

Rapid Genotyping of FecB Mutation in Sheep Using CRISPR-Cas12a Integrated with DNA Nanotree Biosensing Platform

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
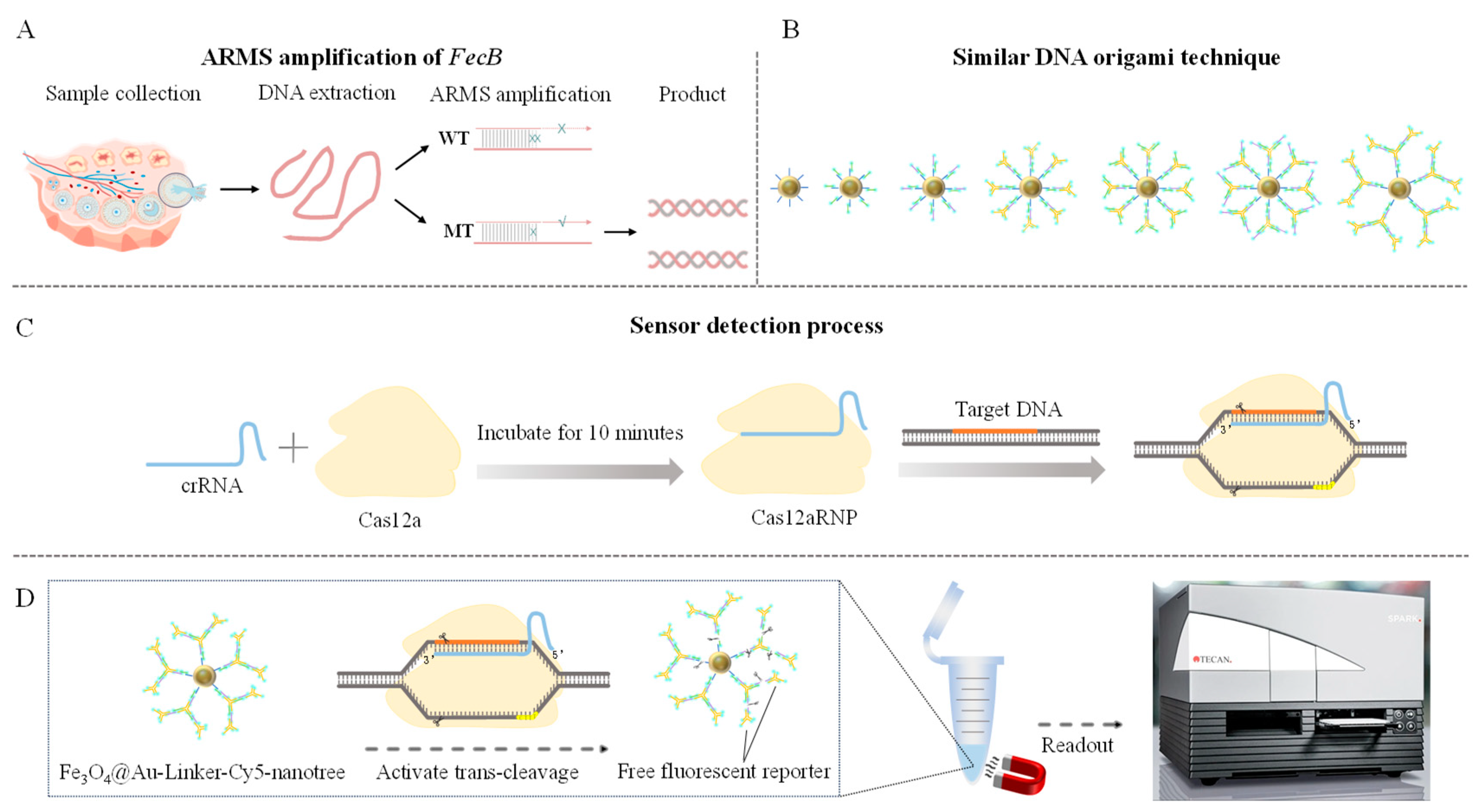
2.2. Genomic DNA Extraction and Genotype Detection
2.3. Optimization and Detection of ARMS Primers
2.4. DNA Extraction and Validation of the Test Sample
2.5. Design and Preparation of Gold Nanomagnetic Bead (Fe3O4@Au)-Fluorescent Probe Complex
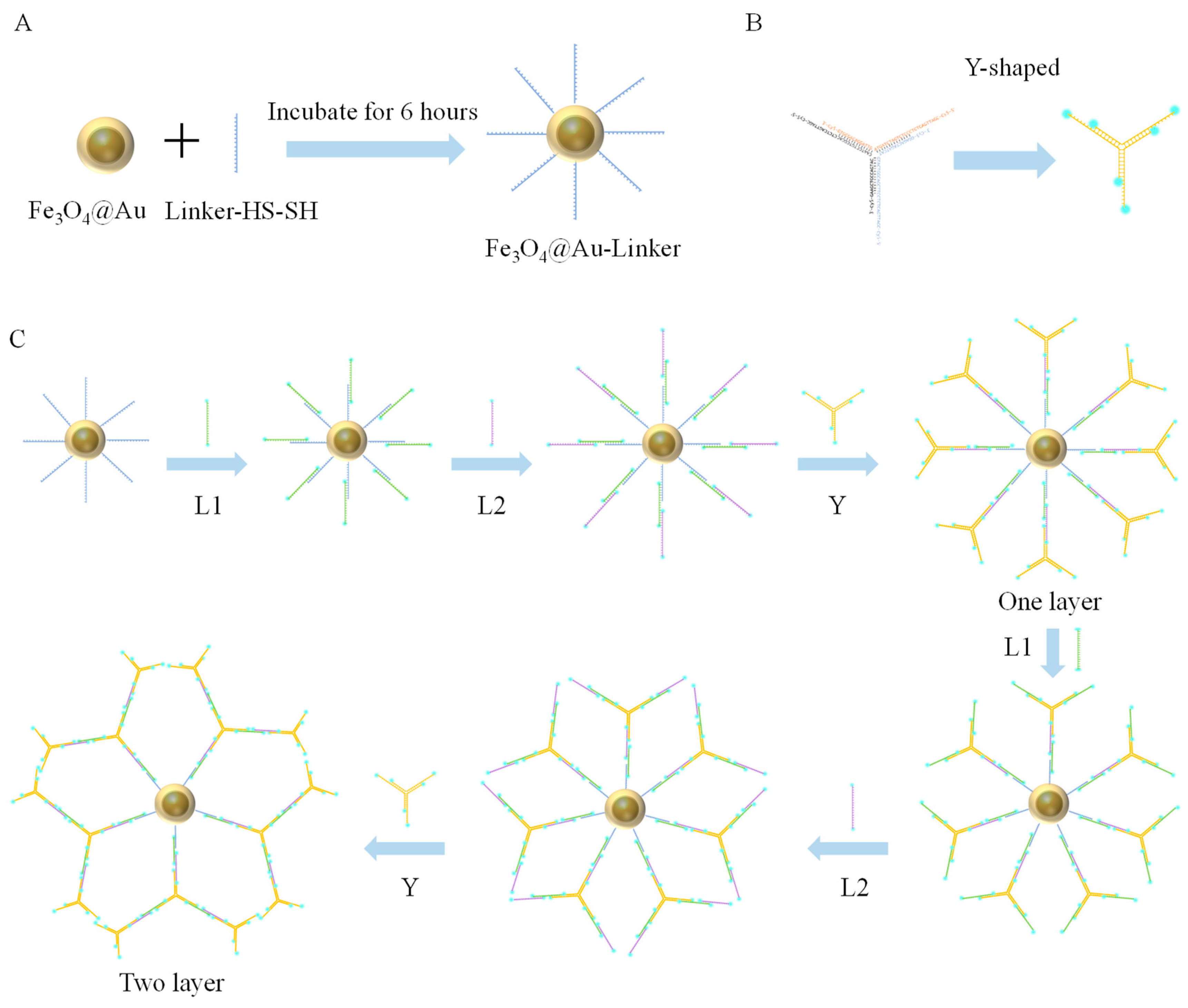
2.5.1. Assembly of Gold Nanomagnetic Beads (Fe3O4@Au) with Linker-HS-SH
2.5.2. Y-Shaped Branches
2.5.3. Fe3O4@Au-Linker-Cy5 Construction of Nanotree
2.6. Establishment of CRISPR-Cas12a Detection Method
2.7. Accuracy Detection of CRISPR-ARMS Detection System
3. Results
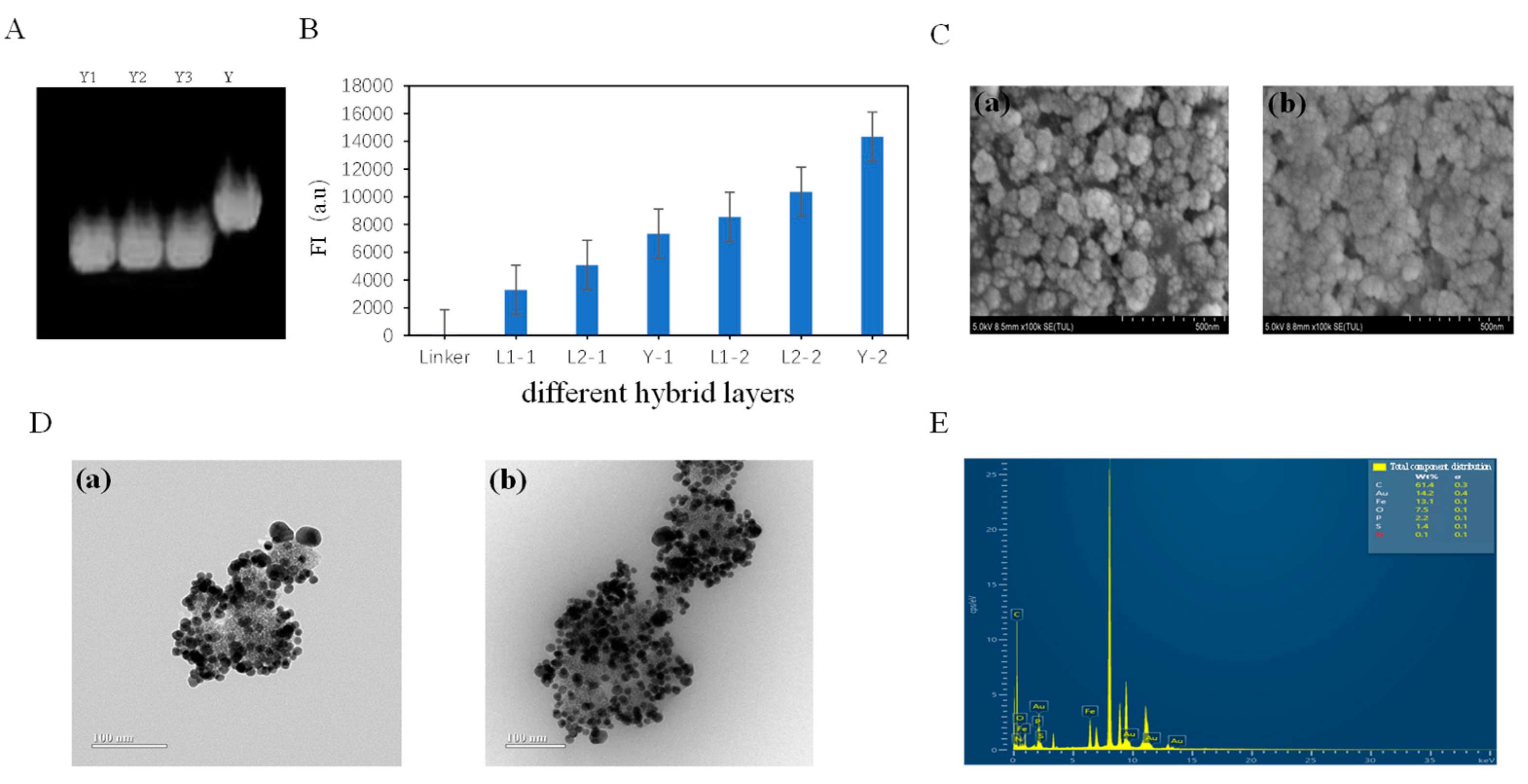
3.1. Fe3O4@Au-Linker-Cy5 Characterization of Nanotree
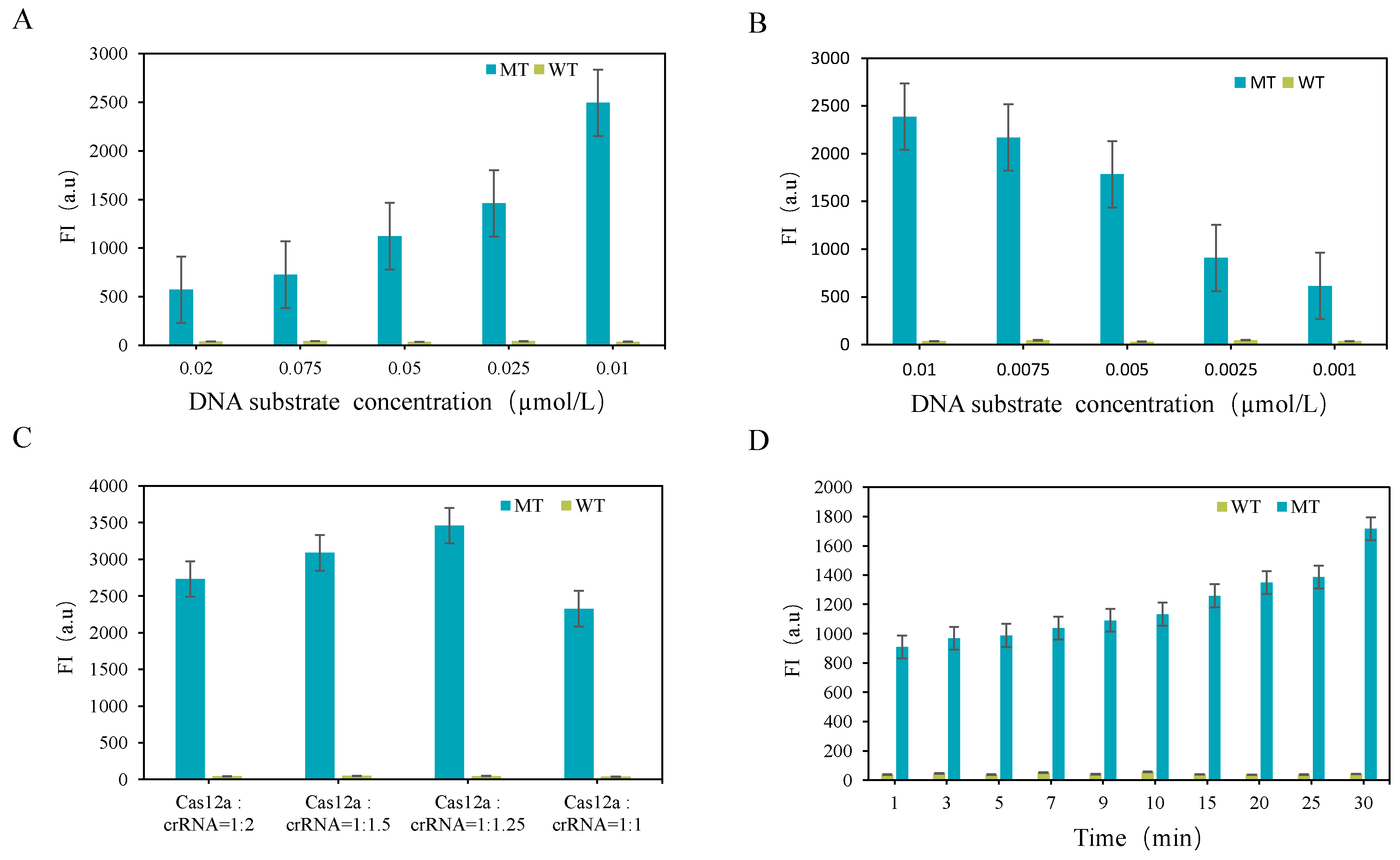
3.2. CRISPR-ARMS System Optimization
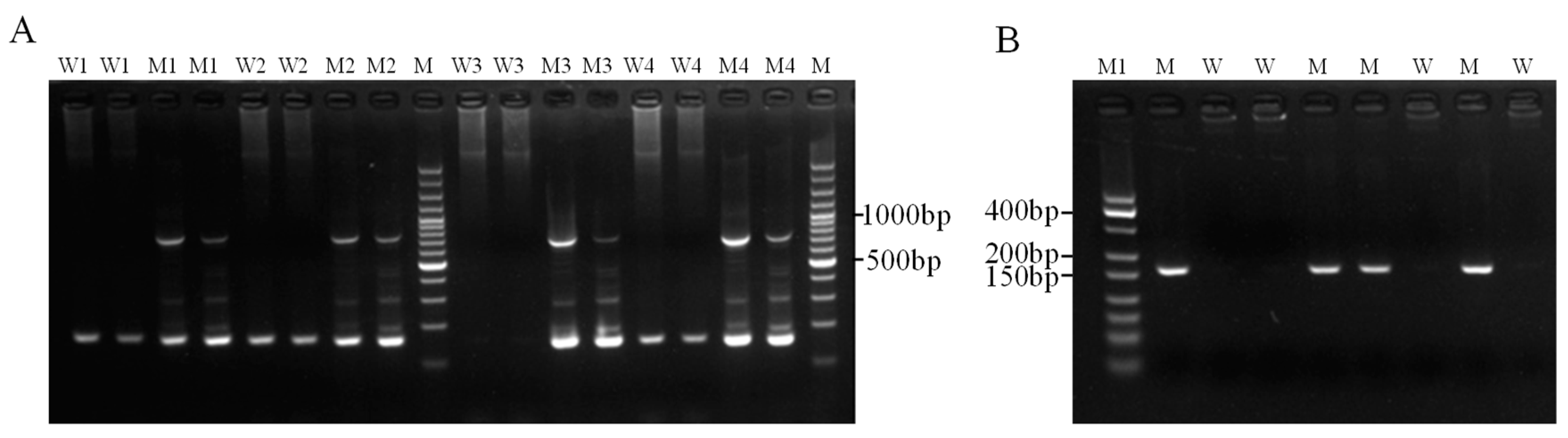
3.2.1. Optimization and Detection of ARMS Primers
3.2.2. Optimization of CRISPR-Cas12a System
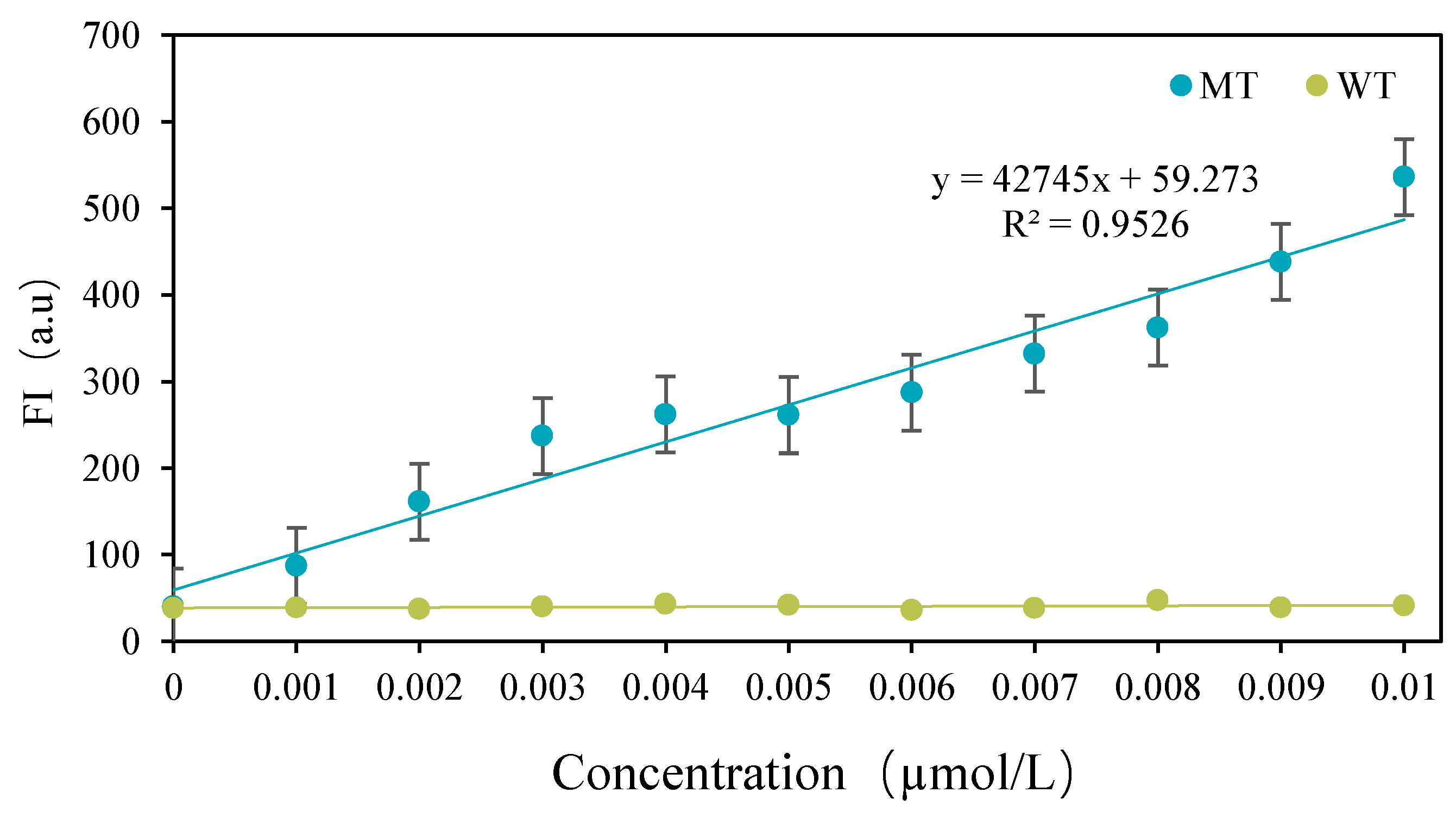
3.3. Detection Results of CRISPR-ARMS System
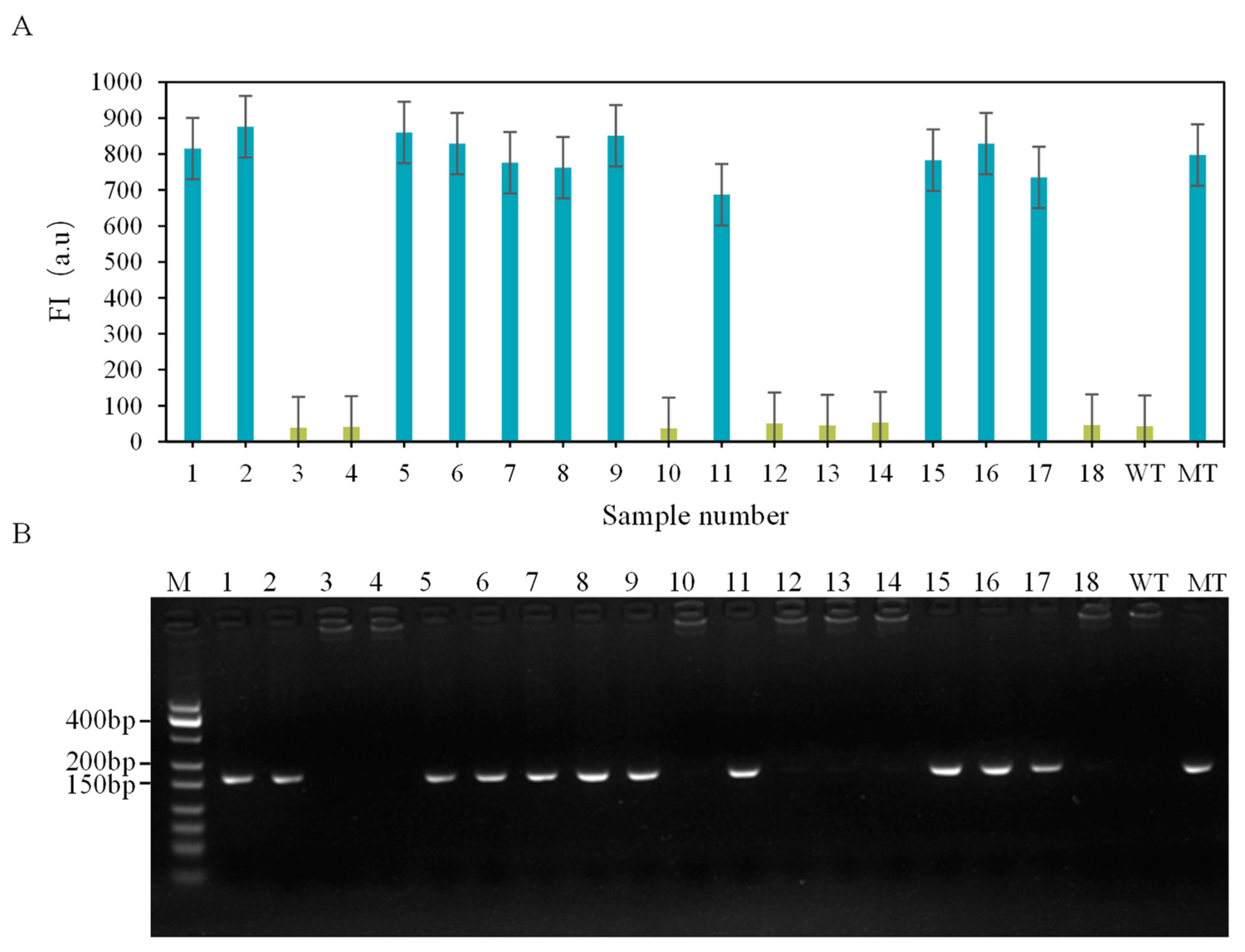
3.4. Actual Sample Testing and Verification
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, H. Identification of Key Regulatory Genes for Litter Size Trait in Chuanzhong Black Goat by Genome Resequencing. Master’s Thesis, Southwest Minzu University, Chengdu, China, 2021. [Google Scholar]
- Hua, G.H.; Yang, L.G. A review of research progress of FecB gene in Chinese breeds of sheep. Anim. Reprod. Sci. 2009, 116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, X.; Liu, G.; Deng, M.; Sun, B.; Guo, Y.; Liu, D.; Li, Y. Polymorphisms in BMPR-IB gene and their association with litter size trait in Chinese Hu sheep. Anim. Biotechnol. 2020, 33, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Sun, W.; Zhang, L.; Zhang, X.; La, Y.; Xiao, F.; Jia, J.; Jin, J. Polymorphisms in BMPRIB gene affect litter size in Chinese indigenous sheep breed. Anim. Biotechnol. 2023, 34, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z. Confirmation and Application of Chalkbrood Resistance-Associated SNP Molecular Markers in Honey Bee, Apis Mellifera Ligustica. Master’s Thesis, Fujian Agriculture and Forestry University, Fuzhou, China, 2016. [Google Scholar]
- Liu, Q.Y.; Hu, W.P.; He, X.Y.; Pan, Z.Y.; Guo, X.F.; Feng, T.; Cao, G.L.; Huang, D.W.; He, J.N.; Di, R.; et al. Establishment of high-throughput molecular detection methods for ovine high fecundity major gene FecB and their application. Chin. J. Anim. Vet. Sci. 2017, 48, 39–51. [Google Scholar]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Fonfara, I.; Richter, H.; Bratovič, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Joung, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Yang, J.; Wang, Z.; Wang, H.; Wang, H. CRISPR/Cas12a-based biosensing platform for the on-site detection of single-base mutants in gene-edited rice. Front. Plant. Sci. 2022, 13, 944295. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, L.; Long, Y.; Liu, Z.; Meng, G.; Zhao, H.; Su, X.; Shi, S. Multiplexed CRISPR-based nucleic acid detection using a single Cas protein. Anal. Chem. 2023, 95, 16089–16097. [Google Scholar] [CrossRef]
- Benvidi, A.; Tezerjani, M.D.; Jahanbani, S.; Mazloum Ardakani, M.; Moshtaghioun, S.M. Comparison of impedimetric detection of DNA hybridization on the various biosensors based on modified glassy carbon electrodes with PANHS and nanomaterials of RGO and MWCNTs. Talanta 2016, 147, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Torres-sangiao, E.; Holban, A.M.; Gestal, M.C. Advanced Nanobiomaterials: Vaccines, Diagnosis and Treatment of Infectious Diseases. Molecules 2016, 21, 867. [Google Scholar] [CrossRef]
- Singh, N.; Dahiya, B.; Radhakrishnan, V.S.; Prasad, T.; Mehta, P.K. Detection of Mycobacterium tuberculosis purified ESAT-6 (Rv3875) by magnetic bead-coupled gold nanoparticle-based immuno-PCR assay. Int. J. Nanomed. 2018, 13, 8523–8535. [Google Scholar] [CrossRef] [PubMed]
- Kaewwonglom, N.; Oliver, M.; Cocovi-solberg, D.J.; Zirngibl, K.; Knopp, D.; Jakmunee, J.; Miró, M. Reliable sensing platform for plasmonic enzyme-linked immunosorbent assays based on automatic flow-based methodology. Anal. Chem. 2019, 91, 13260–13267. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, N.; Thiruvengadam, V.; Sushil, S.N. Nanoparticle-mediated dsRNA delivery for precision insect pest control: A comprehensive review. Mol. Biol. Rep. 2024, 51, 355. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Xu, D.; Henzie, J.; Lopes, A.; Yamauchi, Y.; Liu, B. Mesoporous gold nanospheres via thiolate-Au(i) intermediates. Chem. Sci. 2019, 10, 6423–6430. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.X.; Mei, Z.C.; Cheng, L.J.; He, Y.Q. Exonuclease I assisted purification of DNA origami nanostructures. Mod. Chem. Ind. 2023, 43, 163–166+172. [Google Scholar]
- Yao, G.; Zhang, F.; Wang, F.; Peng, T.; Liu, H.; Poppleton, E.; Šulc, P.; Jiang, S.; Liu, L.; Gong, C.; et al. Meta-DNA structures. Nat. Chem. 2020, 12, 1067–1075. [Google Scholar] [CrossRef]
- Sun, Y.; Yan, L.; Sun, J.; Xiao, M.; Lai, W.; Song, G.; Li, L.; Li, L.; Fan, C.; Pei, H. Nanoscale organization of two-dimensional multimeric pMHC reagents with DNA origami for CD8(+) T cell detection. Nat. Commun. 2022, 13, 3916. [Google Scholar] [CrossRef]
- Zhang, T.T. Application of DNA Origami in Combinatorial Optimization Problems. Master’s Thesis, Anhui University of Science and Technology, Huainan, China, 2024. [Google Scholar]
- Yao, G.; Li, J.; Li, Q.; Chen, X.; Liu, X.; Wang, F.; Qu, Z.; Ge, Z.; Narayanan, R.P.; Williams, D.; et al. Programming nanoparticle valence bonds with single-stranded DNA encoders. Nat. Mater. 2020, 19, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, J.; Jiang, S.; Xie, M.; Zhai, T.; Guo, L.; Cao, S.; Xing, S.; Qu, Z.; Zhao, Y.; et al. Encoding quantized fluorescence states with fractal DNA frameworks. Nat. Commun. 2020, 11, 2185. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Liu, M.; Yan, L.; Deng, M.; Li, F.; Li, M.; Wang, F.; Li, J.; Wang, L.; Tian, Y.; et al. Programming biomimetically confined aptamers with DNA frameworks. ACS Nano 2020, 14, 8776–8783. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, J.G.; Yun, G.; Lee, C.; Kim, Y.J.; Kim, K.S.; Kim, T.H.; Kim, D.N. Rapid computational analysis of DNA origami assemblies at near-atomic resolution. ACS Nano 2021, 15, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Shen, J.; Ye, D.; Dong, B.; Wang, F.; Pei, H.; Wang, J.; Shi, J.; Wang, L.; Xue, W.; et al. Programming bulk enzyme heterojunctions for biosensor development with tetrahedral DNA framework. Nat. Commun. 2020, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu , M.; Deng , R.; Rizwan Khan , M.; Deng , S.; Wang , X.; Busquets , R.; Deng , W.; Luo , A. Amplification Refractory Mutation System (ARMS)-PCR for Waxy Sorghum Authentication with Single-Nucleotide Resolution. Foods 2021, 10, 2218. [Google Scholar]
- Wang, M.; Zhang, R.; Li, J. CRISPR/cas systems redefine nucleic acid detection: Principles and methods. Biosens Bioelectron. 2020, 165, 112430. [Google Scholar] [CrossRef]
- Yu, L.; Teng, F.; Jiang, M.; Guo, J. Establishment of a novel TB-ARMS qPCR method for kras mutation detection. Chin. J. Biotechnol. 2019, 35, 880–891. [Google Scholar]
- Yu, D.; Zhong, Q.; Xiao, Y.; Feng, Z.; Tang, F.; Feng, S.; Cai, Y.; Gao, Y.; Lan, T.; Li, M.; et al. Combination of MRI-based prediction and CRISPR/Cas12a-based detection for IDH genotyping in glioma. NPJ Precis. Oncol. 2024, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Xu, Z.; Zhou, H.; Li, T.; Du, X.; Hu, R.; Zhu, J.; Ou, G.; Li, Y.; Yang, Y. Integration of CRISPR/Cas12a and multiplexed RPA for fast detection of gene doping. Anal. Chem. 2022, 94, 16481–16490. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Ding, R.; Chen, S.; Duan, G. Advances in Field Detection Based on CRISPR/Cas System. ACS Synth. Biol. 2021, 10, 2824–2832. [Google Scholar] [CrossRef]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; Depristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′ → 3′) |
|---|---|
| PCR1-F | AACATTAAACAATCTGTAGTGCCGT |
| PCR1-R | CATAGCGGATCTAGACAGATGAGTT |
| ARMS1-F | GGTTCCGAGAGACAGAAATATATGA |
| ARMS1-R | TATTGTATTTGCATTTCCTGTCCCT |
| ARMS2-F | GGTTCCGAGAGACAGAAATATAACA |
| ARMS2-R | TATTGTATTTGCATTTCCTGTCCCT |
| ARMS3-F | GGTTCCGAGAGACAGAAATATATGG |
| ARMS3-R | TATTGTATTTGCATTTCCTGTCCCT |
| ARMS4-F | GGTTCCGAGAGACAGAAATATAACG |
| ARMS4-R | TATTGTATTTGCATTTCCTGTCCCT |
| Linker-HS-SH | HS-SH-CGATTGACTCTCTTTTTTTT |
| L1 | Cy5-GAGAGTCAATCGTTTTTTTTTCTATTCGCAGTAGAT-Cy5 |
| L2 | Cy5-GAGAGTCAATCGTTTTTTTTATCTACTGCGAATAGA-Cy5 |
| Y1 | Cy5-CGATTGACTCTCCACGCTGTCCTAACCATGACCGTCGAAG-Cy5 |
| Y2 | Cy5-CGATTGACTCTCCTTCGACGGTCATGTACTAGATCAGAGG-Cy5 |
| Y3 | Cy5-CGATTGACTCTCCCTCTGATCTAGTAGTTAGGACAGCGTG-Cy5 |
| crRNA | ATGCCTCATCAACACCGTCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, D.; Mijit, M.; Wang, H.; Sun, C.; Pingcuo, B.; Yu, Z.; Xiong, B.; Tang, X. Rapid Genotyping of FecB Mutation in Sheep Using CRISPR-Cas12a Integrated with DNA Nanotree Biosensing Platform. Biomolecules 2025, 15, 315. https://doi.org/10.3390/biom15030315
Pan D, Mijit M, Wang H, Sun C, Pingcuo B, Yu Z, Xiong B, Tang X. Rapid Genotyping of FecB Mutation in Sheep Using CRISPR-Cas12a Integrated with DNA Nanotree Biosensing Platform. Biomolecules. 2025; 15(3):315. https://doi.org/10.3390/biom15030315
Chicago/Turabian StylePan, Dongxia, Mukaddas Mijit, Hui Wang, Chaoqun Sun, Bantan Pingcuo, Zhixue Yu, Benhai Xiong, and Xiangfang Tang. 2025. "Rapid Genotyping of FecB Mutation in Sheep Using CRISPR-Cas12a Integrated with DNA Nanotree Biosensing Platform" Biomolecules 15, no. 3: 315. https://doi.org/10.3390/biom15030315
APA StylePan, D., Mijit, M., Wang, H., Sun, C., Pingcuo, B., Yu, Z., Xiong, B., & Tang, X. (2025). Rapid Genotyping of FecB Mutation in Sheep Using CRISPR-Cas12a Integrated with DNA Nanotree Biosensing Platform. Biomolecules, 15(3), 315. https://doi.org/10.3390/biom15030315