossRef]
- DeForest, N.; Kavitha, B.; Hu, S.; Isaac, R.; Krohn, L.; Wang, M.; Du, X.; De Arruda Saldanha, C.; Gylys, J.; Merli, E.; et al. Human gain-of-function variants in HNF1A confer protection from diabetes but independently increase hepatic secretion of atherogenic lipoproteins. Cell Genom. 2023, 3, 100339. [Google Scholar] [CrossRef]
- Low, B.S.J.; Lim, C.S.; Ding, S.S.L.; Tan, Y.S.; Ng, N.H.J.; Krishnan, V.G.; Ang, S.F.; Neo, C.W.Y.; Verma, C.S.; Hoon, S.; et al. Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant β cells. Nat. Commun. 2021, 12, 3133. [Google Scholar] [CrossRef]
- Berndt, S.I.; Gustafsson, S.; Mägi, R.; Ganna, A.; Wheeler, E.; Feitosa, M.F.; Justice, A.E.; Monda, K.L.; Croteau-Chonka, D.C.; Day, F.R.; et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat. Genet. 2013, 45, 501–512. [Google Scholar] [CrossRef]
- Hani, E.H.; Suaud, L.; Boutin, P.; Chèvre, J.C.; Durand, E.; Philippi, A.; Demenais, F.; Vionnet, N.; Furuta, H.; Velho, G.; et al. A missense mutation in hepatocyte nuclear factor-4 alpha, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1998, 101, 521–526. [Google Scholar] [CrossRef]
- Üstay, Ö.; Apaydın, T.; Elbasan, O.; Polat, H.; Günhan, G.; Dinçer, C.; Şeker, L.; Ateş, E.A.; Yabacı, A.; Güney, A.; et al. When do we need to suspect maturity onset diabetes of the young in patients with type 2 diabetes mellitus? Arch. Endocrinol. Metab. 2022, 66, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Chagoya, A.; Schroeder, P.; Mandla, R.; Li, J.; Morris, L.; Vora, M.; Alkanaq, A.; Nagy, D.; Szczerbinski, L.; Madsen, J.G.S.; et al. Rare variant analyses in 51,256 type 2 diabetes cases and 370,487 controls reveal the pathogenicity spectrum of monogenic diabetes genes. Nat. Genet. 2024, 56, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Haghvirdizadeh, P.; Mohamed, Z.; Abdullah, N.A.; Haghvirdizadeh, P.; Haerian, M.S.; Haerian, B.S. KCNJ11: Genetic Polymorphisms and Risk of Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 908152. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Raza, S.T.; Mahdi, F.; Singh, S.P.; Rajput, M.; Rahman, Q. Genetic polymorphisms in KCNJ11 (E23K, rs5219) and SDF-1β (G801A, rs1801157) genes are associated with the risk of type 2 diabetes mellitus. Br. J. Biomed. Sci. 2018, 75, 139–144. [Google Scholar] [CrossRef]
- Vedovato, N.; Salguero, M.V.; Greeley, S.A.W.; Yu, C.H.; Philipson, L.H.; Ashcroft, F.M. A loss-of-function mutation in KCNJ11 causing sulfonylurea-sensitive diabetes in early adult life. Diabetologia 2024, 67, 940–951. [Google Scholar] [CrossRef]
- Alqadri, N. Independent case-control study in KCNJ11 gene polymorphism with Type 2 diabetes Mellitus. Saudi J. Biol. Sci. 2022, 29, 2794–2799. [Google Scholar] [CrossRef]
- del Bosque-Plata, L.; Hernández-Cortés, E.P.; Gragnoli, C. The broad pathogenetic role of TCF7L2 in human diseases beyond type 2 diabetes. J. Cell. Physiol. 2022, 237, 301–312. [Google Scholar] [CrossRef]
- Jan, A.; Zakiullah; Khuda, F.; Akbar, R. Validation of Genome-Wide Association Studies (GWAS)-Identified Type 2 Diabetes Mellitus Risk Variants in Pakistani Pashtun Population. J. ASEAN Fed. Endocr. Soc. 2022, 38, 55–61. [Google Scholar] [CrossRef]
- Hansen, A.L.; Andersen, M.K.; Engelhard, L.M.; Brøns, C.; Hansen, T.; Nielsen, J.S.; Vestergaard, P.; Højlund, K.; Jessen, N.; Olsen, M.H.; et al. Impact of TCF7L2 rs7903146 on clinical presentation and risk of complications in patients with type 2 diabetes. Diabetes Obes. Metab. 2025, 27, 2002–2011. [Google Scholar] [CrossRef]
- Farid, M.M.M.; Abdel-Mageed, A.I.; El-sherbini, A.; Mohamed, N.R.; Mohsen, M. Study of the association between GLIS3 rs10758593 and type 2 diabetes mellitus in Egyptian population. Egypt. J. Med. Hum. Genet. 2022, 23, 44. [Google Scholar] [CrossRef]
- Meulebrouck, S.; Scherrer, V.; Boutry, R.; Toussaint, B.; Vaillant, E.; Dechaume, A.; Loiselle, H.; Balkau, B.; Charpentier, G.; Franc, S.; et al. Pathogenic monoallelic variants in GLIS3 increase type 2 diabetes risk and identify a subgroup of patients sensitive to sulfonylureas. Diabetologia 2024, 67, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Hur, H.J.; Yang, H.J.; Kim, M.J.; Lee, K.-H.; Kim, M.-S.; Park, S. Association of Polygenic Variants with Type 2 Diabetes Risk and Their Interaction with Lifestyles in Asians. Nutrients 2022, 14, 3222. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Dankel, S.N.; Klocke, B.; Grallert, H.; Glunk, V.; Berulava, T.; Lee, H.; Oskolkov, N.; Fadista, J.; Ehlers, K.; et al. Leveraging cross-species transcription factor binding site patterns: From diabetes risk loci to disease mechanisms. Cell 2014, 156, 343–358. [Google Scholar] [CrossRef]
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.-A.; Fontanillas, P.; Gabriel, S.B.; GoT2D Consortium; NHGRI JHS/FHS Allelic Spectrum Project; SIGMA T2D Consortium; et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 13127–13132. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Niu, T.; Ma, Y.; You, N.C.; Song, Y.; Sobel, E.M.; Hsu, Y.H.; Balasubramanian, R.; Qiao, Y.; Tinker, L.; et al. Common genetic variants in peroxisome proliferator-activated receptor-gamma (PPARG) and type 2 diabetes risk among Women’s Health Initiative postmenopausal women. J. Clin. Endocrinol. Metab. 2013, 98, E600–E604. [Google Scholar] [CrossRef]
- Ali, L.A.; Jemon, K.; Latif, N.A.; Bakar, S.A.; Alwi, S.S.S. LEP G2548A polymorphism is associated with increased serum leptin and insulin resistance among T2DM Malaysian patients. BioMedicine 2022, 12, 31–39. [Google Scholar] [CrossRef]
- Flouris, A.D.; Shidlovskii, Y.V.; Shaposhnikov, A.V.; Yepiskoposyan, L.; Nadolnik, L.; Karabon, L.; Kowalska, A.; Carrillo, A.E.; Metsios, G.S.; Sakellariou, P. Role of UCP1 Gene Variants in Interethnic Differences in the Development of Cardio-Metabolic Diseases. Front. Genet. 2017, 8, 7. [Google Scholar] [CrossRef]
- Chathoth, S.; Ismail, M.H.; Vatte, C.; Cyrus, C.; Al Ali, Z.; Ahmed, K.A.; Acharya, S.; Al Barqi, A.M.; Al Ali, A. Association of Uncoupling Protein 1 (UCP1) gene polymorphism with obesity: A case-control study. BMC Med. Genet. 2018, 19, 203. [Google Scholar] [CrossRef]
- Brondani, L.A.; Assmann, T.S.; Duarte, G.C.; Gross, J.L.; Canani, L.H.; Crispim, D. The role of the uncoupling protein 1 (UCP1) on the development of obesity and type 2 diabetes mellitus. Arq. Bras. Endocrinol. Metabol. 2012, 56, 215–225. [Google Scholar] [CrossRef]
- Sailaja, A.N.; Nanda, N.; Suryanarayana, B.S.; Pal, G.K. Association of rs2073618 polymorphism and osteoprotegerin levels with hypertension and cardiovascular risks in patients with type 2 diabetes mellitus. Sci. Rep. 2023, 13, 17451. [Google Scholar] [CrossRef]
- Tosur, M.; Philipson, L.H. Precision diabetes: Lessons learned from maturity-onset diabetes of the young (MODY). J. Diabetes Investig. 2022, 13, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, A.; Pryse, R.; Schuman, A.; Owen, K.R. When to consider a diagnosis of MODY at the presentation of diabetes: Aetiology matters for correct management. Br. J. Gen. Pract. 2016, 66, e457–e459. [Google Scholar] [CrossRef] [PubMed]
- Fajans, S.S.; Bell, G.I. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef]
- Törn, C.; Landin-Olsson, M.; Ostman, J.; Scherstén, B.; Arnqvist, H.; Blohmé, G.; Björk, E.; Bolinder, J.; Eriksson, J.; Littorin, B.; et al. Glutamic acid decarboxylase antibodies (GADA) is the most important factor for prediction of insulin therapy within 3 years in young adult diabetic patients not classified as Type 1 diabetes on clinical grounds. Diabetes/Metab. Res. Rev. 2000, 16, 442–447. [Google Scholar] [CrossRef]
- McDonald, T.J.; Colclough, K.; Brown, R.; Shields, B.; Shepherd, M.; Bingley, P.; Williams, A.; Hattersley, A.T.; Ellard, S. Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from Type 1 diabetes. Diabet. Med. 2011, 28, 1028–1033. [Google Scholar] [CrossRef]
- Zhang, H.; Colclough, K.; Gloyn, A.L.; Pollin, T.I. Monogenic diabetes: A gateway to precision medicine in diabetes. J. Clin. Investig. 2021, 131, e142244. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chan, L. Monogenic Diabetes: What It Teaches Us on the Common Forms of Type 1 and Type 2 Diabetes. Endocr. Rev. 2016, 37, 190–222. [Google Scholar] [CrossRef]
- Gardner, D.S.; Tai, E.S. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab. Syndr. Obes. Targets Ther. 2012, 5, 101–108. [Google Scholar] [CrossRef]
- Schiabor Barrett, K.M.; Telis, N.; McEwen, L.M.; Burrows, E.K.; Khuder, B.; Judge, D.P.; Pawloski, P.A.; Grzymski, J.J.; Washington, N.L.; Bolze, A.; et al. Underestimated risk of secondary complications in pathogenic and glucose-elevating GCK variant carriers with type 2 diabetes. Commun. Med. 2024, 4, 239. [Google Scholar] [CrossRef]
- Ferre, T.; Riu, E.; Bosch, F.; Valera, A. Evidence from transgenic mice that glucokinase is rate limiting for glucose utilization in the liver. FASEB J. 1996, 10, 1213–1218. [Google Scholar] [CrossRef]
- Christesen, H.B.T.; Jacobsen, B.B.; Odili, S.; Buettger, C.; Cuesta-Munoz, A.; Hansen, T.; Brusgaard, K.; Massa, O.; Magnuson, M.A.; Shiota, C.; et al. The Second Activating Glucokinase Mutation (A456V): Implications for Glucose Homeostasis and Diabetes Therapy. Diabetes 2002, 51, 1240–1246. [Google Scholar] [CrossRef]
- Hulín, J.; Škopková, M.; Valkovičová, T.; Mikulajova, S.; Rosoľanková, M.; Papcun, P.; Gašperíková, D.; Stanik, J. Clinical implications of the glucokinase impaired function–Gck-Mody today. Physiol. Res. 2020, 69, 995. [Google Scholar] [CrossRef] [PubMed]
- Abu Aqel, Y.; Alnesf, A.; Aigha, I.I.; Islam, Z.; Kolatkar, P.R.; Teo, A.; Abdelalim, E.M. Glucokinase (GCK) in diabetes: From molecular mechanisms to disease pathogenesis. Cell. Mol. Biol. Lett. 2024, 29, 120. [Google Scholar] [CrossRef]
- Chakera, A.J.; Steele, A.M.; Gloyn, A.L.; Shepherd, M.H.; Shields, B.; Ellard, S.; Hattersley, A.T. Recognition and Management of Individuals With Hyperglycemia Because of a Heterozygous Glucokinase Mutation. Diabetes Care 2015, 38, 1383–1392. [Google Scholar] [CrossRef]
- Gersing, S.; Cagiada, M.; Gebbia, M.; Gjesing, A.P.; Coté, A.G.; Seesankar, G.; Li, R.; Tabet, D.; Weile, J.; Stein, A.; et al. A comprehensive map of human glucokinase variant activity. Genome Biol. 2023, 24, 97. [Google Scholar] [CrossRef]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Chakera, A.J.; Spyer, G.; Vincent, N.; Ellard, S.; Hattersley, A.T.; Dunne, F.P. The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: The Atlantic Diabetes in Pregnancy cohort. Diabetes Care 2014, 37, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Han, X.; Zhou, X.; Li, Y.; Gong, S.; Zhang, S.; Cai, X.; Zhou, L.; Luo, Y.; Li, M.; et al. A new clinical screening strategy and prevalence estimation for glucokinase variant-induced diabetes in an adult Chinese population. Genet. Med. 2019, 21, 939–947. [Google Scholar] [CrossRef]
- Shields, B.M.; Shepherd, M.; Hudson, M.; McDonald, T.J.; Colclough, K.; Peters, J.; Knight, B.; Hyde, C.; Ellard, S.; Pearson, E.R.; et al. Population-Based Assessment of a Biomarker-Based Screening Pathway to Aid Diagnosis of Monogenic Diabetes in Young-Onset Patients. Diabetes Care 2017, 40, 1017–1025. [Google Scholar] [CrossRef]
- da Silva Santos, T.; Fonseca, L.; Santos Monteiro, S.; Borges Duarte, D.; Martins Lopes, A.; Couto de Carvalho, A.; Oliveira, M.J.; Borges, T.; Laranjeira, F.; Couce, M.L.; et al. MODY probability calculator utility in individuals’ selection for genetic testing: Its accuracy and performance. Endocrinol. Diabetes Metab. 2022, 5, e00332. [Google Scholar] [CrossRef]
- Kreienkamp, R.J.; Shields, B.M.; Pollin, T.I.; Shah, A.S.; Liese, A.D.; Bellatorre, A.; Pihoker, C.; Tosur, M.; Florez, J.C.; Srinivasan, S.; et al. MODY Calculator and Clinical Features Routinely Used to Distinguish MODY From Type 2 Diabetes in Adults Perform Poorly for Youth Clinically Diagnosed With Type 2 Diabetes. Diabetes Care 2024, 48, e3–e5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, Y.; Ma, F.; Shu, H.; Zheng, L.; Liu, Y.; Li, X.; Xu, T.; Zhou, Z.; Zhou, K. MODY Probability Calculator Is Suitable for MODY Screening in China: A Population-based Study. J. Endocr. Soc. 2024, 8, bvae047. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; Carlsson, A.; Patel, K.; Knupp, J.; Kaur, A.; Johnston, D.; Colclough, K.; Larsson, H.E.; Forsander, G.; Samuelsson, U.; et al. Development of a clinical calculator to aid the identification of MODY in pediatric patients at the time of diabetes diagnosis. Sci. Rep. 2024, 14, 10589. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.H.J.; Ghosh, S.; Bok, C.M.; Ching, C.; Low, B.S.J.; Chen, J.T.; Lim, E.; Miserendino, M.C.; Tan, Y.S.; Hoon, S.; et al. HNF4A and HNF1A exhibit tissue specific target gene regulation in pancreatic beta cells and hepatocytes. Nat. Commun. 2024, 15, 4288. [Google Scholar] [CrossRef]
- Ferrè, S.; Igarashi, P. New insights into the role of HNF-1β in kidney (patho)physiology. Pediatr. Nephrol. 2019, 34, 1325–1335. [Google Scholar] [CrossRef]
- St-Jean, M.; Boudreau, F.; Carpentier, A.C.; Hivert, M.-F. HNF1α defect influences post-prandial lipid regulation. PLoS ONE 2017, 12, e0177110. [Google Scholar] [CrossRef]
- Li, L.-M.; Jiang, B.-G.; Sun, L.-L. HNF1A: From Monogenic Diabetes to Type 2 Diabetes and Gestational Diabetes Mellitus. Front. Endocrinol. 2022, 13, 829565. [Google Scholar] [CrossRef]
- Peixoto-Barbosa, R.; Reis, A.F.; Giuffrida, F.M.A. Update on clinical screening of maturity-onset diabetes of the young (MODY). Diabetol. Metab. Syndr. 2020, 12, 50. [Google Scholar] [CrossRef]
- Steele, A.M.; Shields, B.M.; Shepherd, M.; Ellard, S.; Hattersley, A.T.; Pearson, E.R. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet. Med. 2010, 27, 157–161. [Google Scholar] [CrossRef]
- Qian, M.F.; Bevacqua, R.J.; Coykendall, V.M.N.; Liu, X.; Zhao, W.; Chang, C.A.; Gu, X.; Dai, X.-Q.; MacDonald, P.E.; Kim, S.K. HNF1α maintains pancreatic α and β cell functions in primary human islets. JCI Insight 2023, 8, e170884. [Google Scholar] [CrossRef]
- Pontoglio, M.; Sreenan, S.; Roe, M.; Pugh, W.; Ostrega, D.; Doyen, A.; Pick, A.J.; Baldwin, A.; Velho, G.; Froguel, P.; et al. Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficient mice. J. Clin. Investig. 1998, 101, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Cujba, A.M.; Alvarez-Fallas, M.E.; Pedraza-Arevalo, S.; Laddach, A.; Shepherd, M.H.; Hattersley, A.T.; Watt, F.M.; Sancho, R. An HNF1α truncation associated with maturity-onset diabetes of the young impairs pancreatic progenitor differentiation by antagonizing HNF1β function. Cell Rep. 2022, 38, 110425. [Google Scholar] [CrossRef]
- Wang, H.; Antinozzi, P.A.; Hagenfeldt, K.A.; Maechler, P.; Wollheim, C.B. Molecular targets of a human HNF1 alpha mutation responsible for pancreatic beta-cell dysfunction. EMBO J. 2000, 19, 4257–4264. [Google Scholar] [CrossRef]
- Miyachi, Y.; Miyazawa, T.; Ogawa, Y. HNF1A Mutations and Beta Cell Dysfunction in Diabetes. Int. J. Mol. Sci. 2022, 23, 3222. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, L.; Guo, H.; Fan, X.; Liu, T.; Xu, C.; He, Z.; Song, Y.; Gao, L.; Shao, S.; et al. Dominant-negative HNF1α mutant promotes liver steatosis and inflammation by regulating hepatic complement factor D. iScience 2023, 26, 108018. [Google Scholar] [CrossRef]
- Lehto, M.; Tuomi, T.; Mahtani, M.M.; Widén, E.; Forsblom, C.; Sarelin, L.; Gullström, M.; Isomaa, B.; Lehtovirta, M.; Hyrkkö, A.; et al. Characterization of the MODY3 phenotype. Early-onset diabetes caused by an insulin secretion defect. J. Clin. Investig. 1997, 99, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhu, X.; Jiang, X.; Li, S.; Lv, Y. Transcriptional control by HNF-1: Emerging evidence showing its role in lipid metabolism and lipid metabolism disorders. Genes Dis. 2022, 9, 1248–1257. [Google Scholar] [CrossRef]
- Yamagata, K.; Yang, Q.; Yamamoto, K.; Iwahashi, H.; Miyagawa, J.; Okita, K.; Yoshiuchi, I.; Miyazaki, J.; Noguchi, T.; Nakajima, H.; et al. Mutation P291fsinsC in the transcription factor hepatocyte nuclear factor-1alpha is dominant negative. Diabetes 1998, 47, 1231–1235. [Google Scholar] [CrossRef]
- Lee, Y.H.; Sauer, B.; Gonzalez, F.J. Laron dwarfism and non-insulin-dependent diabetes mellitus in the Hnf-1alpha knockout mouse. Mol. Cell. Biol. 1998, 18, 3059–3068. [Google Scholar] [CrossRef]
- Shih, D.Q.; Bussen, M.; Sehayek, E.; Ananthanarayanan, M.; Shneider, B.L.; Suchy, F.J.; Shefer, S.; Bollileni, J.S.; Gonzalez, F.J.; Breslow, J.L.; et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 2001, 27, 375–382. [Google Scholar] [CrossRef]
- Estrada, K.; Aukrust, I.; Bjørkhaug, L.; Burtt, N.P.; Mercader, J.M.; García-Ortiz, H.; Huerta-Chagoya, A.; Moreno-Macías, H.; Walford, G.; Flannick, J.; et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 2014, 311, 2305–2314. [Google Scholar] [CrossRef]
- Locke, J.M.; Saint-Martin, C.; Laver, T.W.; Patel, K.A.; Wood, A.R.; Sharp, S.A.; Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T.; Harries, L.W.; et al. The Common HNF1A Variant I27L Is a Modifier of Age at Diabetes Diagnosis in Individuals with HNF1A-MODY. Diabetes 2018, 67, 1903–1907. [Google Scholar] [CrossRef] [PubMed]
- Ludwig-Słomczyńska, A.H.; Seweryn, M.T.; Radkowski, P.; Kapusta, P.; Machlowska, J.; Pruhova, S.; Gasperikova, D.; Bellanne-Chantelot, C.; Hattersley, A.; Kandasamy, B.; et al. Variants influencing age at diagnosis of HNF1A-MODY. Mol. Med. 2022, 28, 113. [Google Scholar] [CrossRef] [PubMed]
- Valkovicova, T.; Skopkova, M.; Stanik, J.; Gasperikova, D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr. Regul. 2019, 53, 110–134. [Google Scholar] [CrossRef] [PubMed]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009, 41, 56–65. [Google Scholar] [CrossRef]
- Terryn, S.; Tanaka, K.; Lengelé, J.P.; Olinger, E.; Dubois-Laforgue, D.; Garbay, S.; Kozyraki, R.; Van Der Smissen, P.; Christensen, E.I.; Courtoy, P.J.; et al. Tubular proteinuria in patients with HNF1α mutations: HNF1α drives endocytosis in the proximal tubule. Kidney Int. 2016, 89, 1075–1089. [Google Scholar] [CrossRef]
- Pontoglio, M.; Prié, D.; Cheret, C.; Doyen, A.; Leroy, C.; Froguel, P.; Velho, G.; Yaniv, M.; Friedlander, G. HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep. 2000, 1, 359–365. [Google Scholar] [CrossRef]
- Acosta-Montalvo, A.; Saponaro, C.; Thevenet, J.; Chiral, M.; Piron, A.; Delalleau, N.; Pasquetti, G.; Coddeville, A.; Moreno, M.; Gmyr, V.; et al. 149-OR: Genetic Modulation of HNF1A Activity via SGLT2 Deficiency Leads to Transient Intermittent Hyperglycemia: Consequences for HNF1A-MODY. Diabetes 2022, 71, 149-OR. [Google Scholar] [CrossRef]
- Ellard, S.; Colclough, K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Hum. Mutat. 2006, 27, 854–869. [Google Scholar] [CrossRef]
- O’Brien, V.P.; Bokelmann, K.; Ramírez, J.; Jobst, K.; Ratain, M.J.; Brockmöller, J.; Tzvetkov, M.V. Hepatocyte nuclear factor 1 regulates the expression of the organic cation transporter 1 via binding to an evolutionary conserved region in intron 1 of the OCT1 gene. J. Pharmacol. Exp. Ther. 2013, 347, 181–192. [Google Scholar] [CrossRef]
- Maher, J.M.; Slitt, A.L.; Callaghan, T.N.; Cheng, X.; Cheung, C.; Gonzalez, F.J.; Klaassen, C.D. Alterations in transporter expression in liver, kidney, and duodenum after targeted disruption of the transcription factor HNF1α. Biochem. Pharmacol. 2006, 72, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, K.L.R.; Evers, R.; Hayden, E.; Hu, S.; Li, C.Y.; Meyer Zu Schwabedissen, H.E.; Neuhoff, S.; Oswald, S.; Piquette-Miller, M.; Saran, C.; et al. Regulation of Drug Transport Proteins-From Mechanisms to Clinical Impact: A White Paper on Behalf of the International Transporter Consortium. Clin. Pharmacol. Ther. 2022, 112, 461–484. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef]
- Kawoosa, F.; Shah, Z.A.; Masoodi, S.R.; Amin, A.; Rasool, R.; Fazili, K.M.; Dar, A.H.; Lone, A.; ul Bashir, S. Role of human organic cation transporter-1 (OCT-1/SLC22A1) in modulating the response to metformin in patients with type 2 diabetes. BMC Endocr. Disord. 2022, 22, 140. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.C.; Sobti, M.; Quinn, A.; Smith, N.J.; Brown, S.H.J.; Vandenberg, J.I.; Ryan, R.M.; O’Mara, M.L.; Stewart, A.G. Structural basis of promiscuous substrate transport by Organic Cation Transporter 1. Nat. Commun. 2023, 14, 6374. [Google Scholar] [CrossRef]
- Stride, A.; Vaxillaire, M.; Tuomi, T.; Barbetti, F.; Njølstad, P.R.; Hansen, T.; Costa, A.; Conget, I.; Pedersen, O.; Søvik, O.; et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002, 45, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Arya, V.B.; Rahman, S.; Senniappan, S.; Flanagan, S.E.; Ellard, S.; Hussain, K. HNF4A mutation: Switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diabet. Med. 2014, 31, e11–e15. [Google Scholar] [CrossRef]
- Colclough, K.; Saint-Martin, C.; Timsit, J.; Ellard, S.; Bellanne-Chantelot, C. Clinical utility gene card for: Maturity-onset diabetes of the young. Eur. J. Hum. Genet. 2014, 22, 1153. [Google Scholar] [CrossRef]
- Kaci, A.; Solheim, M.H.; Silgjerd, T.; Hjaltadottir, J.; Hornnes, L.H.; Molnes, J.; Madsen, A.; Sjøholt, G.; Bellanné-Chantelot, C.; Caswell, R.; et al. Functional characterization of HNF4A gene variants identify promoter and cell line specific transactivation effects. Hum. Mol. Genet. 2024, 33, 894–904. [Google Scholar] [CrossRef]
- Qu, N.; Luan, T.; Liu, N.; Kong, C.; Xu, L.; Yu, H.; Kang, Y.; Han, Y. Hepatocyte nuclear factor 4 a (HNF4α): A perspective in cancer. Biomed. Pharmacother. 2023, 169, 115923. [Google Scholar] [CrossRef]
- Sladek, F.M.; Zhong, W.M.; Lai, E.; Darnell, J.E., Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990, 4, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Gaál, Z.; Szűcs, Z.; Kántor, I.; Luczay, A.; Tóth-Heyn, P.; Benn, O.; Felszeghy, E.; Karádi, Z.; Madar, L.; Balogh, I. A Comprehensive Analysis of Hungarian MODY Patients-Part I: Gene Panel Sequencing Reveals Pathogenic Mutations in HNF1A, HNF1B, HNF4A, ABCC8 and INS Genes. Life 2021, 11, 755. [Google Scholar] [CrossRef]
- Fajans, S.S.; Bell, G.I.; Polonsky, K.S. Molecular Mechanisms and Clinical Pathophysiology of Maturity-Onset Diabetes of the Young. N. Engl. J. Med. 2001, 345, 971–980. [Google Scholar] [CrossRef]
- Girard, R.; Darsigny, M.; Jones, C.; Maloum-Rami, F.; Gélinas, Y.; Carpentier, A.C.; Laplante, M.; Perreault, N.; Boudreau, F. HNF4α is a novel regulator of intestinal glucose-dependent insulinotropic polypeptide. Sci. Rep. 2019, 9, 4200. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, M.; Duncan, S.A. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4α regulates expression of genes required for glucose transport and metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 13209–13214. [Google Scholar] [CrossRef]
- Vemuri, K.; Radi, S.H.; Sladek, F.M.; Verzi, M.P. Multiple roles and regulatory mechanisms of the transcription factor HNF4 in the intestine. Front. Endocrinol. 2023, 14, 1232569. [Google Scholar] [CrossRef]
- Taraviras, S.; Monaghan, A.P.; Schütz, G.; Kelsey, G. Characterization of the mouse HNF-4 gene and its expression during mouse embryogenesis. Mech. Dev. 1994, 48, 67–79. [Google Scholar] [CrossRef]
- Tanaka, T.; Jiang, S.; Hotta, H.; Takano, K.; Iwanari, H.; Sumi, K.; Daigo, K.; Ohashi, R.; Sugai, M.; Ikegame, C.; et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J. Pathol. 2006, 208, 662–672. [Google Scholar] [CrossRef]
- Girard, R.; Tremblay, S.; Noll, C.; St-Jean, S.; Jones, C.; Gélinas, Y.; Maloum-Rami, F.; Perreault, N.; Laplante, M.; Carpentier, A.C.; et al. The transcription factor hepatocyte nuclear factor 4A acts in the intestine to promote white adipose tissue energy storage. Nat. Commun. 2022, 13, 224. [Google Scholar] [CrossRef]
- Dzhemileva, L.U.; Zakharova, E.N.; Goncharenko, A.O.; Vorontsova, M.V.; Rumyantsev, S.A.; Mokrysheva, N.G.; Loguinova, M.Y.; Chekhonin, V.P. Current views on etiology, diagnosis, epidemiology and gene therapy of maturity onset diabetes in the young. Front. Endocrinol. 2024, 15, 1497298. [Google Scholar] [CrossRef]
- Yin, L.; Ma, H.; Ge, X.; Edwards, P.A.; Zhang, Y. Hepatic hepatocyte nuclear factor 4α is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Özsu, E.; Çetinkaya, S.; Bolu, S.; Hatipoğlu, N.; Savaş Erdeve, Ş.; Evliyaoğlu, O.; Baş, F.; Çayır, A.; Dündar, İ.; Akbaş, E.D.; et al. Clinical and Laboratory Characteristics of MODY Cases, Genetic Mutation Spectrum and Phenotype-genotype Relationship. J. Clin. Res. Pediatr. Endocrinol. 2024, 16, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Pihoker, C.; Gilliam, L.K.; Ellard, S.; Dabelea, D.; Davis, C.; Dolan, L.M.; Greenbaum, C.J.; Imperatore, G.; Lawrence, J.M.; Marcovina, S.M.; et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: Results from the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab. 2013, 98, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Aarthy, R.; Aston-Mourney, K.; Amutha, A.; Mikocka-Walus, A.; Anjana, R.M.; Unnikrishnan, R.; Jebarani, S.; Venkatesan, U.; Gopi, S.; Radha, V.; et al. Prevalence, clinical features and complications of common forms of Maturity Onset Diabetes of the Young (MODY) seen at a tertiary diabetes centre in south India. Prim. Care Diabetes 2023, 17, 401–407. [Google Scholar] [CrossRef]
- Yorifuji, T.; Higuchi, S.; Kawakita, R.; Hosokawa, Y.; Aoyama, T.; Murakami, A.; Kawae, Y.; Hatake, K.; Nagasaka, H.; Tamagawa, N. Genetic basis of early-onset, maturity-onset diabetes of the young-like diabetes in Japan and features of patients without mutations in the major MODY genes: Dominance of maternal inheritance. Pediatr. Diabetes 2018, 19, 1164–1172. [Google Scholar] [CrossRef]
- Billings, L.K.; Shi, Z.; Resurreccion, W.K.; Wang, C.H.; Wei, J.; Pollin, T.I.; Udler, M.S.; Xu, J. Statistical evidence for high-penetrance MODY-causing genes in a large population-based cohort. Endocrinol. Diabetes Metab. 2022, 5, e372. [Google Scholar] [CrossRef]
- Shankar, R.K.; Ellard, S.; Standiford, D.; Pihoker, C.; Gilliam, L.K.; Hattersley, A.; Dolan, L.M. Digenic heterozygous HNF1A and HNF4A mutations in two siblings with childhood-onset diabetes. Pediatr. Diabetes 2013, 14, 535–538. [Google Scholar] [CrossRef]
- Laver, T.W.; Colclough, K.; Shepherd, M.; Patel, K.; Houghton, J.A.; Dusatkova, P.; Pruhova, S.; Morris, A.D.; Palmer, C.N.; McCarthy, M.I.; et al. The Common p.R114W HNF4A Mutation Causes a Distinct Clinical Subtype of Monogenic Diabetes. Diabetes 2016, 65, 3212–3217. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef]
- McTaggart, J.S.; Clark, R.H.; Ashcroft, F.M. The role of the KATP channel in glucose homeostasis in health and disease: More than meets the islet. J. Physiol. 2010, 588 Pt 17, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Riveline, J.P.; Rousseau, E.; Reznik, Y.; Fetita, S.; Philippe, J.; Dechaume, A.; Hartemann, A.; Polak, M.; Petit, C.; Charpentier, G.; et al. Clinical and metabolic features of adult-onset diabetes caused by ABCC8 mutations. Diabetes Care 2012, 35, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Diatloff-Zito, C.; Edghill, E.L.; Bellanné-Chantelot, C.; Nivot, S.; Coutant, R.; Ellard, S.; Hattersley, A.T.; Robert, J.J. KCNJ11 activating mutations are associated with developmental delay, epilepsy and neonatal diabetes syndrome and other neurological features. Eur. J. Hum. Genet. 2006, 14, 824–830. [Google Scholar] [CrossRef]
- Bankura, B.; Pattanayak, A.K.; Ghosh, S.; Guria, S.; Sinha, A.; Das, M. Implication of KCNJ11 and TCF7L2 gene variants for the predisposition of type 2 diabetes mellitus in West Bengal, India. Diabetes Epidemiol. Manag. 2022, 6, 100066. [Google Scholar] [CrossRef]
- Edghill, E.L.; Gloyn, A.L.; Goriely, A.; Harries, L.W.; Flanagan, S.E.; Rankin, J.; Hattersley, A.T.; Ellard, S. Origin of de Novo KCNJ11 Mutations and Risk of Neonatal Diabetes for Subsequent Siblings. J. Clin. Endocrinol. Metab. 2007, 92, 1773–1777. [Google Scholar] [CrossRef]
- Phani, N.M.; Guddattu, V.; Bellampalli, R.; Seenappa, V.; Adhikari, P.; Nagri, S.K.; D′Souza, S.C.; Mundyat, G.P.; Satyamoorthy, K.; Rai, P.S. Population Specific Impact of Genetic Variants in KCNJ11 Gene to Type 2 Diabetes: A Case-Control and Meta-Analysis Study. PLoS ONE 2014, 9, e107021. [Google Scholar] [CrossRef]
- Gallardo-Blanco, H.L.; Villarreal-Perez, J.Z.; Cerda-Flores, R.M.; Figueroa, A.; Sanchez-Dominguez, C.N.; Gutierrez-Valverde, J.M.; Torres-Muñoz, I.C.; Lavalle-Gonzalez, F.J.; Gallegos-Cabriales, E.C.; Martinez-Garza, L.E. Genetic variants in KCNJ11, TCF7L2 and HNF4A are associated with type 2 diabetes, BMI and dyslipidemia in families of Northeastern Mexico: A pilot study. Exp. Ther. Med. 2017, 13, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Javorsky, M.; Klimcakova, L.; Schroner, Z.; Zidzik, J.; Babjakova, E.; Fabianova, M.; Kozarova, M.; Tkacova, R.; Salagovic, J.; Tkac, I. KCNJ11 gene E23K variant and therapeutic response to sulfonylureas. Eur. J. Intern. Med. 2012, 23, 245–249. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Inoue, H.; Keshavarz, P.; Miyawaki, K.; Yamaguchi, Y.; Moritani, M.; Kunika, K.; Nakamura, N.; Yoshikawa, T.; Yasui, N.; et al. SNPs in the KCNJ11-ABCC8 gene locus are associated with type 2 diabetes and blood pressure levels in the Japanese population. J. Hum. Genet. 2007, 52, 781–793. [Google Scholar] [CrossRef]
- Koo, B.K.; Cho, Y.M.; Park, B.L.; Cheong, H.S.; Shin, H.D.; Jang, H.C.; Kim, S.Y.; Lee, H.K.; Park, K.S. Polymorphisms of KCNJ11 (Kir6.2 gene) are associated with Type 2 diabetes and hypertension in the Korean population. Diabet. Med. 2007, 24, 178–186. [Google Scholar] [CrossRef]
- Moazzam-Jazi, M.; Najd-Hassan-Bonab, L.; Masjoudi, S.; Tohidi, M.; Hedayati, M.; Azizi, F.; Daneshpour, M.S. Risk of type 2 diabetes and KCNJ11 gene polymorphisms: A nested case-control study and meta-analysis. Sci. Rep. 2022, 12, 20709. [Google Scholar] [CrossRef]
- Mao, H.; Li, Q.; Gao, S. Meta-analysis of the relationship between common type 2 diabetes risk gene variants with gestational diabetes mellitus. PLoS ONE 2012, 7, e45882. [Google Scholar] [CrossRef]
- Lauenborg, J.; Grarup, N.; Damm, P.; Borch-Johnsen, K.; Jørgensen, T.; Pedersen, O.; Hansen, T. Common type 2 diabetes risk gene variants associate with gestational diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 145–150. [Google Scholar] [CrossRef]
- Majcher, S.; Ustianowski, P.; Malinowski, D.; Czerewaty, M.; Tarnowski, M.; Safranow, K.; Dziedziejko, V.; Pawlik, A. KCNJ11 and KCNQ1 Gene Polymorphisms and Placental Expression in Women with Gestational Diabetes Mellitus. Genes 2022, 13, 1315. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Na, R.; Xu, R.; Wang, S.; Sheng, H.; Wu, W.; Qu, Y. Quantitative assessment of the effect of KCNJ11 gene polymorphism on the risk of type 2 diabetes. PLoS ONE 2014, 9, e93961. [Google Scholar] [CrossRef] [PubMed]
- Muftin, N.Q.; Jubair, S. KCNJ11 polymorphism is associated with type 2 diabetes mellitus in Iraqi patients. Gene Rep. 2019, 17, 100480. [Google Scholar] [CrossRef]
- Keshavarz, P.; Habibipour, R.; Ghasemi, M.; Kazemnezhad, E.; Alizadeh, M.; Omami, M.H. Lack of genetic susceptibility of KCNJ11 E23K polymorphism with risk of type 2 diabetes in an Iranian population. Endocr. Res. 2014, 39, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Makhzoom, O.; Kabalan, Y.; Al-Quobaili, F. Association of KCNJ11 rs5219 gene polymorphism with type 2 diabetes mellitus in a population of Syria: A case-control study. BMC Med. Genet. 2019, 20, 107. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, D.; Liu, Y.; Zhao, T.; Chen, Z.; Liu, Z.; Yu, L.; Zhang, Z.; Xu, H.; He, L. The E23K variation in the KCNJ11 gene is associated with type 2 diabetes in Chinese and East Asian population. J. Hum. Genet. 2009, 54, 433–435. [Google Scholar] [CrossRef]
- Gonen, M.S.; Arikoglu, H.; Erkoc Kaya, D.; Ozdemir, H.; Ipekci, S.H.; Arslan, A.; Kayis, S.A.; Gogebakan, B. Effects of single nucleotide polymorphisms in K(ATP) channel genes on type 2 diabetes in a Turkish population. Arch. Med. Res. 2012, 43, 317–323. [Google Scholar] [CrossRef]
- Elzehery, R.; El-Hafez, H.A.; Elsehely, I.; Barakat, A.; Foda, E.A.E.; Hendawy, S.R.; Gameil, M.A.; Nada, H.S.; El-Sebaie, A. Association of the E23K (rs5219) polymorphism in the potassium channel (KCNJ11) gene with diabetic neuropathy in type 2 diabetes. Gene 2024, 921, 148525. [Google Scholar] [CrossRef] [PubMed]
- Shorokhova, P.; Baranov, V.; Vorokhobina, N. Abstract #1001320: Effects of KCNJ11 RS5219 Variant on Metformin Pharmacodynamics in Patients With Newly Diagnosed Type 2 Diabetes Mellitus. Endocr. Pract. 2021, 27, S39. [Google Scholar] [CrossRef]
- Ahmed, A.; Elsadek, H.M.; Shalaby, S.M.; Elnahas, H.M. Association of SLC22A1, SLC47A1, and KCNJ11 polymorphisms with efficacy and safety of metformin and sulfonylurea combination therapy in Egyptian patients with type 2 diabetes. Res. Pharm. Sci. 2023, 18, 614–625. [Google Scholar] [CrossRef]
- Lie, X.; Fang, Y.A.O.; Limin, J.I.N.; Fan, N.; Hanqiang, S.H.I.; Shuqin, D.U.; Yanbo, S.H.I. Study of KCNJ11 rs5219 Gene Polymorphism on the Efficacy of Metformin Combined with Gliclazide in Newly Diagnosed Diabetes Mellitus Type 2 Patients. Chin. J. Mod. Appl. Pharm. 2023, 40, 3431–3438. [Google Scholar] [CrossRef]
- Sesti, G.; Laratta, E.; Cardellini, M.; Andreozzi, F.; Del Guerra, S.; Irace, C.; Gnasso, A.; Grupillo, M.; Lauro, R.; Hribal, M.L.; et al. The E23K Variant of KCNJ11 Encoding the Pancreatic β-Cell Adenosine 5′-Triphosphate-Sensitive Potassium Channel Subunit Kir6.2 Is Associated with an Increased Risk of Secondary Failure to Sulfonylurea in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2006, 91, 2334–2339. [Google Scholar] [CrossRef] [PubMed]
- Del Bosque-Plata, L.; Martínez-Martínez, E.; Espinoza-Camacho, M.; Gragnoli, C. The Role of TCF7L2 in Type 2 Diabetes. Diabetes 2021, 70, 1220–1228. [Google Scholar] [CrossRef]
- Tong, Y.; Lin, Y.; Zhang, Y.; Yang, J.; Zhang, Y.; Liu, H.; Zhang, B. Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: A large Human Genome Epidemiology (HuGE) review and meta-analysis. BMC Med. Genet. 2009, 10, 15. [Google Scholar] [CrossRef]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef]
- Ip, W.; Chiang, Y.-t.A.; Jin, T. The involvement of the wnt signaling pathway and TCF7L2 in diabetes mellitus: The current understanding, dispute, and perspective. Cell Biosci. 2012, 2, 28. [Google Scholar] [CrossRef]
- Jin, T. The WNT signalling pathway and diabetes mellitus. Diabetologia 2008, 51, 1771–1780. [Google Scholar] [CrossRef]
- Jin, T. Current Understanding on Role of the Wnt Signaling Pathway Effector TCF7L2 in Glucose Homeostasis. Endocr. Rev. 2016, 37, 254–277. [Google Scholar] [CrossRef] [PubMed]
- Helgason, A.; Pálsson, S.; Thorleifsson, G.; Grant, S.F.; Emilsson, V.; Gunnarsdottir, S.; Adeyemo, A.; Chen, Y.; Chen, G.; Reynisdottir, I.; et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat. Genet. 2007, 39, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hassouna, M.M.; Moustafa, M.S.; Hamdy, M.; Abdelsameea, E.; Abbasy, M.; Naguib, M. Study of transcription factor 7-like 2 (TCF7L2) gene polymorphism in cirrhotic patients with diabetes. Egypt. Liver J. 2023, 13, 54. [Google Scholar] [CrossRef]
- Sakhneny, L.; Rachi, E.; Epshtein, A.; Guez, H.C.; Wald-Altman, S.; Lisnyansky, M.; Khalifa-Malka, L.; Hazan, A.; Baer, D.; Priel, A.; et al. Pancreatic Pericytes Support beta-Cell Function in a TCF7L2-Dependent Manner. Diabetes 2018, 67, 437–447. [Google Scholar] [CrossRef]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef]
- Yang, Y.; Chang, B.H.; Samson, S.L.; Li, M.V.; Chan, L. The Krüppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009, 37, 2529–2538. [Google Scholar] [CrossRef]
- Scoville, D.W.; Kang, H.S.; Jetten, A.M. GLIS1-3: Emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig. 2017, 4, 80. [Google Scholar] [CrossRef]
- Collier, J.B.; Kang, H.S.; Roh, Y.G.; Srivastava, C.; Grimm, S.A.; Jarmusch, A.K.; Jetten, A.M. GLIS3: A novel transcriptional regulator of mitochondrial functions and metabolic reprogramming in postnatal kidney and polycystic kidney disease. Mol. Metab. 2024, 90, 102052. [Google Scholar] [CrossRef]
- Michaud, M.E.; Mota, L.; Bakhtiari, M.; Thomas, B.E.; Tomeo, J.; Pilcher, W.; Contreras, M.; Ferran, C.; Bhasin, S.S.; Pradhan-Nabzdyk, L.; et al. Early Injury Landscape in Vein Harvest by Single-Cell and Spatial Transcriptomics. Circ. Res. 2024, 135, 110–134. [Google Scholar] [CrossRef]
- Nogueira, T.C.; Paula, F.M.; Villate, O.; Colli, M.L.; Moura, R.F.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Cnop, M.; Julier, C.; et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 2013, 9, e1003532. [Google Scholar] [CrossRef]
- Wen, X.; Yang, Y. Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J. Mol. Endocrinol. 2017, 58, R73–R85. [Google Scholar] [CrossRef] [PubMed]
- Pinto, K.; Chetty, R. Gene of the month: GLIS1-3. J. Clin. Pathol. 2020, 73, 527–530. [Google Scholar] [CrossRef]
- Kang, H.S.; Kim, Y.S.; ZeRuth, G.; Beak, J.Y.; Gerrish, K.; Kilic, G.; Sosa-Pineda, B.; Jensen, J.; Pierreux, C.E.; Lemaigre, F.P.; et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol. Cell. Biol. 2009, 29, 6366–6379. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.W.; Jetten, A.M. GLIS3: A Critical Transcription Factor in Islet β-Cell Generation. Cells 2021, 10, 3471. [Google Scholar] [CrossRef]
- De Franco, E.; Flanagan, S.E.; Houghton, J.A.L.; Allen, H.L.; Mackay, D.J.G.; Temple, I.K.; Ellard, S.; Hattersley, A.T. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: An international cohort study. Lancet 2015, 386, 957–963. [Google Scholar] [CrossRef]
- Kang, H.S.; ZeRuth, G.; Lichti-Kaiser, K.; Vasanth, S.; Yin, Z.; Kim, Y.S.; Jetten, A.M. Gli-similar (Glis) Krüppel-like zinc finger proteins: Insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol. Histopathol. 2010, 25, 1481–1496. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Have, C.T.; Hollensted, M.; Grarup, N.; Linneberg, A.; Pedersen, O.; Nielsen, J.S.; Rungby, J.; Christensen, C.; Brandslund, I.; et al. Sequencing reveals protective and pathogenic effects on development of diabetes of rare GLIS3 variants. PLoS ONE 2019, 14, e0220805. [Google Scholar] [CrossRef]
- Nogueira, J.P.; Cusi, K. Role of Insulin Resistance in the Development of Nonalcoholic Fatty Liver Disease in People With Type 2 Diabetes: From Bench to Patient Care. Diabetes Spectr. 2024, 37, 20–28. [Google Scholar] [CrossRef]
- Kim, J.-a.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Shen, Y.; Su, Y.; Silva, F.J.; Weller, A.H.; Sostre-Colón, J.; Titchenell, P.M.; Steger, D.J.; Seale, P.; Soccio, R.E. Shared PPARα/γ Target Genes Regulate Brown Adipocyte Thermogenic Function. Cell Rep. 2020, 30, 3079–3091.e5. [Google Scholar] [CrossRef]
- Ricote, M.; Huang, J.; Fajas, L.; Li, A.; Welch, J.; Najib, J.; Witztum, J.L.; Auwerx, J.; Palinski, W.; Glass, C.K. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 7614–7619. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Fruchart, J.-C. Therapeutic roles of peroxisome proliferator–activated receptor agonists. Diabetes 2005, 54, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, H.F.; Thatcher, T.H.; Kottmann, R.M.; Garcia, T.M.; Phipps, R.P.; Sime, P.J. The Role of PPARs in Lung Fibrosis. PPAR Res. 2007, 2007, 71323. [Google Scholar] [CrossRef]
- Martínez-García, C.; Izquierdo, A.; Velagapudi, V.; Vivas, Y.; Velasco, I.; Campbell, M.; Burling, K.; Cava, F.; Ros, M.; Oresic, M.; et al. Accelerated renal disease is associated with the development of metabolic syndrome in a glucolipotoxic mouse model. Dis. Models Mech. 2012, 5, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, Y.; Gao, L.; Yang, Z.; Lin, J.; Ren, S.; Li, F.; Chen, J.; Wang, Z.; Dong, Z.; et al. PPAR-γ integrates obesity and adipocyte clock through epigenetic regulation of Bmal1. Theranostics 2022, 12, 1589–1606. [Google Scholar] [CrossRef]
- Kökény, G.; Calvier, L.; Hansmann, G. PPARγ and TGFβ-Major Regulators of Metabolism, Inflammation, and Fibrosis in the Lungs and Kidneys. Int. J. Mol. Sci. 2021, 22, 10431. [Google Scholar] [CrossRef]
- Roszer, T.; Menéndez-Gutiérrez, M.P.; Lefterova, M.I.; Alameda, D.; Núñez, V.; Lazar, M.A.; Fischer, T.; Ricote, M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 2011, 186, 621–631. [Google Scholar] [CrossRef]
- Arabshomali, A.; Bazzazzadehgan, S.; Mahdi, F.; Shariat-Madar, Z. Potential Benefits of Antioxidant Phytochemicals in Type 2 Diabetes. Molecules 2023, 28, 7209. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 206. [Google Scholar] [CrossRef]
- Mehta, R.; Birerdinc, A.; Younossi, Z.M. Host Genetic Variants in Obesity-Related Nonalcoholic Fatty Liver Disease. Clin. Liver Dis. 2014, 18, 249–267. [Google Scholar] [CrossRef]
- Semple, R.K.; Chatterjee, V.K.K.; O’Rahilly, S. PPARγ and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Sarhangi, N.; Sharifi, F.; Hashemian, L.; Hassani Doabsari, M.; Heshmatzad, K.; Rahbaran, M.; Jamaldini, S.H.; Aghaei Meybodi, H.R.; Hasanzad, M. PPARG (Pro12Ala) genetic variant and risk of T2DM: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12764. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; He, C.; Nie, H.; Pang, Q.; Wang, R.; Zeng, Z.; Song, Y. G Allele of the rs1801282 Polymorphism in PPARgamma Gene Confers an Increased Risk of Obesity and Hypercholesterolemia, While T Allele of the rs3856806 Polymorphism Displays a Protective Role Against Dyslipidemia: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2022, 13, 919087. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Korman, B.D.; Allanore, Y.; Dieude, P.; Armstrong, L.L.; Rzhetskaya, M.; Hinchcliff, M.; Carns, M.; Podlusky, S.; Shah, S.J.; et al. A candidate gene study reveals association between a variant of the Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ) gene and systemic sclerosis. Arthritis Res. Ther. 2015, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, S.; Jayaprakash, V. Thiazolidinediones and PPAR orchestra as antidiabetic agents: From past to present. Eur. J. Med. Chem. 2017, 126, 879–893. [Google Scholar] [CrossRef]
- Xue, C.Y.; Zhou, M.Q.; Zheng, Q.Y.; Zhang, J.H.; Cheng, W.T.; Bai, X.H.; Zhou, F.; Wu, A.M.; Nie, B.; Liu, W.J.; et al. Thiazolidinediones play a positive role in the vascular endothelium and inhibit plaque progression in diabetic patients with coronary atherosclerosis: A systematic review and meta-analysis. Front. Cardiovasc. Med. 2022, 9, 1043406. [Google Scholar] [CrossRef]
- Zhu, J.; Yu, X.; Zheng, Y.; Li, J.; Wang, Y.; Lin, Y.; He, Z.; Zhao, W.; Chen, C.; Qiu, K.; et al. Association of glucose-lowering medications with cardiovascular outcomes: An umbrella review and evidence map. Lancet Diabetes Endocrinol. 2020, 8, 192–205. [Google Scholar] [CrossRef]
- Yoo, J.; Jeon, J.; Baik, M.; Kim, J. Lobeglitazone, a novel thiazolidinedione, for secondary prevention in patients with ischemic stroke: A nationwide nested case-control study. Cardiovasc. Diabetol. 2023, 22, 106. [Google Scholar] [CrossRef]
- Arnold, S.V.; Inzucchi, S.E.; Echouffo-Tcheugui, J.B.; Tang, F.; Lam, C.S.P.; Sperling, L.S.; Kosiborod, M. Understanding Contemporary Use of Thiazolidinediones. Circ. Heart Fail. 2019, 12, e005855. [Google Scholar] [CrossRef]
- Roughead, E.E.; Chan, E.W.; Choi, N.K.; Kimura, M.; Kimura, T.; Kubota, K.; Lai, E.C.; Man, K.K.; Nguyen, T.A.; Ooba, N.; et al. Variation in Association Between Thiazolidinediones and Heart Failure Across Ethnic Groups: Retrospective analysis of Large Healthcare Claims Databases in Six Countries. Drug Saf. 2015, 38, 823–831. [Google Scholar] [CrossRef]
- Shen, C.; Fan, W.; Xie, H.J.; Wu, M.; Zhou, Z.Y.; Guo, Z.R.; Dong, C. Haplotype Analysis of PPARγ Gene Polymorphisms and the Lipoprotein (a) Level. Iran. J. Public Health 2018, 47, 973–979. [Google Scholar] [PubMed]
- Gallicchio, L.; Kalesan, B.; Huang, H.-Y.; Strickland, P.; Hoffman, S.C.; Helzlsouer, K.J. Genetic Polymorphisms of Peroxisome Proliferator-Activated Receptors and the Risk of Cardiovascular Morbidity and Mortality in a Community-Based Cohort in Washington County, Maryland. PPAR Res. 2008, 2008, 276581. [Google Scholar] [CrossRef] [PubMed]
- Bage, I.; Kamalanathan, S.; Selvarajan, S.; Sahoo, J.; Mathaiyan, J.; Naik, D. Peroxisome Proliferator-Activated Receptor α and γ Gene Polymorphisms among South Indian Patients with Diabetic Dyslipidaemia. Indian J. Endocrinol. Metab. 2023, 27, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Li, Z.; Chen, E.R.; Foong, Y.H.; Benson, K.K.; Dispirito, J.R.; Mullican, S.E.; Emmett, M.J.; Briggs, E.R.; Peed, L.C.; et al. Targeting PPARgamma in the epigenome rescues genetic metabolic defects in mice. J. Clin. Investig. 2017, 127, 1451–1462. [Google Scholar] [CrossRef]
- Mendoza-Herrera, K.; Florio, A.A.; Moore, M.; Marrero, A.; Tamez, M.; Bhupathiraju, S.N.; Mattei, J. The Leptin System and Diet: A Mini Review of the Current Evidence. Front. Endocrinol. 2021, 12, 749050. [Google Scholar] [CrossRef]
- Weigle, D.S.; Breen, P.A.; Matthys, C.C.; Callahan, H.S.; Meeuws, K.E.; Burden, V.R.; Purnell, J.Q. A high-protein diet induces sustained reductions in appetite, ad libitum caloric intake, and body weight despite compensatory changes in diurnal plasma leptin and ghrelin concentrations. Am. J. Clin. Nutr. 2005, 82, 41–48. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- German, J.P.; Wisse, B.E.; Thaler, J.P.; Oh, I.S.; Sarruf, D.A.; Ogimoto, K.; Kaiyala, K.J.; Fischer, J.D.; Matsen, M.E.; Taborsky, G.J., Jr.; et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes 2010, 59, 1626–1634. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Leptin, cardiovascular diseases and type 2 diabetes mellitus. Acta Pharmacol. Sin. 2018, 39, 1176–1188. [Google Scholar] [CrossRef]
- Bonnefond, A.; Semple, R.K. Achievements, prospects and challenges in precision care for monogenic insulin-deficient and insulin-resistant diabetes. Diabetologia 2022, 65, 1782–1795. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of Recombinant Leptin Therapy in a Child with Congenital Leptin Deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.T.; Suk, H.Y.; Yang, X.; Olabisi, O.; Yu, R.Y.; Durand, J.; Jelicks, L.A.; Kim, J.Y.; Scherer, P.E.; Wang, Y.; et al. Role of transcription factor NFAT in glucose and insulin homeostasis. Mol. Cell. Biol. 2006, 26, 7372–7387. [Google Scholar] [CrossRef] [PubMed]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Rahman, T.; Ismail, A.A.; Rashid, A.R. Diabetes-associated macrovasculopathy: Pathophysiology and pathogenesis. Diabetes Obes. Metab. 2007, 9, 767–780. [Google Scholar] [CrossRef]
- Jia, G.; Bai, H.; Mather, B.; Hill, M.A.; Jia, G.; Sowers, J.R. Diabetic Vasculopathy: Molecular Mechanisms and Clinical Insights. Int. J. Mol. Sci. 2024, 25, 804. [Google Scholar] [CrossRef]
- Kwok, T.n.C.; Ramage, L.E.; Kelman, A.; Suchacki, K.J.; Gray, C.; Boyle, L.D.; Semple, S.I.; MacGillivray, T.; MacNaught, G.; Patel, D.; et al. UCP1 expression in human brown adipose tissue is inversely associated with cardiometabolic risk factors. Eur. J. Endocrinol. 2024, 191, 106–115. [Google Scholar] [CrossRef]
- Jones, S.A.; Gogoi, P.; Ruprecht, J.J.; King, M.S.; Lee, Y.; Zögg, T.; Pardon, E.; Chand, D.; Steimle, S.; Copeman, D.M.; et al. Structural basis of purine nucleotide inhibition of human uncoupling protein 1. Sci. Adv. 2023, 9, eadh4251. [Google Scholar] [CrossRef]
- Ledesma, A.; de Lacoba, M.G.; Rial, E. The mitochondrial uncoupling proteins. Genome Biol. 2002, 3, reviews3015.3011. [Google Scholar] [CrossRef]
- Wang, G.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74, 844–857.e7. [Google Scholar] [CrossRef]
- Fisler, J.S.; Warden, C.H. Uncoupling proteins, dietary fat and the metabolic syndrome. Nutr. Metab. 2006, 3, 38. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.S.; Li, R.; Liu, V.Y.; Fu, L.; Moore, D.D.; Ma, K.; Yechoor, V.K. Loss of Bmal1 leads to uncoupling and impaired glucose-stimulated insulin secretion in β-cells. Islets 2011, 3, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Keipert, S.; Kutschke, M.; Ost, M.; Schwarzmayr, T.; van Schothorst, E.M.; Lamp, D.; Brachthäuser, L.; Hamp, I.; Mazibuko, S.E.; Hartwig, S.; et al. Long-Term Cold Adaptation Does Not Require FGF21 or UCP1. Cell Metab. 2017, 26, 437–446.e5. [Google Scholar] [CrossRef]
- Rahbani, J.F.; Bunk, J.; Lagarde, D.; Samborska, B.; Roesler, A.; Xiao, H.; Shaw, A.; Kaiser, Z.; Braun, J.L.; Geromella, M.S.; et al. Parallel control of cold-triggered adipocyte thermogenesis by UCP1 and CKB. Cell Metab. 2024, 36, 526–540.e7. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Jiang, T.; Song, Y.; Li, D.; Zhan, S.; Zhong, T.; Guo, J.; Li, L.; Zhang, H.; Wang, L. Identification of a distal enhancer of Ucp1 essential for thermogenesis and mitochondrial function in brown fat. Commun. Biol. 2025, 8, 31. [Google Scholar] [CrossRef]
- Nagai, N.; Sakane, N.; Fujishita, A.; Fujiwara, R.; Kimura, T.; Kotani, K.; Moritani, T. The −3826 A → G variant of the uncoupling protein-1 gene diminishes thermogenesis during acute cold exposure in healthy children. Obes. Res. Clin. Pract. 2007, 1, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Dominiczak, A.F. Genomics of hypertension: The road to precision medicine. Nat. Rev. Cardiol. 2021, 18, 235–250. [Google Scholar] [CrossRef]
- Pandey, K.N. Genetic and Epigenetic Mechanisms Regulating Blood Pressure and Kidney Dysfunction. Hypertension 2024, 81, 1424–1437. [Google Scholar] [CrossRef]
- Wilmot, E.; Idris, I. Early onset type 2 diabetes: Risk factors, clinical impact and management. Ther. Adv. Chronic Dis. 2014, 5, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Forman, J.P.; Jensen, M.K.; Flint, A.; Curhan, G.C.; Rimm, E.B.; Hu, F.B.; Qi, L. Genetic predisposition to high blood pressure associates with cardiovascular complications among patients with type 2 diabetes: Two independent studies. Diabetes 2012, 61, 3026–3032. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Shoily, S.S.; Ahsan, T.; Fatema, K.; Sajib, A.A. Common genetic variants and pathways in diabetes and associated complications and vulnerability of populations with different ethnic origins. Sci. Rep. 2021, 11, 7504. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Shu, Y.; Cai, Y.D. Genetic differences among ethnic groups. BMC Genom. 2015, 16, 1093. [Google Scholar] [CrossRef]
- Harper, E.; Forde, H.; Davenport, C.; Rochfort, K.D.; Smith, D.; Cummins, P.M. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vasc. Pharmacol. 2016, 82, 30–40. [Google Scholar] [CrossRef]
- Kalra, S.S.; Shanahan, C.M. Vascular calcification and hypertension: Cause and effect. Ann. Med. 2012, 44 (Suppl. S1), S85–S92. [Google Scholar] [CrossRef]
- Duan, P.; Tu, P.; Si, L.; Hu, W.; Liu, M.; Liu, J.; Xue, Y. Gene Polymorphisms in the RANKL/RANK/OPG Pathway Are Associated with Type 2 Diabetes Mellitus in Southern Han Chinese Women. Genet. Test. Mol. Biomark. 2016, 20, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, K.; Kolodgie, F.D.; Lutter, C.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 191–204. [Google Scholar] [CrossRef]
- Kamimura, D.; Suzuki, T.; Furniss, A.L.; Griswold, M.E.; Kullo, I.J.; Lindsey, M.L.; Winniford, M.D.; Butler, K.R.; Mosley, T.H.; Hall, M.E. Elevated serum osteoprotegerin is associated with increased left ventricular mass index and myocardial stiffness. J. Cardiovasc. Med. 2017, 18, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Blazquez-Medela, A.M.; Garcia-Ortiz, L.; Gomez-Marcos, M.A.; Recio-Rodriguez, J.I.; Sanchez-Rodriguez, A.; Lopez-Novoa, J.M.; Martinez-Salgado, C. Osteoprotegerin is associated with cardiovascular risk in hypertension and/or diabetes. Eur. J. Clin. Investig. 2012, 42, 548–556. [Google Scholar] [CrossRef]
- Jasmine, M.R.; Nanda, N.; Sahoo, J.; Velkumary, S.; Pal, G.K. Increased osteoprotegerin level is associated with impaired cardiovagal modulation in type-2 diabetic patients treated with oral antidiabetic drugs. BMC Cardiovasc. Disord. 2020, 20, 453. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes—2025. Diabetes Care 2024, 48 (Suppl. S1), S181–S206. [Google Scholar] [CrossRef]
- Lemkes, B.A.; Hermanides, J.; Devries, J.H.; Holleman, F.; Meijers, J.C.; Hoekstra, J.B. Hyperglycemia: A prothrombotic factor? J. Thromb. Haemost. 2010, 8, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Bryk, A.H.; Prior, S.M.; Plens, K.; Konieczynska, M.; Hohendorff, J.; Malecki, M.T.; Butenas, S.; Undas, A. Predictors of neutrophil extracellular traps markers in type 2 diabetes mellitus: Associations with a prothrombotic state and hypofibrinolysis. Cardiovasc. Diabetol. 2019, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- Salavati, M.; Arabshomali, A.; Nouranian, S.; Shariat-Madar, Z. Overview of Venous Thromboembolism and Emerging Therapeutic Technologies Based on Nanocarriers-Mediated Drug Delivery Systems. Molecules 2024, 29, 4883. [Google Scholar] [CrossRef]
- Picard, F.; Adjedj, J.; Varenne, O. Diabetes Mellitus, a prothrombotic disease. Ann. Cardiol. D’angeiologie 2017, 66, 385–392. [Google Scholar] [CrossRef]
- Baye, A.M.; Fanta, T.G.; Siddiqui, M.K.; Dawed, A.Y. The Genetics of Adverse Drug Outcomes in Type 2 Diabetes: A Systematic Review. Front. Genet. 2021, 12, 675053. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Hirschhorn, J.N.; Klannemark, M.; Lindgren, C.M.; Vohl, M.C.; Nemesh, J.; Lane, C.R.; Schaffner, S.F.; Bolk, S.; Brewer, C.; et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000, 26, 76–80. [Google Scholar] [CrossRef]
- Vella, A. Pharmacogenetics for type 2 diabetes: Practical considerations for study design. J. Diabetes Sci. Technol. 2009, 3, 705–709. [Google Scholar] [CrossRef]
- Vella, A.; Camilleri, M. Pharmacogenetics: Potential role in the treatment of diabetes and obesity. Expert Opin. Pharmacother. 2008, 9, 1109–1119. [Google Scholar] [CrossRef]
- Zhou, K.; Yee, S.W.; Seiser, E.L.; van Leeuwen, N.; Tavendale, R.; Bennett, A.J.; Groves, C.J.; Coleman, R.L.; van der Heijden, A.A.; Beulens, J.W.; et al. Variation in the glucose transporter gene SLC2A2 is associated with glycemic response to metformin. Nat. Genet. 2016, 48, 1055–1059. [Google Scholar] [CrossRef]
- Williams, L.K.; Padhukasahasram, B.; Ahmedani, B.K.; Peterson, E.L.; Wells, K.E.; González Burchard, E.; Lanfear, D.E. Differing effects of metformin on glycemic control by race-ethnicity. J. Clin. Endocrinol. Metab. 2014, 99, 3160–3168. [Google Scholar] [CrossRef]
- Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 1 March 2024).
- Fruh, S.M. Obesity: Risk factors, complications, and strategies for sustainable long-term weight management. J. Am. Assoc. Nurse Pract. 2017, 29, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Moini, J.; Ahangari, R.; Miller, C.; Samsam, M. Chapter 18—Perspective on economics and obesity. In Global Health Complications of Obesity; Moini, J., Ahangari, R., Miller, C., Samsam, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 411–423. [Google Scholar] [CrossRef]
- Evans, M.; de Courcy, J.; de Laguiche, E.; Faurby, M.; Haase, C.L.; Matthiessen, K.S.; Moore, A.; Pearson-Stuttard, J. Obesity-related complications, healthcare resource use and weight loss strategies in six European countries: The RESOURCE survey. Int. J. Obes. 2023, 47, 750–757. [Google Scholar] [CrossRef]
- Haslam, D.W.; James, W.P. Obesity. Lancet 2005, 366, 1197–1209. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Jones, M.R.; Ankener, W.; Stewart, D.R.; Urbanek, M.; Dunaif, A.; Legro, R.S.; Chua, A.; Azziz, R.; Spielman, R.S.; et al. FTO and MC4R gene variants are associated with obesity in polycystic ovary syndrome. PLoS ONE 2011, 6, e16390. [Google Scholar] [CrossRef]
- Wojciechowski, P.; Lipowska, A.; Rys, P.; Ewens, K.G.; Franks, S.; Tan, S.; Lerchbaum, E.; Vcelak, J.; Attaoua, R.; Straczkowski, M.; et al. Impact of FTO genotypes on BMI and weight in polycystic ovary syndrome: A systematic review and meta-analysis. Diabetologia 2012, 55, 2636–2645. [Google Scholar] [CrossRef]
- Cauchi, S.; Stutzmann, F.; Cavalcanti-Proenca, C.; Durand, E.; Pouta, A.; Hartikainen, A.L.; Marre, M.; Vol, S.; Tammelin, T.; Laitinen, J.; et al. Combined effects of MC4R and FTO common genetic variants on obesity in European general populations. J. Mol. Med. 2009, 87, 537–546. [Google Scholar] [CrossRef]
- Castillo, J.J.; Orlando, R.A.; Garver, W.S. Gene-nutrient interactions and susceptibility to human obesity. Genes Nutr. 2017, 12, 29. [Google Scholar] [CrossRef]
- Trang, K.; Grant, S.F.A. Genetics and epigenetics in the obesity phenotyping scenario. Rev. Endocr. Metab. Disord. 2023, 24, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Lalle, G.; Lautraite, R.; Bouherrou, K.; Plaschka, M.; Pignata, A.; Voisin, A.; Twardowski, J.; Perrin-Niquet, M.; Stéphan, P.; Durget, S.; et al. NF-κB subunits RelA and c-Rel selectively control CD4+ T cell function in multiple sclerosis and cancer. J. Exp. Med. 2024, 221, e20231348. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, W.; Wang, X. Studies on the fat mass and obesity-associated (FTO) gene and its impact on obesity-associated diseases. Genes Dis. 2023, 10, 2351–2365. [Google Scholar] [CrossRef]
- Saqlain, M.; Khalid, M.; Fiaz, M.; Saeed, S.; Mehmood Raja, A.; Mobeen Zafar, M.; Fatima, T.; Bosco Pesquero, J.; Maglio, C.; Valadi, H.; et al. Risk variants of obesity associated genes demonstrate BMI raising effect in a large cohort. PLoS ONE 2022, 17, e0274904. [Google Scholar] [CrossRef] [PubMed]
- Müssig, K. Novel Treatment Options in Patients with Maturity-Onset Diabetes of the Young. Exp. Clin. Endocrinol. Diabetes 2025, 133, 51–58. [Google Scholar] [CrossRef]
- Gonçalves, J.; Ferreira, H.U.; Ribeiro, S.; Fernandes da Rocha, D.; Souto, S.B.; Pedro, J.; Freitas, P.; Queirós, J. Challenges in diagnosis and treatment of KCNJ11-MODY. Endocrinol. Diabetes Metab. Case Rep. 2024, 2024, e240048. [Google Scholar] [CrossRef]
- Fajans, S.S.; Brown, M.B. Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care 1993, 16, 1254–1261. [Google Scholar] [CrossRef]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef]
- Suzuki, S.; Kokumai, T.; Furuya, A.; Takahashi, S. SGLT2i as a Useful Adjunctive Medication for HNF4A-MODY. Diabetes Care 2023, 46, e74–e75. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, Y.; Ba, T.; Han, X.; Ren, Q.; Ji, L. Hypoglycemic Response to Dorzagliatin in a Patient With GCK-MODY. Diabetes Care 2024, 47, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Almutair, A.; Almulhem, B. Semaglutide as a potential therapeutic alternative for HNF1B-MODY: A case study. Front. Endocrinol. 2024, 15, 1294264. [Google Scholar] [CrossRef]
- Pearson, E.R.; Pruhova, S.; Tack, C.J.; Johansen, A.; Castleden, H.A.; Lumb, P.J.; Wierzbicki, A.S.; Clark, P.M.; Lebl, J.; Pedersen, O.; et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 2005, 48, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.; Dharia, A.; Alrowiyti, I.; Cherney, D.Z.I. Prescribing SGLT2 Inhibitors in Patients with CKD: Expanding Indications and Practical Considerations. Kidney international reports 2022, 7, 1463–1476. [Google Scholar] [CrossRef]
- Lv, R.; Xu, L.; Che, L.; Liu, S.; Wang, Y.; Dong, B. Cardiovascular-renal protective effect and molecular mechanism of finerenone in type 2 diabetic mellitus. Front. Endocrinol. 2023, 14, 1125693. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.L.; Lee, S.E.; Kim, N.H. Renal Protection of Mineralocorticoid Receptor Antagonist, Finerenone, in Diabetic Kidney Disease. Endocrinol. Metab. 2023, 38, 43–55. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pearson, E.R.; Starkey, B.J.; Powell, R.J.; Gribble, F.M.; Clark, P.M.; Hattersley, A.T. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003, 362, 1275–1281. [Google Scholar] [CrossRef]
- Radtke, M.A.; Nermoen, I.; Kollind, M.; Skeie, S.; Sørheim, J.I.; Svartberg, J.; Hals, I.; Moen, T.; Dørflinger, G.H.; Grill, V. Six months of diazoxide treatment at bedtime in newly diagnosed subjects with type 1 diabetes does not influence parameters of {beta}-cell function and autoimmunity but improves glycemic control. Diabetes Care 2010, 33, 589–594. [Google Scholar] [CrossRef]
- Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. [Google Scholar] [CrossRef]
- Chung, W.K.; Erion, K.; Florez, J.C.; Hattersley, A.T.; Hivert, M.F.; Lee, C.G.; McCarthy, M.I.; Nolan, J.J.; Norris, J.M.; Pearson, E.R.; et al. Precision Medicine in Diabetes: A Consensus Report From the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020, 43, 1617–1635. [Google Scholar] [CrossRef] [PubMed]
- Mapes, B.M.; Foster, C.S.; Kusnoor, S.V.; Epelbaum, M.I.; AuYoung, M.; Jenkins, G.; Lopez-Class, M.; Richardson-Heron, D.; Elmi, A.; Surkan, K.; et al. Diversity and inclusion for the All of Us research program: A scoping review. PLoS ONE 2020, 15, e0234962. [Google Scholar] [CrossRef]
- Hill, J.O.; Galloway, J.M.; Goley, A.; Marrero, D.G.; Minners, R.; Montgomery, B.; Peterson, G.E.; Ratner, R.E.; Sanchez, E.; Aroda, V.R. Scientific statement: Socioecological determinants of prediabetes and type 2 diabetes. Diabetes Care 2013, 36, 2430–2439. [Google Scholar] [CrossRef]
- Bepo, L.; Nguyen, O.K.; Makam, A.N. Disparities in Use of Novel Diabetes Medications by Insurance: A Nationally Representative Cohort Study. J. Gen. Intern. Med. 2024, 39, 2987–2994. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.A.; Finertie, H.; Neugebauer, R.S.; Gosiker, B.; Thomas, T.W.; Karter, A.J.; Gilliam, L.K.; Oshiro, C.; An, J.; Simonson, G.; et al. Race and ethnicity and pharmacy dispensing of SGLT2 inhibitors and GLP-1 receptor agonists in type 2 diabetes. Lancet Reg. Health-Am. 2024, 34, 100759. [Google Scholar] [CrossRef] [PubMed]
- Hill-Briggs, F.; Adler, N.E.; Berkowitz, S.A.; Chin, M.H.; Gary-Webb, T.L.; Navas-Acien, A.; Thornton, P.L.; Haire-Joshu, D. Social Determinants of Health and Diabetes: A Scientific Review. Diabetes Care 2020, 44, 258–279. [Google Scholar] [CrossRef]
- Mohsen, F.; Al-Absi, H.R.H.; Yousri, N.A.; El Hajj, N.; Shah, Z. A scoping review of artificial intelligence-based methods for diabetes risk prediction. NPJ Digit. Med. 2023, 6, 197. [Google Scholar] [CrossRef]
- Khan, S.; Mohsen, F.; Shah, Z. Genetic biomarkers and machine learning techniques for predicting diabetes: Systematic review. Artif. Intell. Rev. 2024, 58, 41. [Google Scholar] [CrossRef]
- Li, L.; Cheng, W.Y.; Glicksberg, B.S.; Gottesman, O.; Tamler, R.; Chen, R.; Bottinger, E.P.; Dudley, J.T. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Transl. Med. 2015, 7, 311ra174. [Google Scholar] [CrossRef]
- Bonnell, J.; Alcazar, O.; Watts, B.; Buchwald, P.; Abdulreda, M.H.; Ogihara, M. Supervised Parametric Learning in the Identification of Composite Biomarker Signatures of Type 1 Diabetes in Integrated Parallel Multi-Omics Datasets. Biomedicines 2024, 12, 492. [Google Scholar] [CrossRef]
- Lyons, T.J.; Basu, A. Biomarkers in diabetes: Hemoglobin A1c, vascular and tissue markers. Transl. Res. 2012, 159, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Kaur, V.; Chamarthi, B.; Littleton, K.R.; Chen, L.; Manning, A.K.; Merino, J.; Thomas, M.K.; Hudson, M.; Goldfine, A.; et al. TCF7L2 Genetic Variation Augments Incretin Resistance and Influences Response to a Sulfonylurea and Metformin: The Study to Understand the Genetics of the Acute Response to Metformin and Glipizide in Humans (SUGAR-MGH). Diabetes Care 2018, 41, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Weeda, E.R.; Muraoka, A.K.; Brock, M.D.; Cannon, J.M. Medication adherence to injectable glucagon-like peptide-1 (GLP-1) receptor agonists dosed once weekly vs once daily in patients with type 2 diabetes: A meta-analysis. Int. J. Clin. Pract. 2021, 75, e14060. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.; Blalock, D.V.; Miller, J.; McDermott, J.; Wessler, H.; Oakes, M.M.; Reed, S.D.; Bosworth, H.B.; Zullig, L.L. Impact of financial medication assistance on medication adherence: A systematic review. J. Manag. Care Spec. Pharm. 2021, 27, 924–935. [Google Scholar] [CrossRef]
- Elwyn, G.; Dehlendorf, C.; Epstein, R.M.; Marrin, K.; White, J.; Frosch, D.L. Shared decision making and motivational interviewing: Achieving patient-centered care across the spectrum of health care problems. Ann. Fam. Med. 2014, 12, 270–275. [Google Scholar] [CrossRef]
- Aleppo, G.; Gal, R.L.; Raghinaru, D.; Kruger, D.; Beck, R.W.; Bergenstal, R.M.; Cushman, T.; Hood, K.K.; Johnson, M.L.; McArthur, T.; et al. Comprehensive Telehealth Model to Support Diabetes Self-Management. JAMA Netw. Open 2023, 6, e2336876. [Google Scholar] [CrossRef]
- Sharma, V.; Feldman, M.; Sharma, R. Telehealth Technologies in Diabetes Self-management and Education. J. Diabetes Sci. Technol. 2024, 18, 148–158. [Google Scholar] [CrossRef]
- Aremu, T.O.; Oluwole, O.E.; Adeyinka, K.O.; Schommer, J.C. Medication Adherence and Compliance: Recipe for Improving Patient Outcomes. Pharmacy 2022, 10, 106. [Google Scholar] [CrossRef]
- Blonde, L.; Aschner, P.; Bailey, C.; Ji, L.; Leiter, L.A.; Matthaei, S. Gaps and barriers in the control of blood glucose in people with type 2 diabetes. Diabetes Vasc. Dis. Res. 2017, 14, 172–183. [Google Scholar] [CrossRef]
- Gold, R.S.; Unkart, J.T.; McClelland, R.L.; Bertoni, A.G.; Allison, M.A. Health insurance status and type associated with varying levels of glycemic control in the US: The multi-ethnic study of atherosclerosis (MESA). Prim. Care Diabetes 2021, 15, 378–384. [Google Scholar] [CrossRef]
- Manov, A.E.; Chauhan, S.; Dhillon, G.; Dhaliwal, A.; Antonio, S.; Donepudi, A.; Jalal, Y.N.; Nazha, J.; Banal, M.; House, J. The Effectiveness of Continuous Glucose Monitoring Devices in Managing Uncontrolled Diabetes Mellitus: A Retrospective Study. Cureus 2023, 15, e42545. [Google Scholar] [CrossRef] [PubMed]
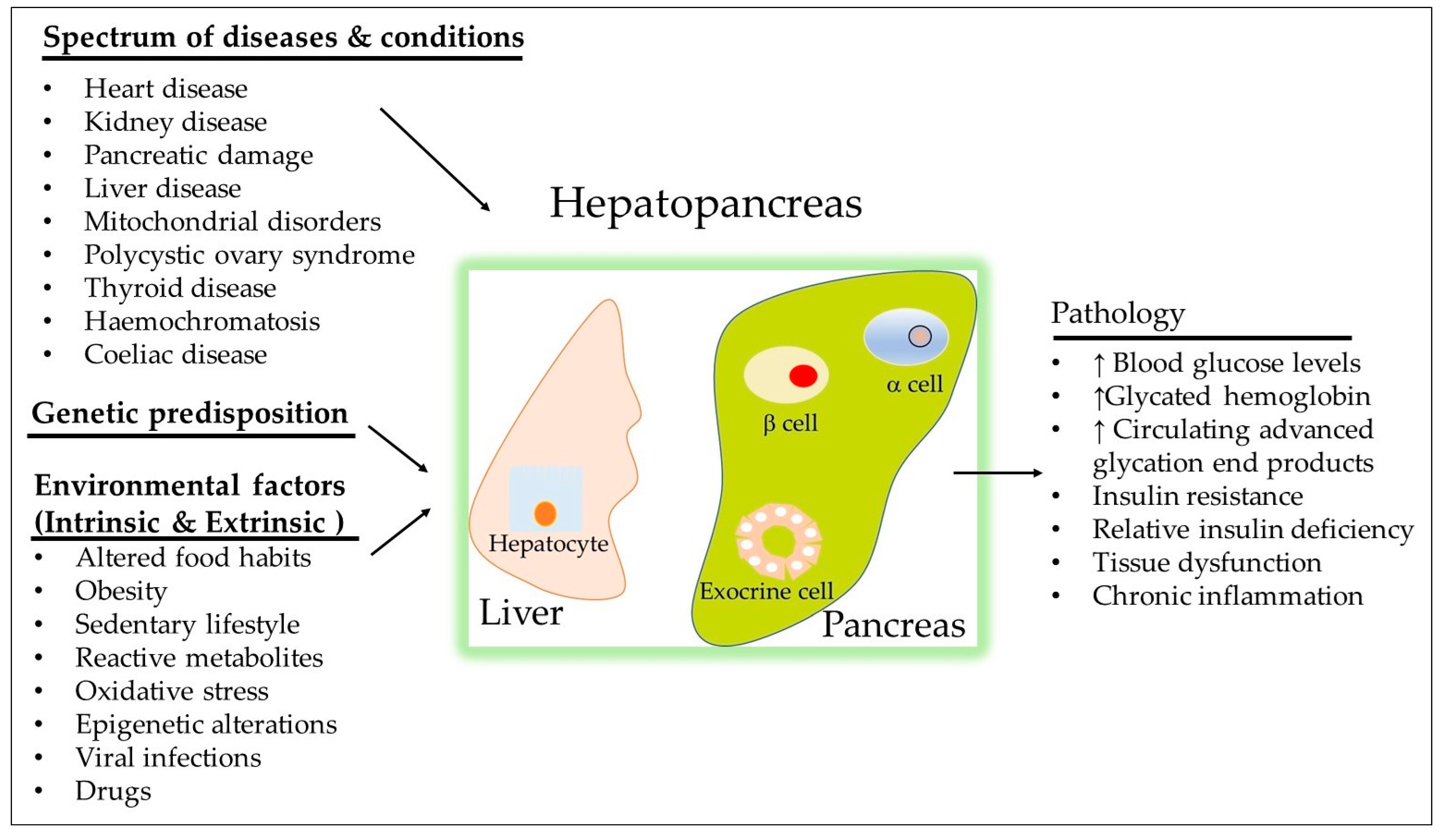
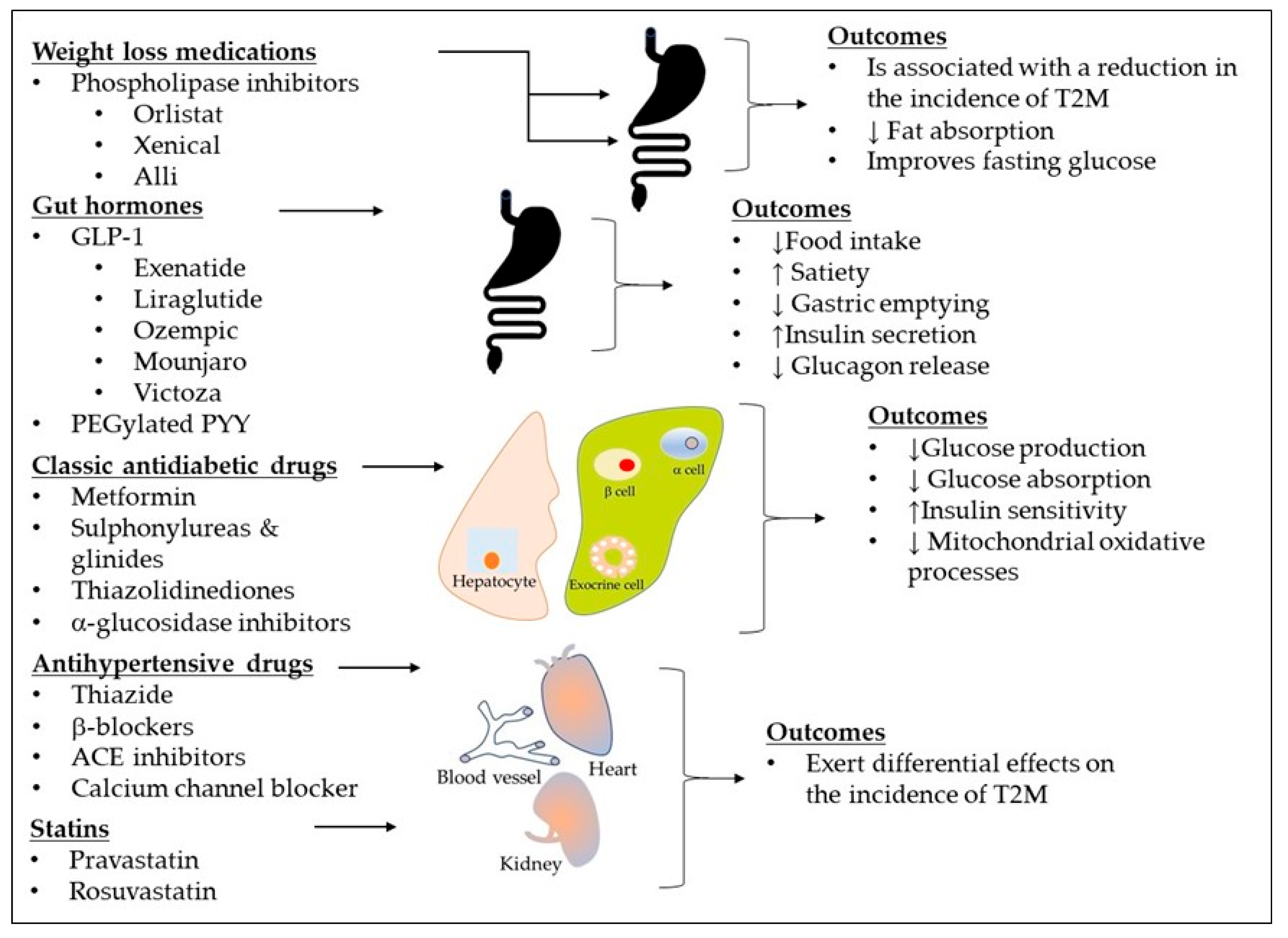
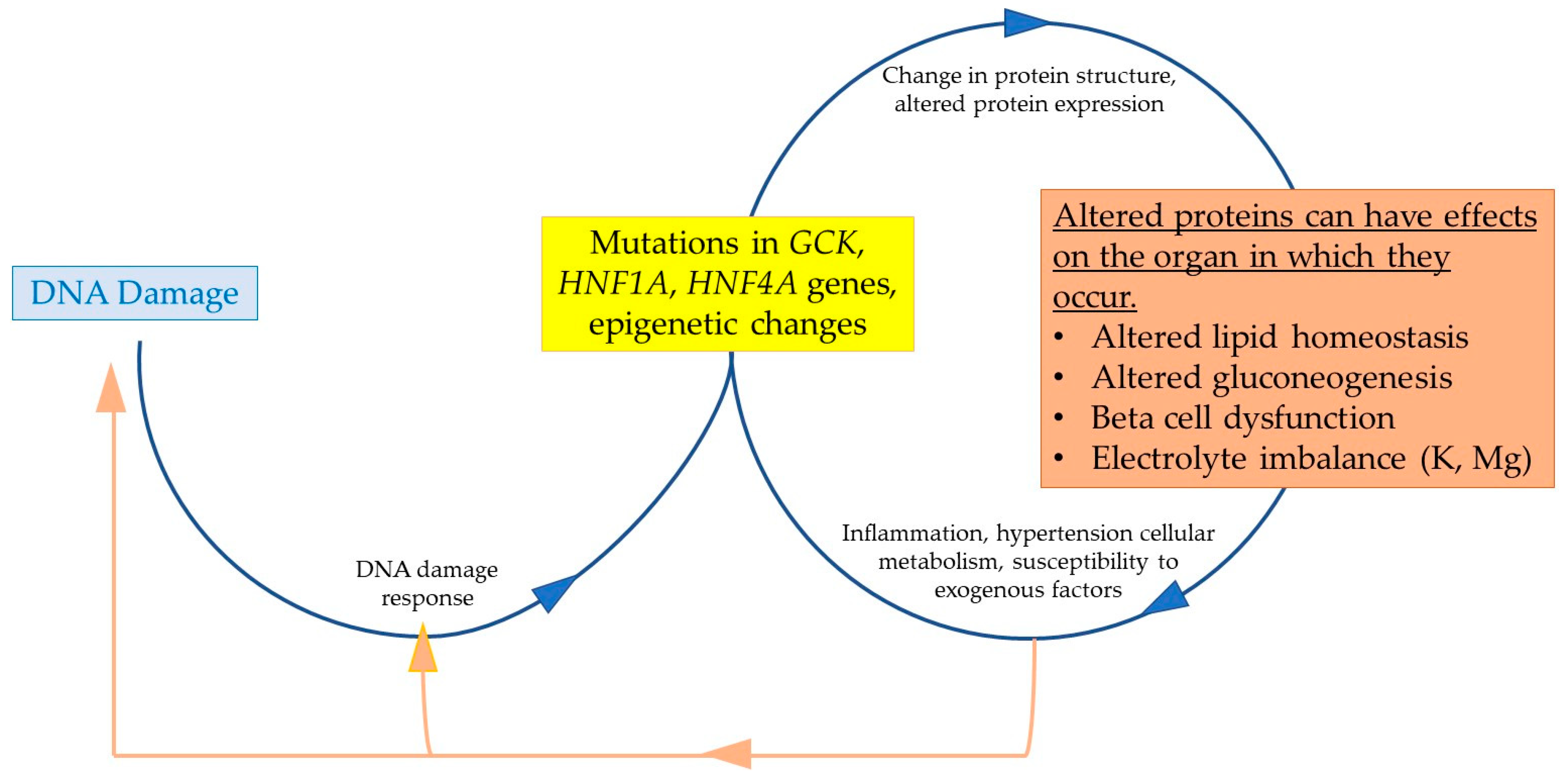
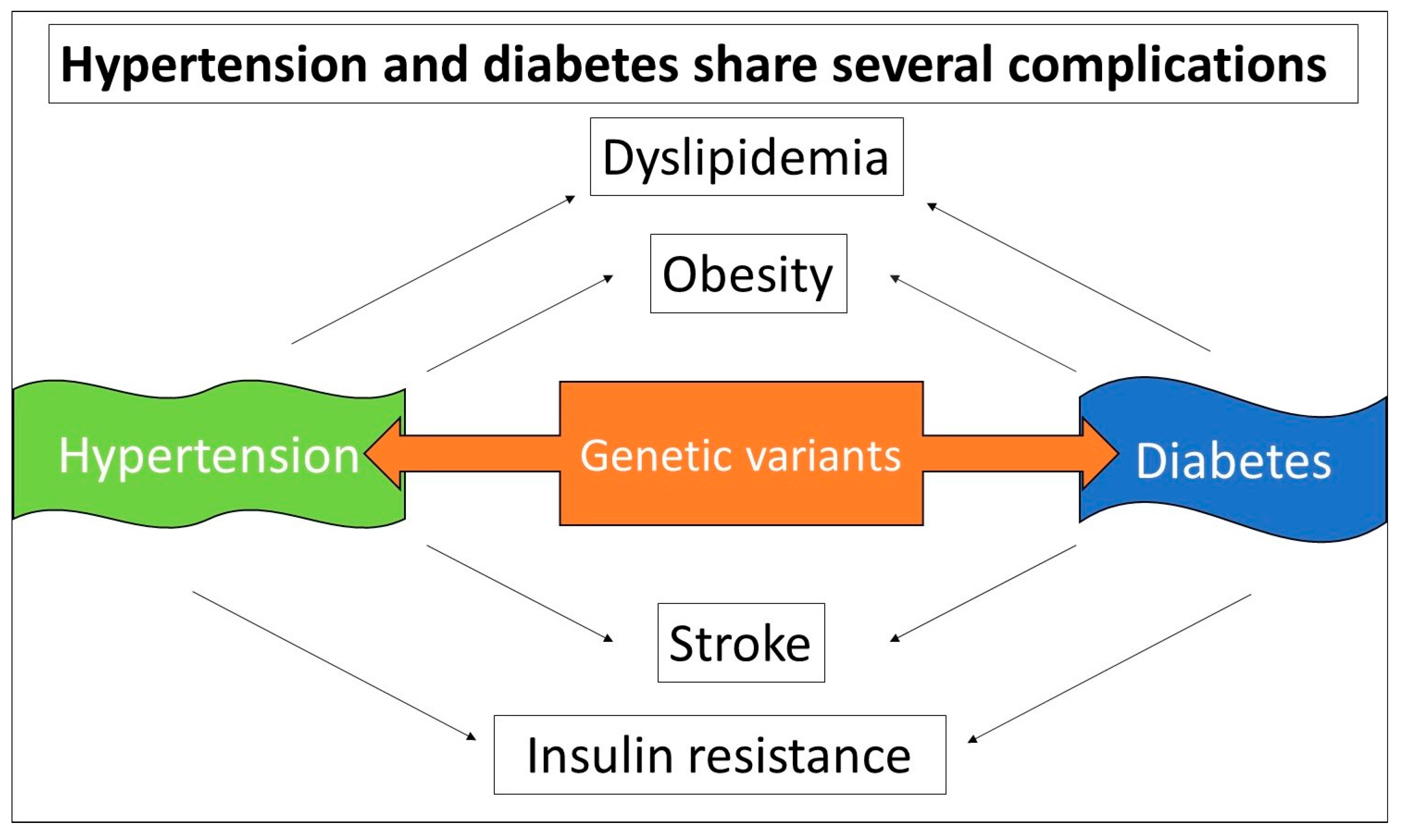

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Role of Gene Product | Rare Variants | Phenotypic Abnormalities | References |
|---|---|---|---|---|
| The most potential pathogenic genes in MODY | ||||
| Gck | Glucokinase catalyzes the rate-limiting step of the glycolysis pathway, functions as the glucose sensor in pancreatic beta cells | c.1015del, p.(glu339Argfs*14) p.G72R, C220Y, p.L58P, p.F123S | Mild hyperglycemia, overweight Glycemic | [32,33,34] |
| Hnf1a | Hepatic nuclear factor 1-alpha is critical for the growth and development of beta cells in the pancreas, controls genes involved in liver development, regulates cell growth and survival, and acts as a tumor suppressor. | R200C T196A P291fsinsC H126D | Decreased insulin secretion Increased hepatogenic secretion of atherogenic lipoproteins Liver steatosis Decreased glucose transporter GLUT2 expression, reduced ATP production | [35,36,37,38] |
| Hnf4a | Hepatic nuclear factor 4-alpha controls genes that are important for the development and function of beta cells in the pancreas. | rs4735692 V393I c.110T>c, c.1097C>G p.R114W | Associated with obesity Reduced transcriptional activity and insulin secretion Increases risk of diabetes | [39,40,41,42] |
| Kcnj11 | Encodes pore-forming inwardly-rectifying potassium channel subunits (Kir6.2) | rs5215, rs5218, and rs5219 | MODY, T2DM, elevated FBS, high BMI | [43,44,45,46] |
| The most significant genes that might play an essential role in late-onset T2DM | ||||
| tcf7l2 | A key regulator of insulin secretion in pancreatic beta-cells | rs7903146 | T2DM, less obesity, lower insulin secretion and higher insulin action at diabetes onset | [47,48,49] |
| glis3 | A transcription factor: Regulator of islet development, insulin gene transcription, and obesity-induced compensatory β-cell proliferation | P/LP GLIS3, rs10758593 rs 7034200 | Neonatal diabetes mellitus, T2DM congenital hypothyroidism and polycystic kidney | [50,51,52] |
| pparγ | A transcription factor: master regulator of adipogenesis, energy balance, lipid biosynthesis, and insulin sensitivity; cellular target of TZDs | rs4684847 rs1801282 | T2DM, impaired insulin sensitivity, partial lipodystrophy | [53,54,55] |
| lep | A regulator of appetite and energy balance | rs147287548 G2548A | T2DM, severe obesity, hyperglycemia | [42,56] |
| ucp1 | A regulator of energy balance and mitochondrial-induced oxidative stress | rs45539933 | T2DM, increased body fat accumulation, risk of insulin resistance | [57,58,59] |
| opg | A decoy receptor for receptor-activator for NF-κB ligand (RANKL) | rs2073618 | T2DM, inflammation, hypertension | [60] |
| Conditions | Medicines | MAO | Outcomes | Side Effects | Comments |
|---|---|---|---|---|---|
| Adjunctive medications for MODY | |||||
| GCK-MODY | Dorzagliatin | A glucokinase activator | Improves glycemic control | Potential gastrointestinal issues, hypoglycemia, and headaches | |
| Heterozygous loss of function in Hnf1a | Low-dose sulfonylurea | Stimulate insulin secretion from the pancreas | Improves glycemic control | Hypoglycemia | |
| Homomozygous Hnf1a mutations | There are currently no approved treatments. | ||||
| HNF1B-MODY | SGLT2 inhibitors Semaglutide Insulin | Increase glycosuria A GLP-1 receptor agonist A hormone | Lowers blood glucose levels Helps manage blood sugar levels Regulates blood sugar levels | Increased urination frequency and urinary tract infection Diarrhea hypoglycemia | |
| HNF4A-MODY | Empagliflozin | SGLT2 inhibitor | Improves glycemic control | ||
| The FDA approved new treatments for adults with pre-diabetes and obesity or overweight | |||||
| T2DM | Brenzavvy (bexagliflozin) | A dual agonist for the glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) receptors | Improves hyperglycemia and promotes weight loss | It may cause ketoacidosis, a serious, potentially life-threatening complication that occurs when the body produces high levels of acids in the blood | It works similarly in Asian, Black or African American, and White adults |
| T2DM | Mounjaro (tirzepatide) | Selectively binds to and activates both the GIP and GLP-1 receptors to target GIP and GLP-1, the native incretin hormones | Improves blood sugar control, promotes weight loss | It may cause serious side effects including inflammation of the pancreas (pancreatitis), low blood sugar, allergic reactions, kidney problems (kidney failure), severe stomach problems, and complications of diabetes-related eye disease (diabetic retinopathy) | It works similarly in Asian, Black or African American, and White adults |
| CKD associated T2DM | Kerendia (finerenone) | Blocks the mineralocorticoid receptor | It reduced the risk of kidney failure associated with T2DM, having a heart attack or stroke, being hospitalized for heart failure, and dying from cardiovascular disease | The most common side effects included high potassium levels in the blood (hyperkalemia), low blood pressure (hypotension), and low sodium levels in the blood (hyponatremia). | No notable difference in side effects was observed by racial subgroups. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzazzadehgan, S.; Shariat-Madar, Z.; Mahdi, F. Distinct Roles of Common Genetic Variants and Their Contributions to Diabetes: MODY and Uncontrolled T2DM. Biomolecules 2025, 15, 414. https://doi.org/10.3390/biom15030414
Bazzazzadehgan S, Shariat-Madar Z, Mahdi F. Distinct Roles of Common Genetic Variants and Their Contributions to Diabetes: MODY and Uncontrolled T2DM. Biomolecules. 2025; 15(3):414. https://doi.org/10.3390/biom15030414
Chicago/Turabian StyleBazzazzadehgan, Shadi, Zia Shariat-Madar, and Fakhri Mahdi. 2025. "Distinct Roles of Common Genetic Variants and Their Contributions to Diabetes: MODY and Uncontrolled T2DM" Biomolecules 15, no. 3: 414. https://doi.org/10.3390/biom15030414
APA StyleBazzazzadehgan, S., Shariat-Madar, Z., & Mahdi, F. (2025). Distinct Roles of Common Genetic Variants and Their Contributions to Diabetes: MODY and Uncontrolled T2DM. Biomolecules, 15(3), 414. https://doi.org/10.3390/biom15030414