1. Introduction
In recent years, chronic kidney disease (CKD) has increased the incidence of cardiovascular diseases (CVD) significantly. CKD induces cardiac remodeling [
1], including phenomena such as myocardial hypertrophy, fibrosis, and cardiomyocyte apoptosis. The heart is a high-metabolic-rate organ that primarily relies on fatty acid oxidation to supply energy [
2]. CKD and CVD share many common mechanisms of metabolic remodeling [
3], including changes in fatty acid and glucose utilization as well as mitochondrial dysfunction. Studies have shown that cardiovascular events, particularly heart failure, significantly increase the risk of CKD patients progressing to kidney failure requiring replacement therapy (KFRT) [
4,
5]. Other studies have also revealed the important roles of gut microbiota metabolites, such as N,N,N-trimethyl-5-aminovaleric acid TMAVA [
6], in myocardial hypertrophy and heart failure. The important role of uremic toxins in myocardial hypertrophy and fibrosis was also demonstrated [
7,
8].
Cardiac lipid peroxidation refers to myocardial dysfunction caused by the excessive accumulation of fatty acids and their metabolic products in cardiomyocytes. It plays a significant role in heart failure, particularly in heart failure with preserved ejection fraction [
9,
10], HFpEF, and diabetic cardiomyopathy DCM, but its impact on cardiac function in the context of uremia remains unclear. Studies have shown that adipsin improves fatty acid β-oxidation by inhibiting the mitochondrial translocation of Irak2 [
11], the DPP-4 inhibitor [
12] evogliptin alleviates lipotoxicity and improves mitochondrial function, Sirt5 [
13] promotes fatty acid oxidation through desuccinylation, and novel ERR [
14] agonists enhance cardiac metabolism, all of them significantly improving cardiac function by improving fatty acid metabolism in different models. Further research [
15] indicated that cardiac-specific deletion of ACC2 increases fatty acid oxidation, maintaining cardiac health by regulating Parkin-mediated mitophagy. Additionally, some studies have explored the complex role of epicardial adipose tissue (EAT) [
16] and myocardial lipotoxicity in HFpEF and obesity-related heart disease. Regulating lipid metabolism pathways and promoting the efficiency of myocardial lipid metabolism may provide emerging strategies to alleviate cardiac dysfunction in the context of chronic kidney disease.
Forkhead box O4 [
17], i.e., FOXO4, is a member of the FOXO transcription factor family that regulates the expression of various genes involved in several biological processes [
18], including cell cycle, apoptosis, and metabolism. In fatty acid metabolism, FOXO4 plays a role in adipose tissue by influencing the insulin signaling pathway [
19]. The activation of FOXO4 can affect cellular responses to oxidative stress. For example, in the absence of insulin and IGF-1 signaling, sustained activation of FOXO4 may lead to metabolic abnormalities [
19]. Additionally, FOXO4 interacts with other proteins to regulate its activity [
20], modulating the cell cycle and apoptosis. Acetyl-CoA acyltransferase 2 (ACAA2) is an acyl-CoA acyltransferase that participates in the final step of fatty acid β-oxidation. In tumor cells, high expression of ACAA2 is associated with neuroendocrine phenotypes of cancer [
21]. Furthermore, the role of ACAA2 in fatty acid oxidation provides protective effects in organs such as the liver and kidney. For instance [
22], during acetaminophen-induced hepatotoxicity, upregulation of ACAA2 can enhance mitochondrial fatty acid oxidation, thereby reducing liver damage.
S-nitrosylation is an important post-translational modification of proteins, playing a crucial role in regulating cellular signal transduction, protein function, and cellular metabolism [
23]. S-nitrosylation involves the addition of a nitric oxide NO group to cysteine residues within proteins, forming S-nitrosothiols. This modification can significantly influence protein stability, activity, and protein–protein interactions. CSNO, i.e., S-nitroso-L-cysteine, has been shown in previous studies to effectively improve cardiac function in diabetic mice following aerosol inhalation [
24]. Additionally, studies have demonstrated that S-nitrosylation plays a critical role in cardioprotection [
25,
26], particularly in myocardial ischemia–reperfusion injury. Through modulating calcium handling in cardiomyocytes, regulating mitochondrial function, reducing reactive oxygen species ROS production, and mitigating myocardial injury, S-nitrosylation exerts protective effects on the heart. Furthermore, it has been reported that S-nitrosylation can modulate mitochondrial respiration and energy metabolism by modifying subunits of the mitochondrial respiratory chain complex. However, some studies also suggest that nitrosylation modifications in the heart may impair cardiac function, and the specific role of thiol S-nitrosylation remains unclear.
Therefore, as the metabolic mechanism of myocardial lipid peroxidation based on renal insufficiency remains unclear, and no studies have explored the specific role of CSNO in this regard, we propose that the ACAA2 protein exerts a protective role against myocardial lipid peroxidation in renal dysfunction and that CSNO participates in protecting against cardiac dysfunction under renal insufficiency through the FOXO4–ACAA2 axis.
2. Materials and Methods
2.1. Bioinformatics Analysis
To better obtain key data from the gene expression profiles, we downloaded RNA-seq data [
27] (GSE106385) from CKD mice and healthy controls through the GEO database (
https://ncbi.nlm.nih.gov/geo/ (accessed on 8 May 2023)) and performed a series of bioinformatics analyses, including the following: search for differential genes using the online websites (
https://www.networkanalyst.ca/ (accessed on 10 May 2023)); identification of statistically significant differentially expressed genes DEGs based on differences in expression values between samples; construction of the ridgeline graph, enrich net, and volcano graph using the online tool to identify overlapping modules and genes in the dataset; construction of the protein–protein interaction PPI network related to fatty acid β-oxidation by analyzing the differentially expressed genes using the STRING database (
https://cn.string-db.org/ (accessed on 5 June 2023)); visualization of the results with Cytoscape software 3.9.1; prediction of the upstream transcription factors of ACAA2 using the JASPAR database (
https://jaspar.elixir.no/ (accessed on 8 December 2023)); and preliminary construction of a molecular docking model using PyMOL software 2.5.5.
2.2. Animals and Adenine-Induced CKD Model
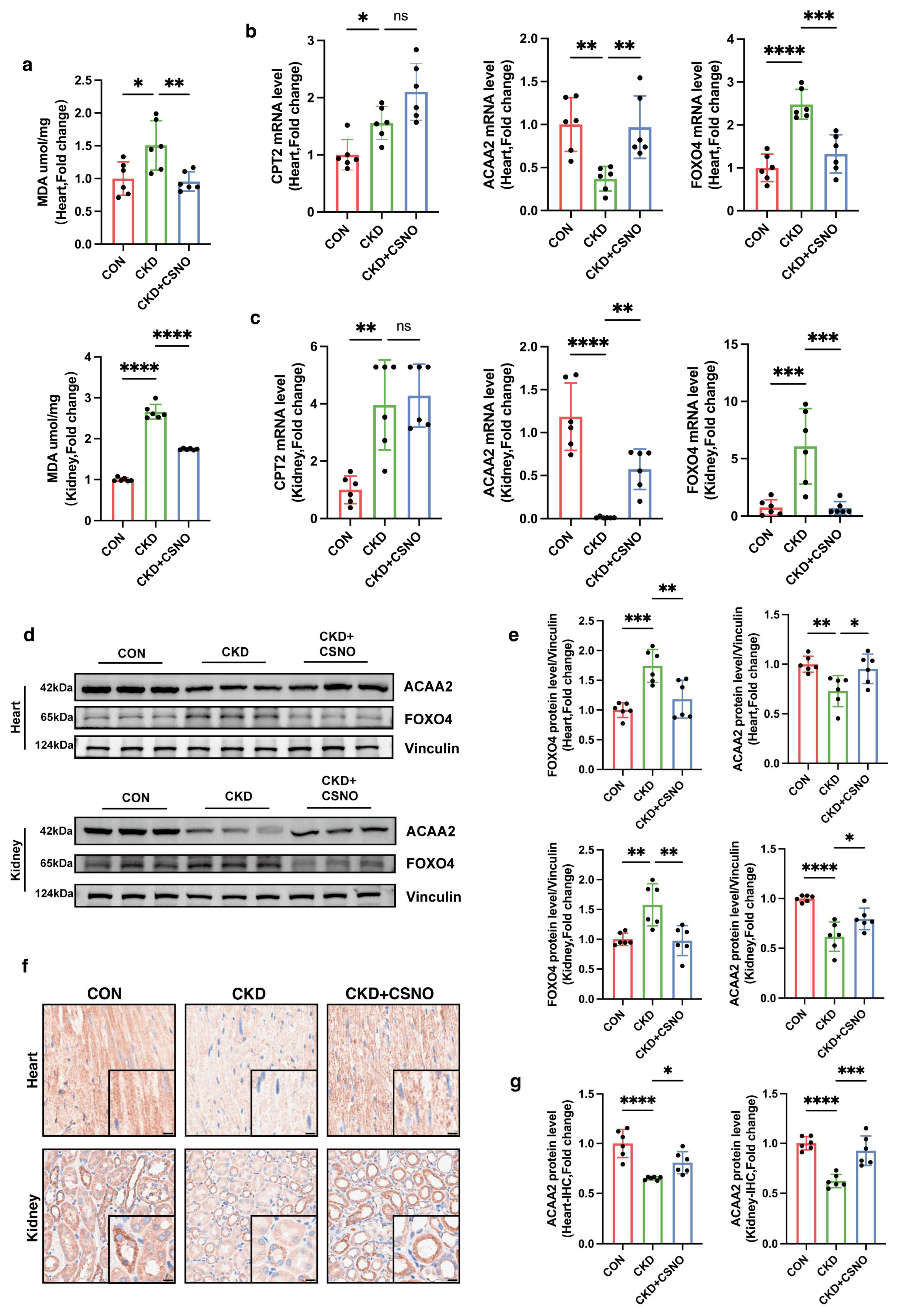
A total of 30 C57BL/6 mice (18–22 g, 6–7 weeks, male) were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China). After one week of adaptation, the C57BL/6 mice were randomly divided into three groups: Control, CKD, and CKD + CSNO groups (n = 10). The random numbers for animal grouping were generated by the “Rand()” function in Microsoft Excel. All animal experiments were approved by the Animal Care and Use Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Approve number: TJH-202306049). Mice had free access to food and water and were housed in a specific pathogen-free room with a 12 h light/dark cycle at a temperature of 25 ± 1. Based on the results of the pre-experiment and the current literature report, for CKD modeling, C57BL/6 mice received an adenine gavage (50 mg/kg/d, in saline, purity ≥ 98.0%, HY-B0152, MCE) for a total of 4 weeks. The CKD + CSNO group mice received nebulized inhalation of CSNO from the week 3 to 8 (from the 15th day) (88 ppm for 20 min per day). The mice in the control group were administered the vehicle in parallel. Dynamic monitoring was performed by non-invasive cardiac ultrasound. After 6 weeks of treatment, the mice were sacrificed after intraperitoneal injection of pentobarbital, and the tissues and blood samples were collected for further data analysis. CKD and CSNO administration methods were based on existing protocols in their respective settings.
2.3. Cell Culture and Treatment
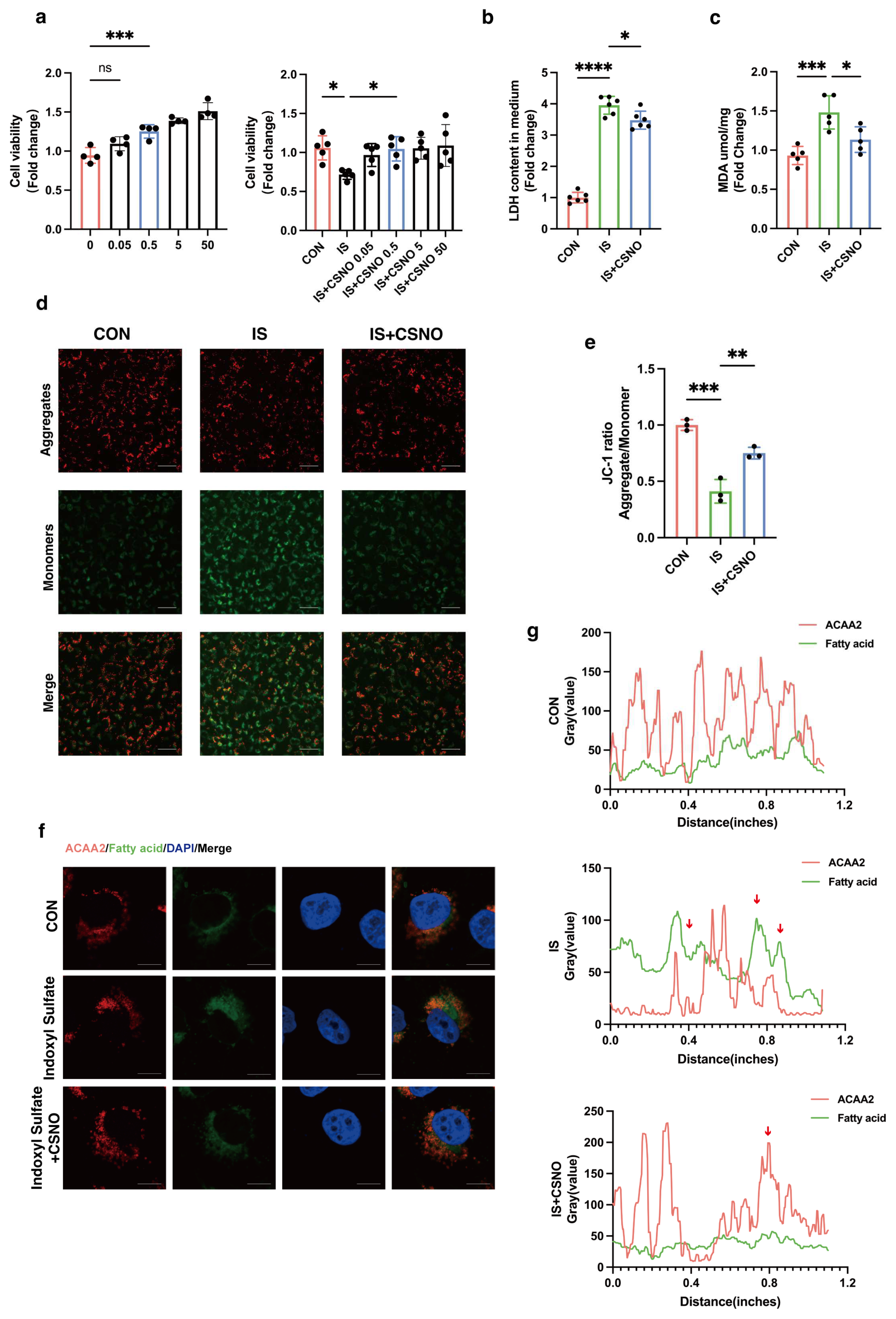
AC16 cells were acquired from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, KeyGEN BioTECH, Nanjing, China) supplemented with 10% fetal bovine serum (FBS, Gibco, Waltham, MA, USA) and 1% penicillin/streptomycin (Sangon, Shanghai, China) at 37 °C with 5% CO2. AC16 cells were treated with different concentration of Indoxyl Sulfate (IS, purity ≥ 98.0%, I3875, Sigma-Aldrich, Saint Louis, MO, USA) to induce in vitro cardiomyocyte injury for 24 h while starving. Meanwhile, CSNO (dissolved and mixed for 5 min until orange) was added to cells with IS treatment. The above experiments were repeated three times independently.
2.4. Real-Time PCR
Total RNA was extracted from tissue samples using the RNA Isolation Kit (Vazyme, Nanjing, China) or from cultured cells using TRIzol (GIBCO Life Technology, Thermo, Waltham, MA, USA). After DNase treatment, first-strand cDNA was prepared from RNA (1.5 μg) using Master Mix (Life Technologies, Waltham, MA, USA). cDNA was diluted and amplified using Powerup SYBR Green qPCR Master Mix on Real-Time PCR instrument. Primers were designed using NCBI/Primer-BLAST (
Supplementary Table S1). Data were normalized to a reference gene (GAPDH) and presented as the fold increase compared with RNA isolated from the control group using the 2
−ΔΔCT method.
2.5. Western Blotting to Detect Protein Expression
Tissues and cells were homogenized with precooled RIPA lysis buffer containing 1 mM protease inhibitor and 1 mM phosphatase inhibitor for 1 h. After being centrifuged at 12,000 rpm for 20 min at 4 °C, the supernatant was collected. A BCA protein assay kit was used to measure the protein concentration. Equal amounts of protein were separated by 10–12% SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 5% skim milk for 1 h and incubated with different primary antibodies at 4 °C overnight. The secondary antibody (1:5000) was incubated for 1 h at room temperature. An ultrahigh sensitivity ECL kit (HY–K1005; MedChemExpress, Princeton, NJ, USA) was used for protein detection, which was performed on a ChemiDoc-It 510 Imager with VisionWorks software 1.6.5 (Ultra-Violet Products Ltd., Cambridge, UK).
2.6. Measurement of ATP Contents
The ATP levels were measured using an enhanced ATP assay kit (S0027, Beyotime, Shanghai, China). After removing the culture medium, we added 200 µL lysis solution to each well of a 6-well plate to lyse the cells and then centrifuged at 4 °C at 12,000× g for 5 min. The ATP standard solution was thawed on ice and diluted with ATP assay lysis solution to create an appropriate concentration gradient (0.01, 0.03, 0.1, 0.3, 1, 3, and 10 µM). Each sample or standard required 100 µL of ATP detection working solution, which was added to a light-shielded 96-well plate after preparation. The plate was left at room temperature for 3–5 min to ensure complete consumption of baseline ATP. Then, 20 µL of either the sample or standard was added to each well, mixed immediately, and measured using the Gen5 software 3.12 on the BioTek multi-function microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). The obtained RLU value was converted into the corresponding ATP (µM) value based on the standard curve. Additionally, calibration using the BCA protein assay kit (mg/mL) was performed to obtain the ATP concentration in nmol/mg protein.
2.7. Measurement of Cell Viability
Cell viability was assessed with the CCK-8 assay kit (RM02823, ABclonal, Wuhan, China). In brief, AC16 cells (5 × 103 cells/well) were plated in medium (100 μL/well) into 96-well plates with six replicate wells. After being attached, they were modeled and administered with the corresponding drugs. Following treatment, AC16 cells were incubated with 10% CCK8 for 1 h in the dark. The absorbance at 450 nm was recorded on a microplate spectrophotometer.
2.8. Assessment Reactive Oxygen Species
To detect the level of intracellular reactive oxygen species (ROS), dihydroethidium (DHE. cat.no.HY-D0079, MedChemExpress, Princeton, NJ, USA), a superoxide indicator, was used. For preparing DHE staining solution, we avoided light and use it immediately after preparation. Aliquoted DHE staining reagent was thawed at room temperature and diluted with DMEM. Next, 1 mL DHE staining solution (1×) was prepared per well on a 6-well plate and thoroughly mixed. It was incubated at 37 °C away from light for 30 min and then observed under a fluorescence microscope (Bx53, Olympus, Tokyo, Japan). The culture medium was removed and the cells washed once with PBS, which was then replaced with fresh DMEM, and observation under the microscope continued. The fluorescence intensity of the same cell mass was quantified using Image J 1.54 to obtain RFU values. The procedure was repeated at least three times.
2.9. Mitochondrial Permeability Transition Pore (mPTP) Detection
Mitochondrial permeability transition pore (mPTP) detection was performed using an mPTP assay kit (C2009S, Beyotime, Haimen, China). The cells were seeded in culture dishes and treated according to the experimental design. The culture medium was removed and the cells washed 1–2 times with PBS. An appropriate volume of mPTP detection working solution (Calcein AM + CoCl2, final concentration 1×) was added and gently shaken to ensure the dye evenly covered all the cells. The mixture was incubated at 37 °C in the dark for 30–45 min. After incubation, the medium replaced with fresh pre-warmed (37 °C) culture medium, and the mixture was incubated again in the dark for another 30 min to ensure intracellular esterases fully hydrolyzed the Calcein AM to generate green fluorescent Calcein. The culture medium was then removed, the cells washed 2–3 times with PBS, and detection buffer added; green fluorescence was observed under a fluorescence microscope (Wuhan, China).
2.10. Immunofluorescence and BODIPY Staining
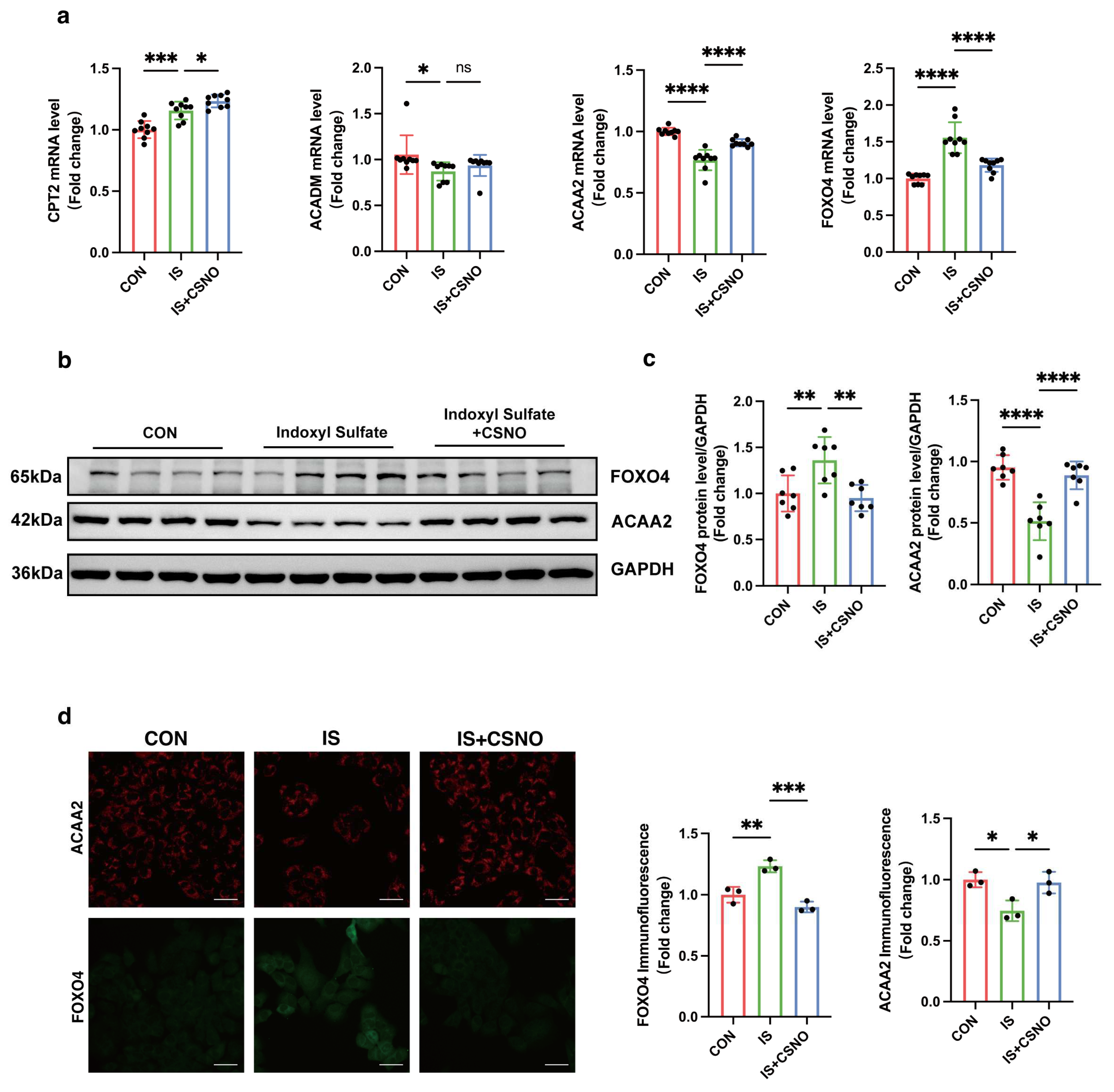
AC16 cells were seeded on coverslips. After treatments, the cells were fixed with methanol for 15 min at room temperature and subsequently permeabilized and blocked with 0.5% Triton-100 and 5% goat serum in PBS for 1 h at room temperature. Then, the cells were incubated with primary antibodies diluted overnight at 4 °C. After washing three times with PBS, the cells were incubated with fluorescence secondary antibody for 1 h. As for the double fluorescence staining of BODIPY and ACAA2, the slides were evenly covered with the green fluorescent fatty acid probe solution (C2055, Beyotime, Haimen, China) after the secondary antibody incubation and incubated at 37 °C for 15 min. Then, the cells were washed three times again, and DAPI-containing anti-fluorescence quencher was used to seal the glass slides. Finally, slides were examined using a confocal microscope (Nikon, Tokyo, Japan). The primary antibodies used in this study targeted endogenous ACAA2 (1:100, A15778, ABclonal, Wuhan, China) and FOXO4 (1:100, 3307, ABclonal, Wuhan, China).
2.11. Transfection
Approximately 18 h before transfection, cells were seeded. Opti-MEM serum-free culture medium was added to a sterile centrifuge tube and gently mixed with an appropriate amount of ExFect. Opti-MEM was added to another sterile centrifuge tube along with an appropriate amount of DNA. ExFect–opti-MEM was added to the DNA–opti-MEM and left sitting at room temperature for 15–20 min before transfection. The ExFect/DNA complex mixture was added into the culture medium, with gentle shaking of the medium to evenly disperse the ExFect/DNA. After overnight cultivation for 24–48 h, the cells were collected for subsequent experiments. Knockout ACAA2 or FOXO4 gene expression by siRNA (RiboBio genOFF™ siRNA, Guangzhou, China) was performed using Lipofectamine 2000. Lipo3000 was used for plasmid transfection of ACAA2 overexpression plasmids (Genomeditech, Shanghai, China). After 6 h, the culture medium was replaced and the cells treated with indoxyl sulfate for 24 h while starving. The efficiency of gene knockout was evaluated through Western blotting and real-time PCR.
2.12. Lactate Dehydrogenase Cytotoxicity Detection
We utilized LDH kits (C0017, Beyotime, Haimen, China) to measure LDH release. We plated an appropriate amount of cells into a 96 well cell culture plate, including cell-free culture medium wells (background blank), untreated control cell wells (sample control), untreated cell wells for calculation (maximum enzyme activity), and drug-treated cell wells (drug-treated). One hour before the detection time, the cell culture plate was removed from the cell culture box and the LDH release reagent added to the maximum enzyme activity wells, in an amount of 10% of the original culture medium volume. After reaching the scheduled time, the cell culture plate was centrifuged at 400× g for 5 min. Then, 60 μL of LDH detection working solution was added to each well, mixed, and incubated at room temperature in the dark for 30 min. Then, we measured the absorbance at 490 nm and used any wavelength of 600 nm or greater as the reference wavelength for dual-wavelength measurement. The absorbance for each group obtained should be subtracted by the absorbance from the background control wells. The percentage of cytotoxicity (%) = (absorbance of sample control/drug treated)/(absorbance to maximum enzymatic activity − absorbance of sample control) × 100.
2.13. Lipid Peroxidation MDA Detection
The level of MDA was measured by an MDA assay kit (S0131M, Beyotime, Haimen, China). After tissue and cell homogenization, samples were centrifuged at 12,000× g for 10 min and the supernatant collected for subsequent measurement. A suitable amount of TBA was weighed and added into a 0.37% TBA stock solution using the TBA-reagent solution. The prepared TBA stock solution was stored at room temperature out of light. The MDA detection working solution was formulated as TBA diluent/TBA stock/antioxidant = 150:50:3. Appropriate standards were diluted to concentrations of 1, 2, 5, 10, 20, and 50 µM for subsequent standard curve preparation. Then, 100 µL of PBS control, standard, and samples were added to each 1.5 mL EP tube, followed by the addition of 200 µL MDA detection working solution. The mixture was then heated at 100 °C for 15 min, cooled in a water bath to room temperature, and centrifuged at 1000× g for 10 min at room temperature. Next, 200 µL supernatant was added to a 96-well plate, and absorbance was measured at 532 nm using a microplate reader. A dual-wavelength measurement was set with 450 nm as the reference wavelength. After determining the protein concentration using the BCA kit (P0009, Beyotime, Haimen, China), the MDA content in the original samples was expressed based on the protein content as umol/mg protein/tissue.
2.14. Measurement of Mitochondrial Membrane Potential (ΔΨm)
The measurement of ΔΨm was based on the JC-1 fluorescent probe (PJC-110; Promotor Biological Co., Ltd., Hangzhou, China). AC16 cells were incubated with JC-1 working solution for 30 min at 37 °C following treatments. Subsequently, the cells were washed at least two times with PBS and resuspended with cell culture medium. The fluorescence signals of JC-1 aggregates (red, 525/590 nm) and JC-1 monomers (green, 485/530 nm) were detected with an MShot fluorescence microscope (Wuhan, China). The ratio of the red/green fluorescence intensity represents the degree of mitochondrial damage.
2.15. Echocardiogram
After completing the model, the mice underwent chest hair removal, followed by anesthesia induction using the gas anesthesia machine. Cardiac ultrasound imaging was then performed using the VINNO6 high-resolution imaging system (VINNO Corporation, Suzhou, China), with anesthesia maintained (n = 10). Echocardiography was conducted to assess cardiac function by recording the left ventricular end-diastolic volume (LEDV) and left ventricular end-systolic volume (LESV).
2.16. HE, Masson Staining, and Immunohistochemistry
After animals were sacrificed, the tissues were fixed with 4% formaldehyde. The formalin-fixed tissue was embedded in paraffin and sectioned for further analysis. Masson staining was used for collagen deposition, and HE staining was used to observe the structure of tissues. As for immunohistochemistry, the fixed paraffin sections were incubated with the primary antibody overnight at 4 °C. After washing three times with PBS, the sections were incubated with secondary antibody for 1 h. Then, after washing three times, they were scanned with a brightfield scanner and observed using the NDP.view 2 software 2.9.29. The primary antibodies used in this study targeted endogenous ACAA2 (1:300, A15778, ABclonal, Wuhan, China).
2.17. Oil Red O Staining
Tissues were prepared with a thickness of 4–8 μm and dried for 15–30 min at room temperature. The sections were fixed with 4% formaldehyde for 10 min, followed by three washes with TBST. The Oil Red O stock solution was diluted with distilled water at a 3:2 ratio and incubated at room temperature in the dark for 20–30 min. The sections were washed three times with TBST and counterstained with hematoxylin, and the slides were mounted using 50% glycerol. After scanning with a brightfield scanner, the lipid droplet aggregation was observed using the NDP.view 2 software 2.9.29.
2.18. Statistical Analysis
All data were assessed for normality, and then, parametric or non-parametric tests were employed for data analysis, as appropriate. The unpaired two-tailed t-test was used to compare data between two groups and one-way ANOVA with Sidak’s correction for multiple testing to compare data between more than two groups. The exact test used for each experiment is noted in the figure legends. Data are expressed as mean ± SEM. Statistical significance was considered when p < 0.05. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; ns means not significant. All statistical analysis was performed using GraphPad Prism 10.1.1.
4. Discussion
Cardiorenal syndrome refers to the complex interplay between the heart and kidneys. Studies have shown that cardiac remodeling in patients with chronic kidney disease (CKD) is closely associated with metabolic abnormalities in cardiomyocytes, particularly alterations in energy metabolism [
28]. Moreover, uremic cardiomyopathy is a common cardiac complication in CKD patients, characterized by myocardial hypertrophy and fibrosis and often accompanied by left ventricular dysfunction. The pathophysiological mechanisms of uremic cardiomyopathy are intricate, involving factors such as electrolyte imbalance, activation of the renin–angiotensin system, and hyperactivity of the sympathetic nervous system. Clinically, despite improvements in adverse factors such as high circulatory load, heart function in patients with both cardiac and renal insufficiency may continue to deteriorate. This observation suggests that renal dysfunction may impair cardiac function through mechanisms other than hemodynamic overload. Therefore, investigating the mechanisms underlying non-hemodynamic cardiac injury in renal dysfunction and identifying effective interventions are of critical importance. Against this backdrop, our study focused on the elevated levels of uremic toxins in patients with renal dysfunction, particularly indoxyl sulfate IS.
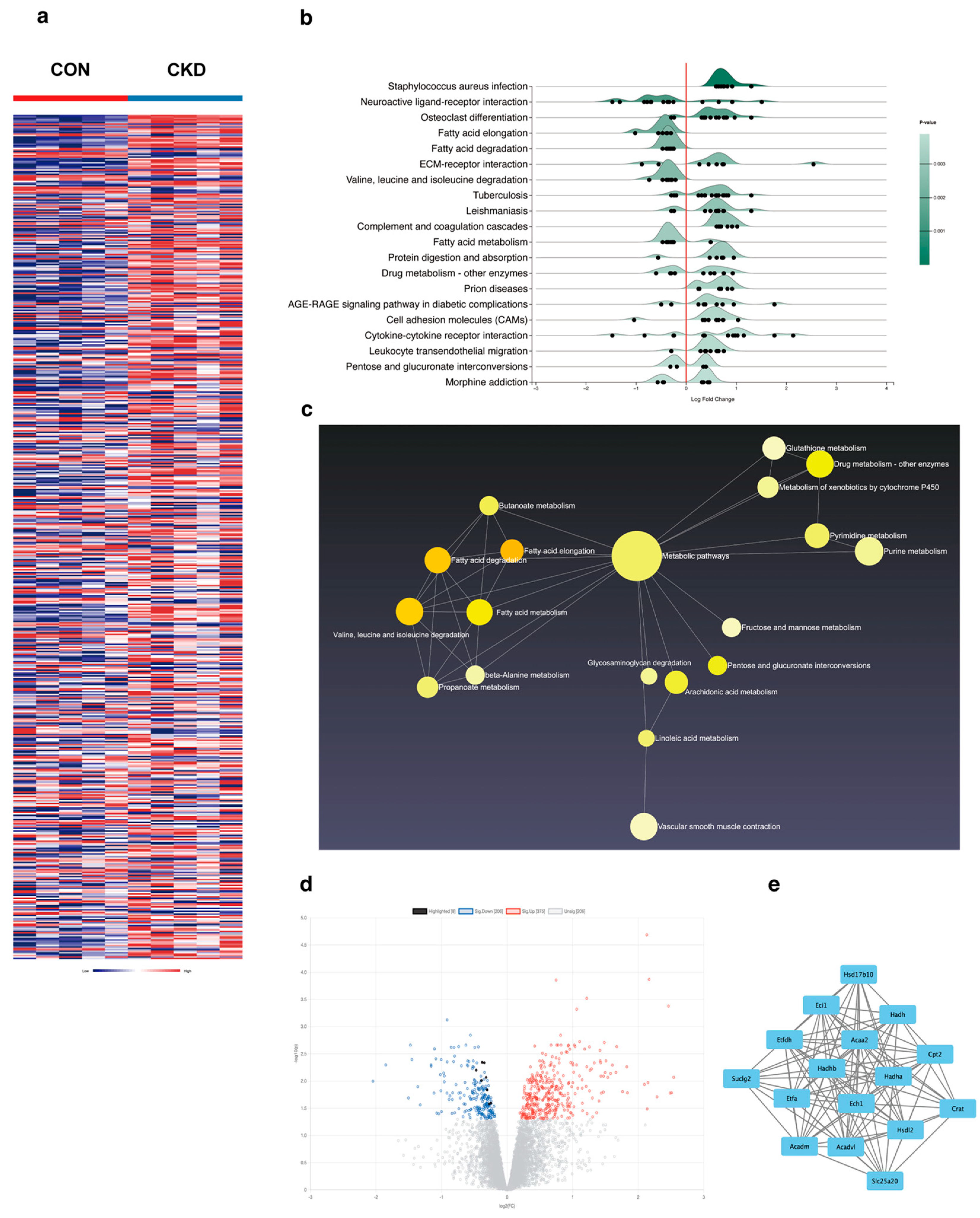
The heart is a highly metabolically active organ that primarily relies on fatty acid oxidation for energy supply. When the uptake of fatty acids exceeds the oxidative capacity of cardiomyocytes, unmetabolized fatty acids accumulate within the cells, forming intermediates such as triglycerides, diacylglycerol, and ceramides [
29,
30]. These intermediates not only directly impact the physiological functions of cardiomyocytes but also activate various intracellular signaling pathways, inducing oxidative stress, inflammatory responses, and apoptosis, thereby exacerbating myocardial injury. Studies have demonstrated that in patients with metabolic disorders such as obesity, type 2 diabetes, and hyperlipidemia, the capacity for myocardial fatty acid oxidation is significantly impaired, leading to abnormal lipid accumulation in the myocardium and resulting in lipid peroxidation [
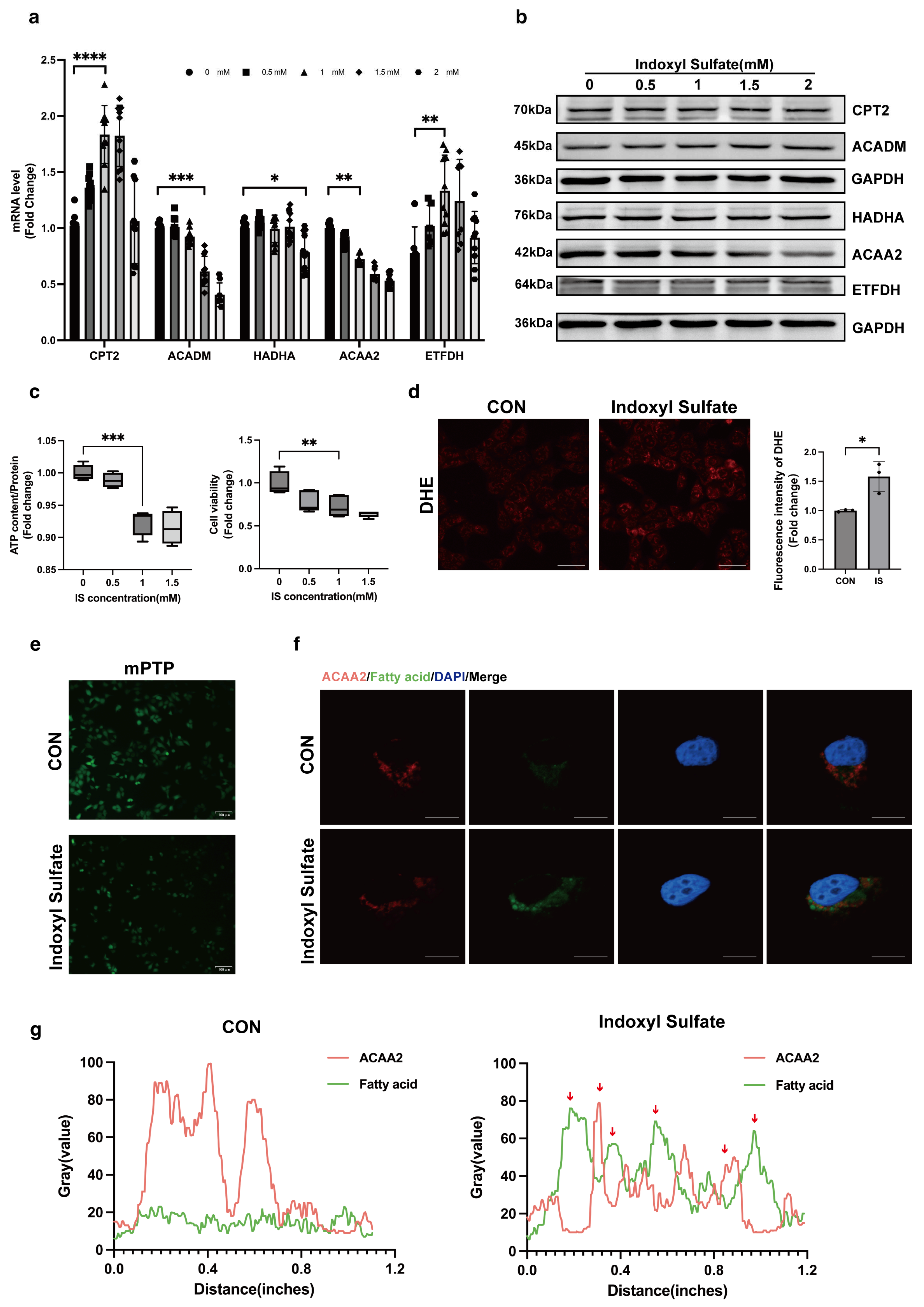
31]. Through GEO data analysis, we found that fatty acid β-oxidation-related proteins in the heart are markedly downregulated under the context of renal dysfunction, with ACAA2 and other related fatty acid metabolic factors playing a critical role in fatty acid metabolism. This suggests that fatty acid oxidation is unable to provide sufficient energy support for cardiomyocytes under renal insufficiency. In AC16 cardiomyocytes stimulated with 1 mM indoxyl sulfate IS, we observed increased cell death rates, decreased mitochondrial ATP levels, elevated mitochondrial oxidative stress, and opening of the mitochondrial permeability transition pore (mPTP), indicating that moderate uremic toxin exposure impairs cardiomyocyte and mitochondrial function. Interestingly, colocalization of ACAA2 protein with lipid droplets showed that indoxyl sulfate reduces ACAA2 expression while increasing lipid droplet accumulation. In addition, IS-stimulated cardiomyocytes exhibited increased mRNA levels of the fatty acid transporter CPT2 and electron transfer protein ETFDH, while β-oxidation-related proteins such as ACADM, Ech1, and ACAA2 were significantly decreased. These findings indicated that, although fatty acid β-oxidation is diminished, fatty acid uptake by cardiomyocytes remains largely unaffected. This suggests that while the myocardium continuously takes up fatty acids for oxidation, the metabolic “fuel factory” fails to operate effectively, resulting in the accumulation of lipid metabolic intermediates within the mitochondria, triggering a cascade of oxidative stress reactions, including lipid peroxidation. However, it is worth noting that there are asymmetric changes in the protein levels and mRNA levels of CPT2, ACADM, and ETFDH. Protein level expression is not only associated with transcription and translation processes but also related to protein degradation. Proteins may be rapidly degraded by the ubiquitin–proteasome system or the autophagy pathway, resulting in lower protein levels despite high mRNA levels. The mechanisms underlying this phenomenon remain to be explored. ACAA2, an acyl-CoA acyltransferase, is involved in the final step of fatty acid β-oxidation and exerts a protective role in various diseases [
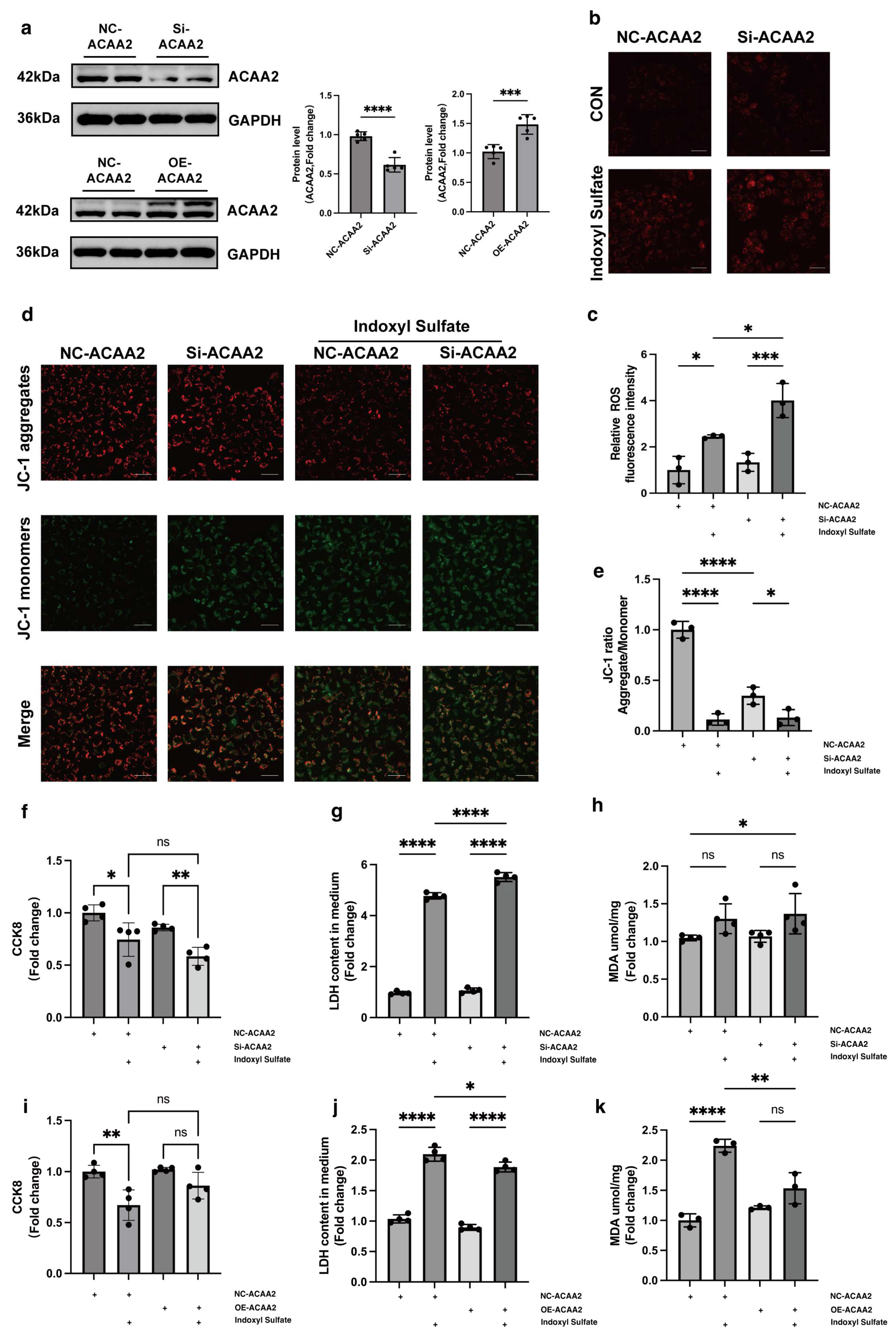
32,
33]. Given this, we questioned whether modulation of ACAA2 might alleviate myocardial injury in renal dysfunction. Therefore, we knocked down and overexpressed ACAA2. Interestingly, simple knockdown of ACAA2 in AC16 cardiomyocytes did not significantly affect ROS levels or LDH in experiments, which suggests that knockdown of ACAA2 alone has limited effects on the accumulation of intracellular oxygen radicals, making it hard to trigger apoptosis or necrosis. However, when stimulating AC16 cells with IS, si-ACAA2 further amplified the toxic effects on cardiomyocytes, which suggests that ACAA2 protein is indispensable in protecting cardiomyocytes from damage induced by urea toxin stimulation. The decline in mitochondrial membrane potential with addition of si-ACAA2 indicated an early-stage damage, suggesting an acute temporal impact of si-ACAA2 on cardiomyocytes. In contrast, simply overexpressing ACAA2 did not significantly restore LDH and MDA levels in AC16 cells. However, under the external stimulus of IS, LDH and MDA levels significantly decreased, indicating that the underlying mechanisms require further investigation. Given that ACAA2 differs from other matrix-associated mitochondrial proteins by possessing a non-cleavable N-terminus, it may exhibit distinct functionalities under certain specific conditions. These results suggest that overexpression of ACAA2 facilitates the management of excess fatty acids within cardiomyocytes, reducing intracellular lipid peroxidation.
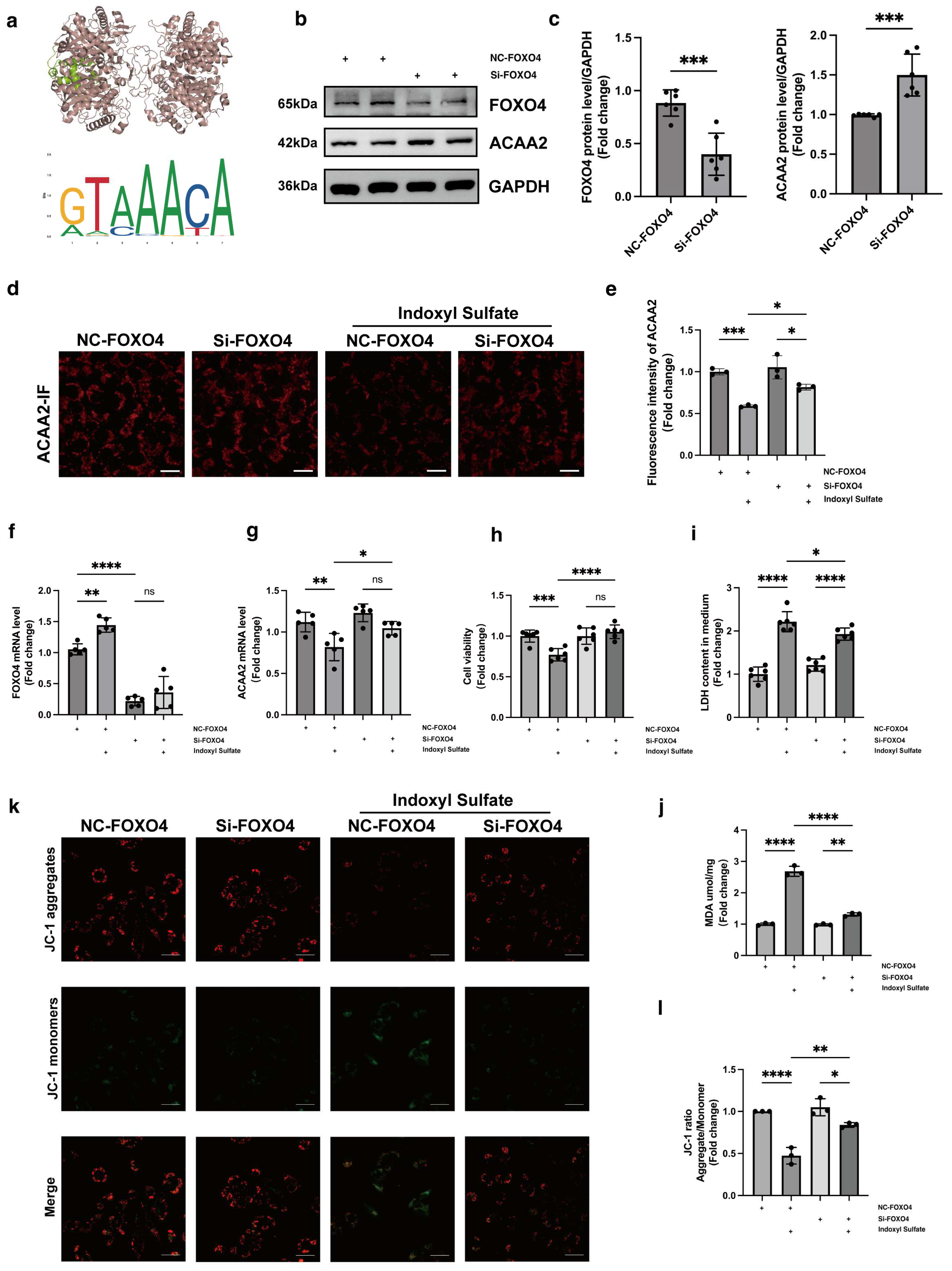
Having established the crucial role of ACAA2 in fatty acid β-oxidation, we further utilized the JASPAR database to predict upstream transcription factors potentially regulating ACAA2. Among these, FOXO4 emerged as a likely candidate. FOXO4, a member of the forkhead box transcription factor family, has received increasing attention in cancer research in recent years [
34]. For instance, the hypoxia-induced FOXO4/LDHA axis was found to play a pivotal role in regulating glycolysis and tumor progression in gastric cancer [
35]. We hypothesized that FOXO4 might exert transcriptional control over ACAA2, and to test this, we knocked down FOXO4 in AC16 cardiomyocytes, followed by stimulation with indoxyl sulfate. From multiple perspectives, including RNA and protein expression levels, we found that FOXO4 knockdown partially restored the reduction in ACAA2 protein levels induced by indoxyl sulfate. Furthermore, knocking down FOXO4 mitigated the effects of indoxyl sulfate on cardiomyocyte apoptosis, mitochondrial lactate dehydrogenase release, mitochondrial membrane potential, and lipid peroxidation levels. In summary, FOXO4 knockdown improved the oxidative stress and mitochondrial dysfunction in cardiomyocytes induced by indoxyl sulfate, partly through the restoration of ACAA2 expression. However, the transcriptional regulation of ACAA2 by FOXO4 did not appear to be highly specific. Our subsequent unpublished results suggest that the regulatory factors controlling ACAA2 expression are multifaceted and complex, extending beyond transcription factors and involving post-translational modifications. Nonetheless, it is unequivocal that FOXO4 is involved in the regulation of ACAA2 expression. However, further studies are needed to clarify how FOXO4 specifically influences lipid metabolism in cardiomyocytes through ACAA2.
Previous studies have demonstrated that CSNO can improve cardiac function in diabetic cardiomyopathy and restore cardiomyocyte viability [
24]. In the cardiovascular system, S-nitrosylation participates in the regulation of cardiac function by modulating key signaling molecules such as CaMKII, JNK [
36], and Hsp90 [
37]. For instance, S-nitrosylation of CaMKII plays an important role in β-adrenergic signaling in the heart, where nitrosylation at specific cysteine residues can trigger autonomous activation, thereby affecting calcium release and contributing to arrhythmogenesis [
38]. However, both excessive and insufficient levels of S-nitrosylation can lead to pathological states [
39,
40]. Excessive S-nitrosylation may lead to abnormal activation or inhibition of protein function, whereas insufficient nitrosylation may impair cellular antioxidant capacity and stress response. Thus, the precise regulation of S-nitrosylation is crucial for maintaining cellular homeostasis and preventing disease [
41]. Can CSNO ameliorate cardiac dysfunction in the context of renal insufficiency? To address this question, we first added various concentrations of CSNO to cardiomyocytes treated with indoxyl sulfate. The results, which included assessments of cell viability, protein levels, mitochondrial membrane potential, and lipid peroxidation MDA levels, indicated that CSNO supplementation partially mitigated the damage induced by indoxyl sulfate. To further verify the effect of lipid peroxidation on cardiomyocytes, ACAA2 was colocalized with BODIPY, and it was observed that CSNO restored ACAA2 protein levels and reduced the accumulation of lipid intermediates. In addition, we investigated the FOXO4–ACAA2 axis and found that CSNO reduced the RNA level of FOXO4, an upstream regulator of ACAA2, while having no significant effect on the fatty acid transporter CPT2 or the common fatty acid transcription factor PPARA. This suggests that FOXO4 exerts some transcriptional regulation on ACAA2, and CSNO exerts its effect through the FOXO4–ACAA2 axis. In a mouse model, daily aerosol inhalation of CSNO for 20 min resulted in an improvement in cardiac function under renal insufficiency, reducing myocardial hypertrophy and lipotoxic accumulation. Additionally, CSNO significantly attenuated renal fibrosis, although the underlying mechanism remains unclear. It is uncertain whether these effects are due to exogenous NO supplementation or the protective effects of S-nitrosylation of thiol groups. Further exploration of the precise mechanisms is required. Nevertheless, it is evident that aerosolized CSNO can partially restore cardiac function in the setting of renal insufficiency and significantly increase ACAA2 protein levels, thereby improving myocardial lipid metabolism.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}