
Efficient Incorporation of DOPA into Proteins Free from Competition with Endogenous Translation Termination Machinery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Genome Editing
2.3. Extract Preparation
2.4. Western Blot
2.5. Expression and Purification of the Proteins
2.6. In Vitro ncAA Incorporation
2.7. sfGFP Quantitative Expression
2.8. Mass Spectrometry Analysis
2.9. Isothermal Titration Calorimetry (ITC)
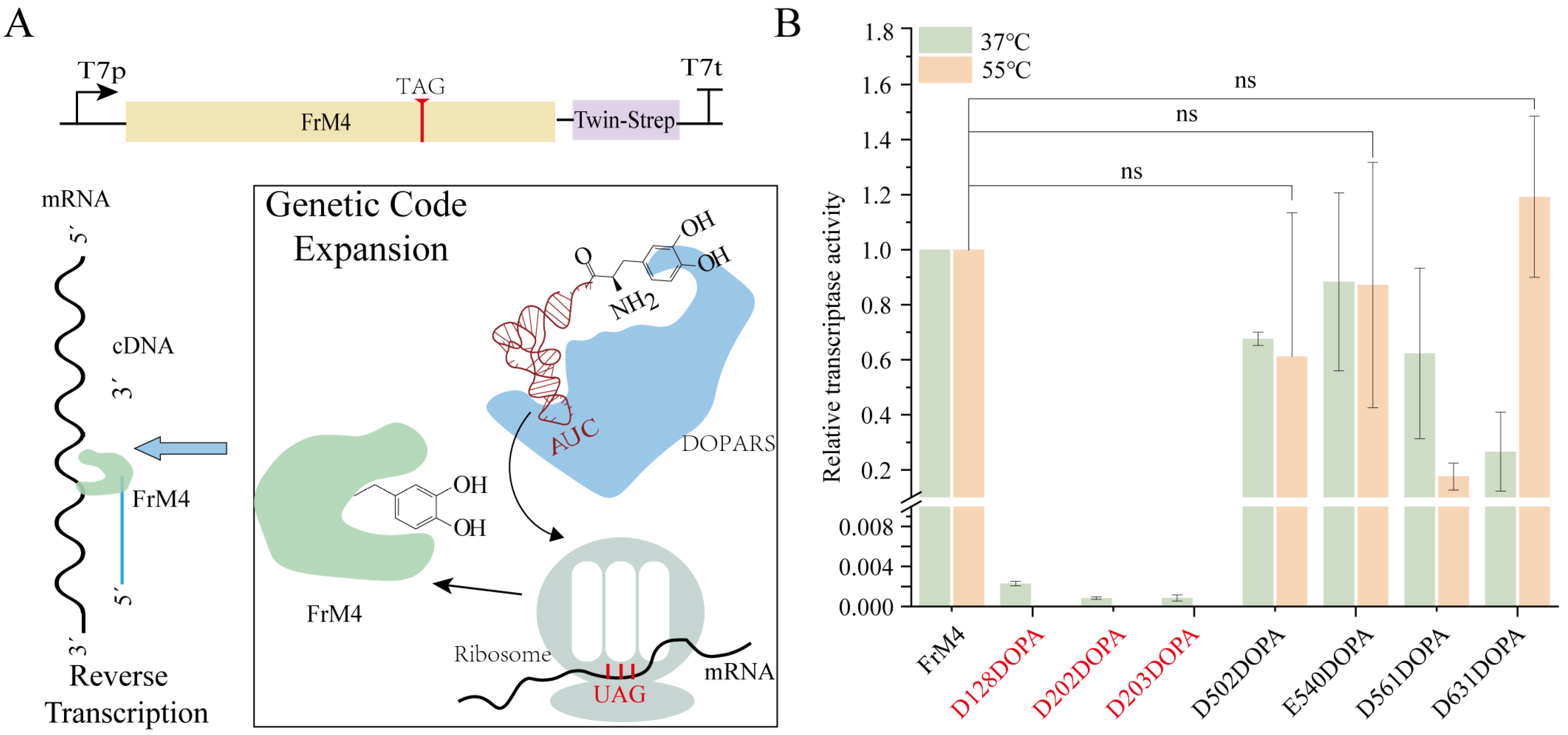
2.10. DOPA-Containing Protein Reverse Transcription Assay
3. Results
3.1. Establishment of CFUPS Without Class I Release Factor
3.2. Optimized Incorporation of DOPA in CFUPS
3.3. DOPA-Containing Protein Expression
3.4. Measurement of the Affinity of DOPARS for DOPA
3.5. Incorporation of L-DOPA into FrM4 RT in CFUPS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CFUPS | Cell-free unnatural protein synthesis |
| ncAAs | Noncanonical amino acids |
| GCE | Genetic code extension |
| ITC | Isothermal titration calorimetry |
| DOPARS | Methanocaldococcus jannaschii Tyrosyl-tRNA Synthetase |
| DOPA | 3,4-Dihydroxy-L-phenylalanine |
References
- Shen, Y.; Su, R.; Hao, D.; Xu, X.; Reches, M.; Min, J.; Chang, H.; Yu, T.; Li, Q.; Zhang, X.; et al. Enzymatic polymerization of enantiomeric L−3, 4-dihydroxyphenylalanine into films with enhanced rigidity and stability. Nat. Commun. 2023, 14, 3054. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Zheng, W.; Zhou, X.; Zhang, C.; Zhang, L. A mussel-inspired chimeric protein as a novel facile antifouling coating. Chem. Commun. 2018, 54, 11328–11331. [Google Scholar] [CrossRef] [PubMed]
- Pinnataip, R.; Lee, B.P. Oxidation chemistry of catechol utilized in designing stimuli-responsive adhesives and antipathogenic biomaterials. ACS Omega 2021, 6, 5113–5118. [Google Scholar] [CrossRef]
- Ayyadurai, N.; Prabhu, N.S.; Deepankumar, K.; Lee, S.G.; Jeong, H.H.; Lee, C.S.; Yun, H. Development of a Selective, Sensitive, and Reversible Biosensor by the Genetic Incorporation of a Metal-Binding Site into Green Fluorescent Protein. Angew. Chem. 2011, 29, 6664–6667. [Google Scholar] [CrossRef]
- Wu, D.; Deepankumar, K.; Ping, Y.; Tang, G.; Saravananprabhu, N.; Miserez, A.; Fang, W. Catechol-modified green fluorescent protein as a specific biosensor for Al ions. Sens. Actuators B Chem. 2017, 251, 326–333. [Google Scholar] [CrossRef]
- Bhagat, A.K.; Buium, H.; Shmul, G.; Alfonta, L. Genetically expanded reactive-oxygen-tolerant alcohol dehydrogenase II. ACS Catal. 2020, 10, 3094–3102. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, Y.; Chung, A.; Yang, S.; Choi, C.H.; Zhang, S.; Han, Y.; Xiao, H. Harnessing Nature-Inspired Catechol Amino Acid to Engineer Sticky Proteins and Bacteria. Small Methods 2024, 8, 2400230. [Google Scholar] [CrossRef]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Möglich, A.; et al. Photoactivatable mussel-based underwater adhesive proteins by an expanded genetic code. ChemBioChem 2017, 18, 1819–1823. [Google Scholar] [CrossRef]
- Koch, N.G.; Baumann, T.; Nickling, J.H.; Dziegielewski, A.; Budisa, N. Engineered bacterial host for genetic encoding of physiologically stable protein nitration. Front. Mol. Biosci. 2022, 9, 992748. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.-R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef]
- Mukai, T.; Hoshi, H.; Ohtake, K.; Takahashi, M.; Yamaguchi, A.; Hayashi, A.; Yokoyama, S.; Sakamoto, K. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep. 2015, 5, 9699. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.E.; Funke, L.F.; de la Torre, D.; Fredens, J.; Elliott, T.S.; Spinck, M.; Christova, Y.; Cervettini, D.; Böge, F.L.; Liu, K.C.; et al. Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 2021, 372, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Gao, W.; Ma, L.; Lu, Y. Mechanism investigation for efficient cell-free unnatural amino acid embedding. Process Biochem. 2022, 122, 306–314. [Google Scholar] [CrossRef]
- Cameron, D.E.; Collins, J.J. Tunable protein degradation in bacteria. Nat. Biotechnol. 2014, 32, 1276–1281. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, M.; Wang, Z.; Yang, Y.; Li, Z.; Liang, S.; Yin, P.; Qi, H. Efficient In Vitro Full-Sense-Codons Protein Synthesis. Adv. Biol. 2022, 6, 2200023. [Google Scholar] [CrossRef]
- Thyer, R.; d’Oelsnitz, S.; Blevins, M.S.; Klein, D.R.; Brodbelt, J.S.; Ellington, A.D. Directed evolution of an improved aminoacyl-tRNA Synthetase for incorporation of L-3,4-Dihydroxyphenylalanine (L-DOPA). Angew. Chem. 2021, 133, 14937–14942. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, T.; Chen, X.; Lu, Y. IRES-mediated Pichia pastoris cell-free protein synthesis. Bioresour. Bioprocess. 2023, 10, 35. [Google Scholar] [CrossRef]
- Matamoros, T.; Barrioluengo, V.; Abia, D.; Menendez-Arias, L. Major groove binding track residues of the connection subdomain of human immunodeficiency virus type 1 reverse transcriptase enhance cDNA synthesis at high temperatures. Biochemistry 2013, 52, 9318–9328. [Google Scholar] [CrossRef]
- Alfonta, L.; Zhang, Z.; Uryu, S.; Loo, J.A.; Schultz, P.G. Site-specific incorporation of a redox-active amino acid into proteins. J. Am. Chem. Soc. 2003, 125, 14662–14663. [Google Scholar] [CrossRef]
- Kim, S.; Sung, B.H.; Kim, S.C.; Lee, H.S. Genetic incorporation of l-dihydroxyphenylalanine (DOPA) biosynthesized by a tyrosine phenol-lyase. Chem. Commun. 2018, 54, 3002–3005. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, H.; Chen, Y.; Zhang, B.; Yang, Y.; Zang, J.; Wu, J.; Lin, S. Chimeric design of pyrrolysyl-tRNA synthetase/tRNA pairs and canonical synthetase/tRNA pairs for genetic code expansion. Nat. Commun. 2020, 11, 3154. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Loredo, A.; Chung, A.; Zhang, M.; Liu, R.; Xiao, H. Biosynthesis and genetic incorporation of 3,4-Dihydroxy-L-phenylalanine into proteins in Escherichia coli. J. Mol. Biol. 2022, 434, 167412. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Sun, S.B.; Furman, J.L.; Xiao, H.; Schultz, P.G. A versatile platform for single-and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 2013, 52, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Duff, M.R.; Grubbs, J.; Howell, E.E. Isothermal titration calorimetry for measuring macromolecule-ligand affinity. J. Vis. Exp. (JoVE) 2011, 55, 2796. [Google Scholar]
- Sheoran, A.; Sharma, G.; First, E.A. Activation of D-tyrosine by Bacillus stearothermophilus tyrosyl-tRNA synthetase: 1. Pre-steady-state kinetic analysis reveals the mechanistic basis for the recognition of D-tyrosine. J. Biol. Chem. 2008, 283, 12960–12970. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, M.; Luo, Z.; Lin, J.; Song, L. Investigation on the site-selective binding of bovine serum albumin by erlotinib hydrochloride. J. Biomol. Struct. Dyn. 2013, 31, 1160–1174. [Google Scholar] [CrossRef]
- Rajan, K.; Mainer, S.; Davis, J. Studies on chelation ofl-DOPA with metal ions and metal-ATP systems. Bioinorg. Chem. 1978, 9, 187–203. [Google Scholar] [CrossRef]
- Fu, Z.; Indrisiunaite, G.; Kaledhonkar, S.; Shah, B.; Sun, M.; Chen, B.; Grassucci, R.A.; Ehrenberg, M.; Frank, J. The structural basis for release factor activation during translation termination revealed by time-resolved cryogenic electron microscopy. Biophys. J. 2019, 116, 574a–575a. [Google Scholar] [CrossRef]
- Huter, P.; Müller, C.; Arenz, S.; Beckert, B.; Wilson, D.N. Structural basis for ribosome rescue in bacteria. Trends Biochem. Sci. 2017, 42, 669–680. [Google Scholar] [CrossRef]
- Keiler, K.C. Mechanisms of ribosome rescue in bacteria. Nat. Rev. Microbiol. 2015, 13, 285–297. [Google Scholar] [CrossRef]
- Loscha, K.V.; Herlt, A.J.; Qi, R.; Huber, T.; Ozawa, K.; Otting, G. Multiple-site labeling of proteins with unnatural amino acids. Angew. Chem. Int. Ed. 2012, 51, 2243–2246. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Wang, Y.; Wang, Z.; Qi, H. Efficient Incorporation of DOPA into Proteins Free from Competition with Endogenous Translation Termination Machinery. Biomolecules 2025, 15, 382. https://doi.org/10.3390/biom15030382
Yang Y, Wang Y, Wang Z, Qi H. Efficient Incorporation of DOPA into Proteins Free from Competition with Endogenous Translation Termination Machinery. Biomolecules. 2025; 15(3):382. https://doi.org/10.3390/biom15030382
Chicago/Turabian StyleYang, Youhui, Yingchen Wang, Zhaoguan Wang, and Hao Qi. 2025. "Efficient Incorporation of DOPA into Proteins Free from Competition with Endogenous Translation Termination Machinery" Biomolecules 15, no. 3: 382. https://doi.org/10.3390/biom15030382
APA StyleYang, Y., Wang, Y., Wang, Z., & Qi, H. (2025). Efficient Incorporation of DOPA into Proteins Free from Competition with Endogenous Translation Termination Machinery. Biomolecules, 15(3), 382. https://doi.org/10.3390/biom15030382