
ANT-Mediated Inhibition of the Permeability Transition Pore Alleviates Palmitate-Induced Mitochondrial Dysfunction and Lipotoxicity

 ,
,  , ,
, ,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. CRISPR/Cas9-Mediated Knockdown of the SLC25A5 Gene in HEK293T Cells
2.3. Modeling of Palmitate-Induced Lipotoxicity
2.4. Cell Viability Assessment
2.5. Mitochondrial Function Assay
2.6. Electrophoresis and Immunoblotting
2.7. Statistical Data Processing
3. Results
3.1. The Effect of Bongkrekic Acid and Carboxyatractyloside on the Viability of Mouse Lung Endothelial Cells under Conditions of Normal and Dyslipidemia
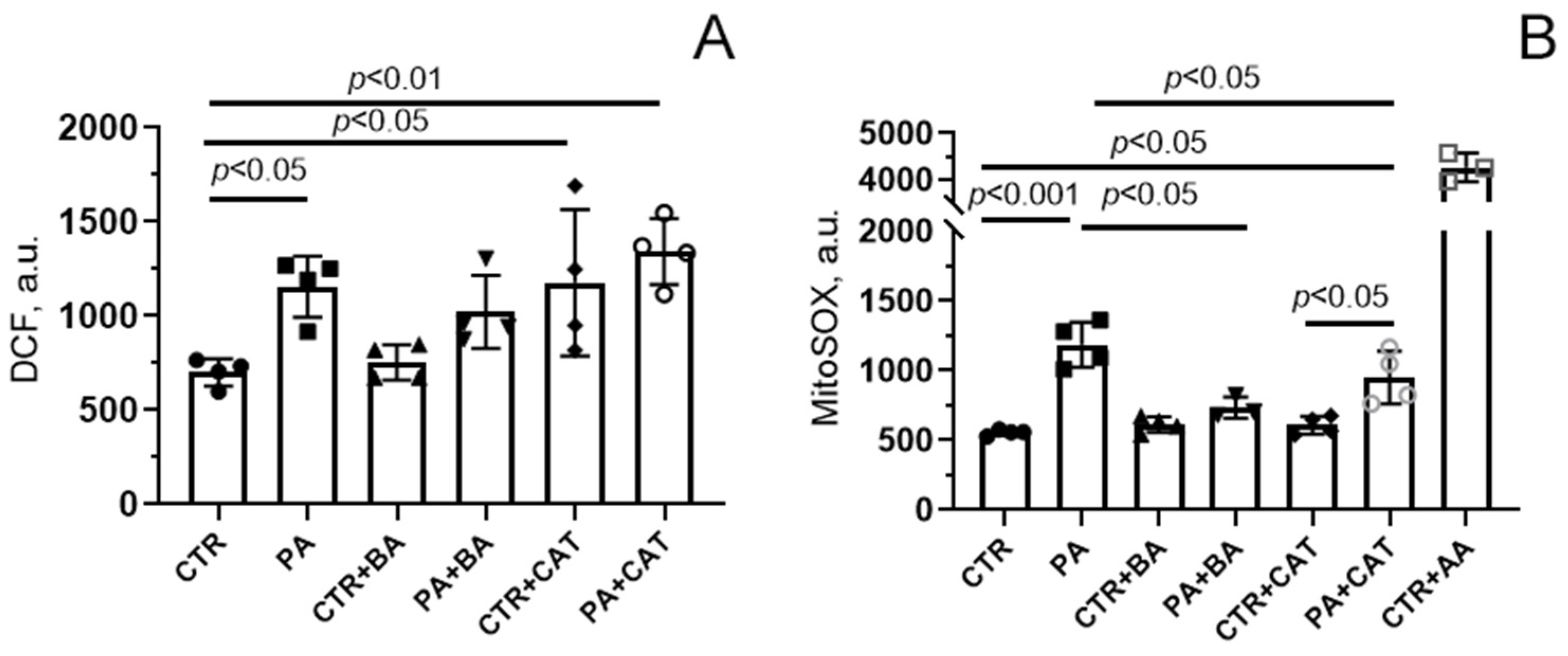
3.2. Effect of Bongkrekic Acid and Carboxyatractyloside on the Development of Oxidative Stress in PA-Induced Lipotoxity
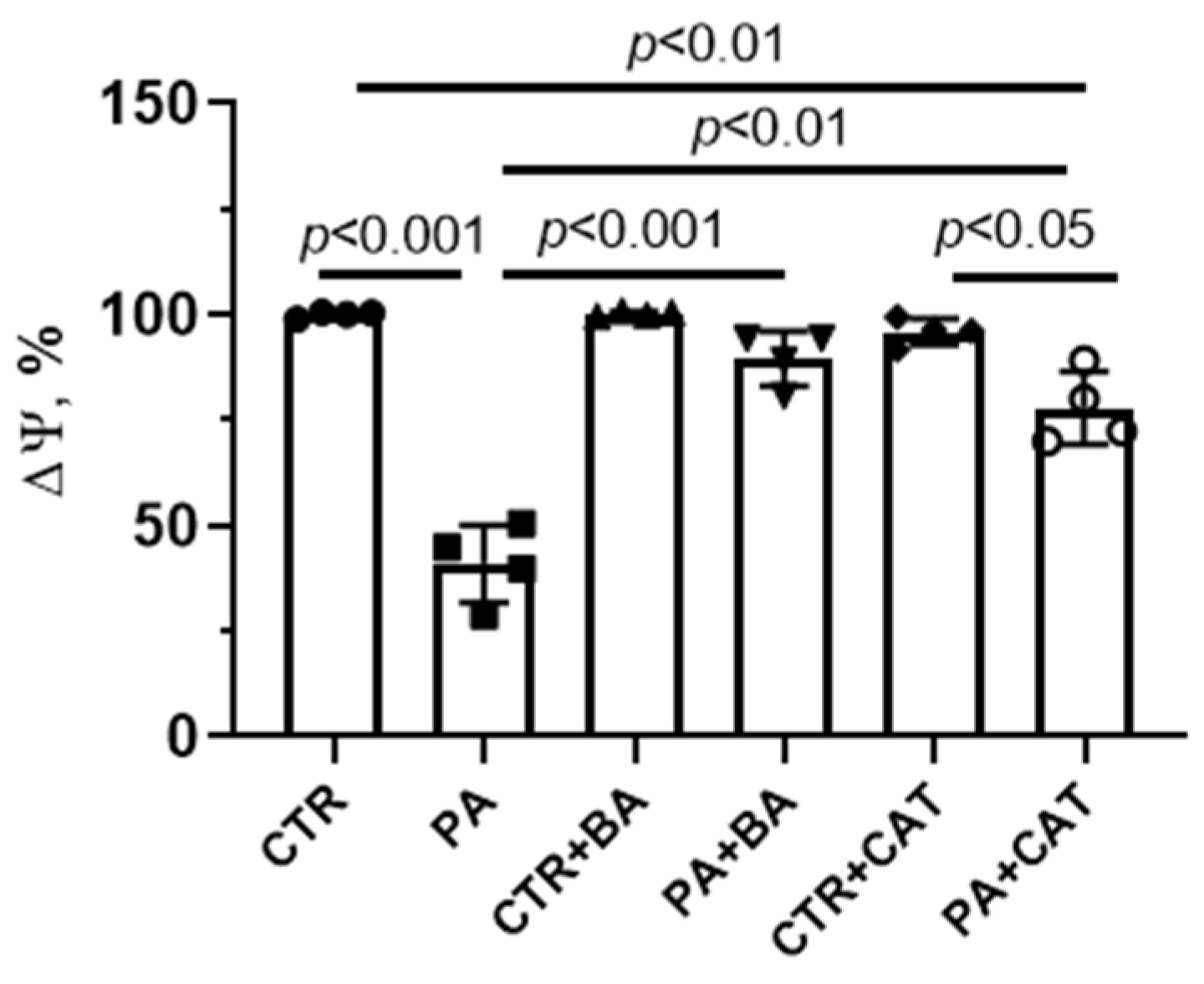
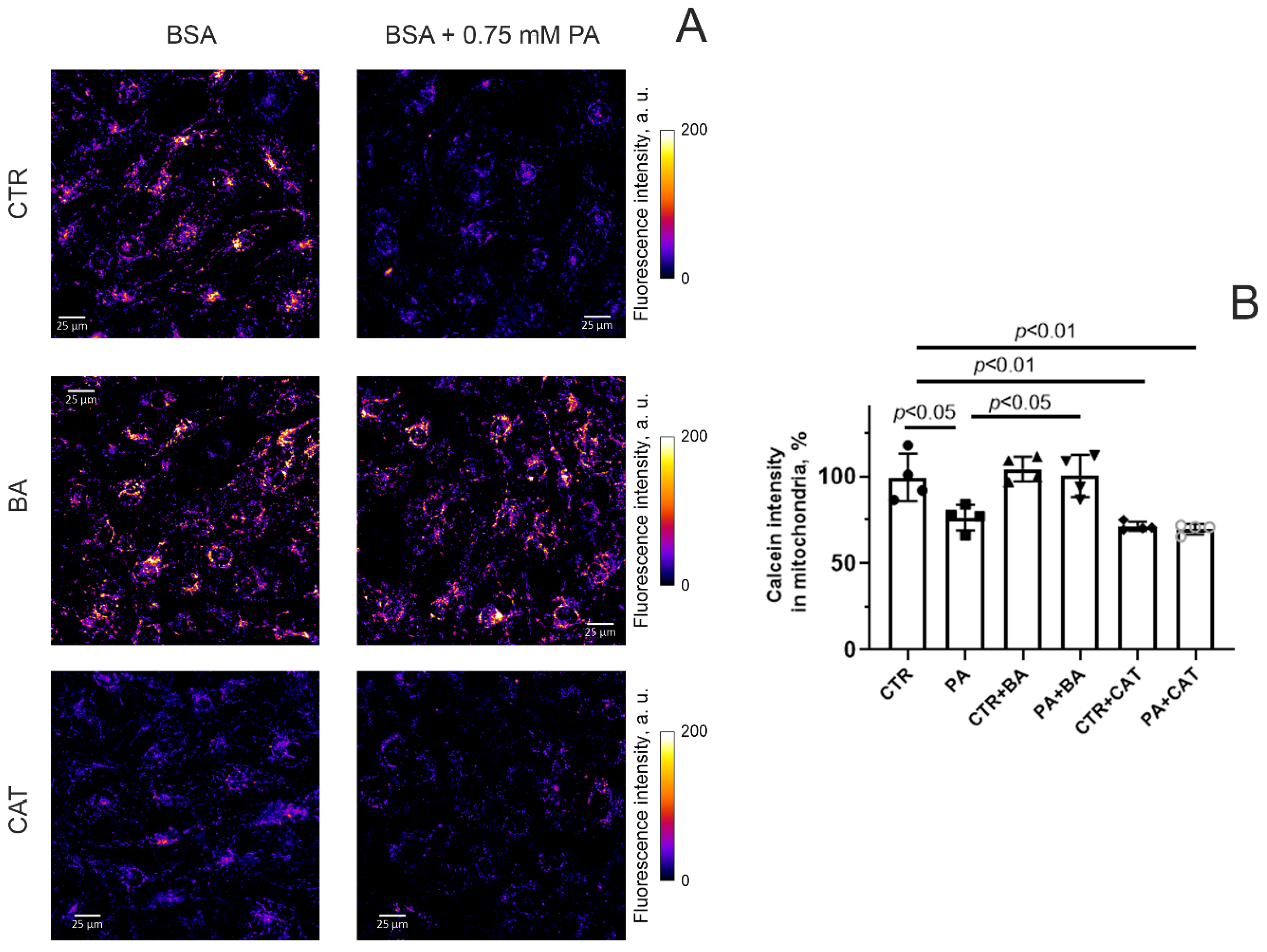
3.3. Effect of Bongkrekic Acid and Carboxyatractyloside on the Development of Mitochondrial Dysfunction in PA-Induced Lipotoxity
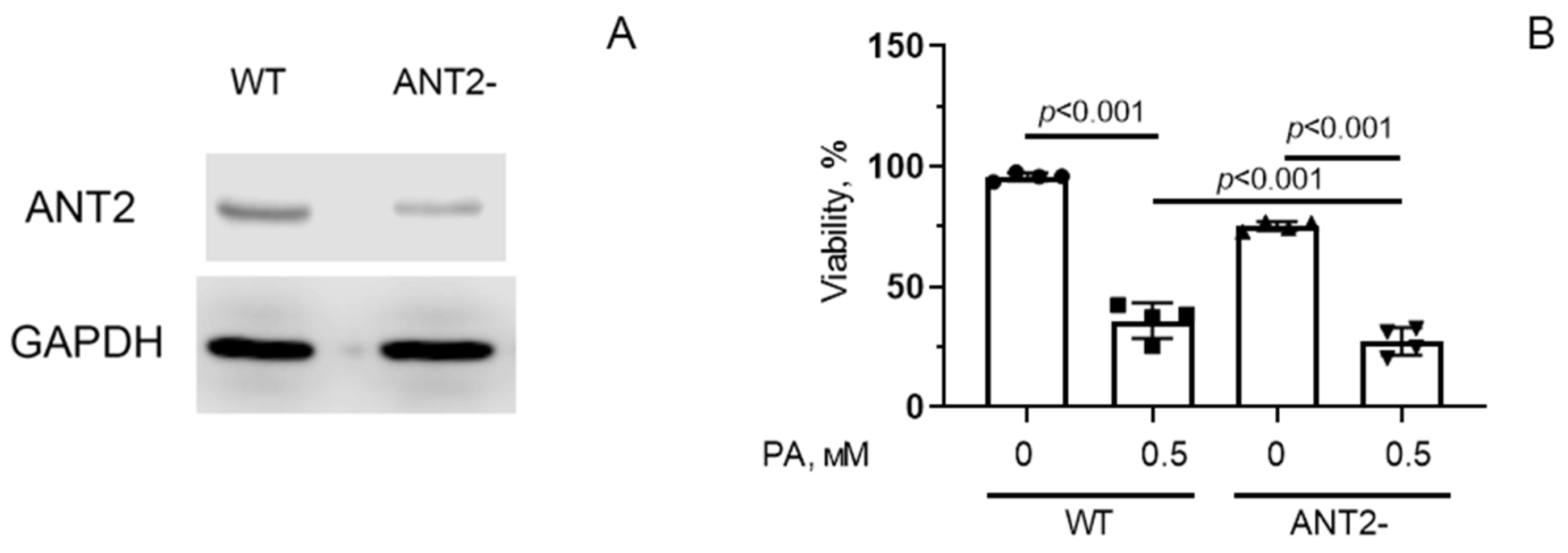
3.4. Suppression of ANT2 Expression Enhances the Development of Oxidative Stress and Death of HEK293T Cells under Conditions of Hyperlipidemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association Professional Practice Committee. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S17–S38. [Google Scholar] [CrossRef]
- Henderson, G.C. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients 2021, 13, 2590. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef]
- Kalinovich, A.V.; Mattsson, C.L.; Youssef, M.R.; Petrovic, N.; Ost, M.; Skulachev, V.P.; Shabalina, I.G. Mitochondria-targeted dodecyltriphenylphosphonium (C12TPP) combats high-fat-diet-induced obesity in mice. Int. J. Obes. 2016, 40, 1864–1874. [Google Scholar] [CrossRef]
- Samsonov, M.V.; Podkuychenko, N.V.; Khapchaev, A.Y.; Efremov, E.E.; Yanushevskaya, E.V.; Vlasik, T.N.; Lankin, V.Z.; Stafeev, I.S.; Skulachev, M.V.; Shestakova, M.V.; et al. AICAR protects vascular endothelial cells from oxidative injury induced by the long-term palmitate excess. Int. J. Mol. Sci. 2021, 23, 211. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Lin, J.; Duan, J.; Wang, Q.; Xu, S.; Zhou, S.; Yao, K. Mitochondrial Dynamics and Mitophagy in Cardiometabolic Disease. Front. Cardiovasc. Med. 2022, 9, 917135. [Google Scholar] [CrossRef]
- Starinets, V.S.; Serov, D.A.; Penkov, N.V.; Belosludtseva, N.V.; Dubinin, M.V.; Belosludtsev, K.N. Alisporivir Normalizes Mitochondrial Function of Primary Mouse Lung Endothelial Cells Under Conditions of Hyperglycemia. Biochemistry (Moscow) 2022, 87, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtseva, N.V. Alisporivir Treatment Alleviates Mitochondrial Dysfunction in the Skeletal Muscles of C57BL/6NCrl Mice with High-Fat Diet/Streptozotocin-Induced Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 9524. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry (Moscow) 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.S.; Soukas, A.A. Identity, structure, and function of the mitochondrial permeability transition pore: Controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef]
- Carraro, M.; Bernardi, P. The mitochondrial permeability transition pore in Ca2+ homeostasis. Cell Calcium 2023, 111, 102719. [Google Scholar] [CrossRef]
- Mironova, G.D.; Pavlov, E.V. Mitochondrial Cyclosporine A-Independent Palmitate/Ca2+-Induced Permeability Transition Pore (PA-mPT Pore) and Its Role in Mitochondrial Function and Protection against Calcium Overload and Glutamate Toxicity. Cells 2021, 10, 125. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell. 2017, 16, 943–955. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Pavlov, E.V.; Sheu, S.S. Electrophysiological properties of the mitochondrial permeability transition pores: Channel diversity and disease implication. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148357. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, L.; Liu, J.; Ma, L.; Zhang, W. Adenine nucleotide translocase: Current knowledge in post-translational modifications, regulations and pathological implications for human diseases. FASEB J. 2023, 37, e22953. [Google Scholar] [CrossRef]
- Maldonado, E.N.; DeHart, D.N.; Patnaik, J.; Klatt, S.C.; Gooz, M.B.; Lemasters, J.J. ATP/ADP Turnover and Import of Glycolytic ATP into Mitochondria in Cancer Cells Is Independent of the Adenine Nucleotide Translocator. J. Biol. Chem. 2016, 291, 19642–19650. [Google Scholar] [CrossRef] [PubMed]
- Permyakova, A.; Hamad, S.; Hinden, L.; Baraghithy, S.; Kogot-Levin, A.; Yosef, O.; Shalev, O.; Tripathi, M.K.; Amal, H.; Basu, A.; et al. Renal Mitochondrial ATP Transporter Ablation Ameliorates Obesity-Induced CKD. J. Am. Soc. Nephrol. 2024, 35, 281–298. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Flierl, A.; Sweeney, K.M.; Angelin, A.; MacGregor, G.R.; Wallace, D.C. Deficiency in the mouse mitochondrial adenine nucleotide translocator isoform 2 gene is associated with cardiac noncompaction. Biochim. Biophys. Acta 2016, 1857, 1203–1212. [Google Scholar] [CrossRef]
- Henderson, P.J.; Lardy, H.A. Bongkrekic acid. An inhibitor of the adenine nucleotide translocase of mitochondria. J. Biol. Chem. 1970, 245, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liang, Z.; Li, Y.; Wen, J.; Zhang, R. Molecular docking and molecular dynamics simulation study on the toxicity mechanism of bongkrekic acid. Toxicon 2023, 223, 107021. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, J.; Škulj, S.; Brkljača, Z.; Bardakji, S.; Vazdar, M.; Pohl, E.E. FA Sliding as the Mechanism for the ANT1-Mediated Fatty Acid Anion Transport in Lipid Bilayers. Int. J. Mol. Sci. 2023, 24, 13701. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar] [CrossRef]
- Huo, H.; Zhou, Z.; Qin, J.; Liu, W.; Wang, B.; Gu, Y. Erastin Disrupts Mitochondrial Permeability Transition Pore (mPTP) and Induces Apoptotic Death of Colorectal Cancer Cells. PLoS ONE 2016, 11, e0154605. [Google Scholar] [CrossRef]
- Ren, Z.; Chen, S.; Seo, J.E.; Guo, X.; Li, D.; Ning, B.; Guo, L. Mitochondrial dysfunction and apoptosis underlie the hepatotoxicity of perhexiline. Toxicol. In Vitro 2020, 69, 104987. [Google Scholar] [CrossRef]
- Stoica, B.A.; Movsesyan, V.A.; Lea, P.M.; Faden, A.I. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol. Cell Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Ali, A.; Katare, R. Molecular complexities underlying the vascular complications of diabetes mellitus—A comprehensive review. J. Diabetes Complicat. 2020, 34, 107613. [Google Scholar] [CrossRef]
- Yamagata, K. Fatty acids act on vascular endothelial cells and influence the development of cardiovascular disease. Prostaglandins Other Lipid Mediat. 2023, 165, 106704. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: Molecular mechanisms and therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 152. [Google Scholar] [CrossRef]
- Vorotnikov, A.V.; Khapchaev, A.Y.; Nickashin, A.V.; Shirinsky, V.P. In Vitro Modeling of Diabetes Impact on Vascular Endothelium: Are Essentials Engaged to Tune Metabolism? Biomedicines 2022, 10, 3181. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Serov, D.A.; Starinets, V.S.; Penkov, N.V.; Belosludtsev, K.N. Alterations in Mitochondrial Morphology and Quality Control in Primary Mouse Lung Microvascular Endothelial Cells and Human Dermal Fibroblasts under Hyperglycemic Conditions. Int. J. Mol. Sci. 2023, 24, 12485. [Google Scholar] [CrossRef]
- Karagyaur, M.N.; Rubtsov, Y.P.; Vasiliev, P.A.; Tkachuk, V.A. Practical Recommendations for Improving Efficiency and Accuracy of the CRISPR/Cas9 Genome Editing System. Biochemistry (Moscow) 2018, 83, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol. Ther. Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef]
- Longo, P.A.; Kavran, J.M.; Kim, M.-S.; Leahy, D.J. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [Google Scholar] [CrossRef]
- Dyikanov, D.T.; Vasiluev, P.A.; Rysenkova, K.D.; Aleksandrushkina, N.A.; Tyurin-Kuzmin, P.A.; Kulebyakin, K.Y.; Rubtsov, Y.P.; Shmakova, A.A.; Evseeva, M.N.; Balatskiy, A.V.; et al. Optimization of CRISPR/Cas9 Technology to Knock Out Genes of Interest in Aneuploid Cell Lines. Tissue Eng. Part. C Methods 2019, 25, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Khapchaev, A.Y.; Vorotnikov, A.V.; Antonova, O.A.; Samsonov, M.V.; Shestakova, E.A.; Sklyanik, I.A.; Tomilova, A.O.; Shestakova, M.V.; Shirinsky, V.P. Shear Stress and the AMP-Activated Protein Kinase Independently Protect the Vascular Endothelium from Palmitate Lipotoxicity. Biomedicines 2024, 12, 339. [Google Scholar] [CrossRef]
- Creed, S.; McKenzie, M. Measurement of Mitochondrial Membrane Potential with the Fluorescent Dye Tetramethylrhodamine Methyl Ester (TMRM). Methods Mol. Biol. 2019, 1928, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Serov, D.A.; Ilzorkina, A.I.; Starinets, V.S.; Dubinin, M.V.; Talanov, E.Y.; Karagyaur, M.N.; Primak, A.L.; Belosludtseva, N.V. Pharmacological and Genetic Suppression of VDAC1 Alleviates the Development of Mitochondrial Dysfunction in Endothelial and Fibroblast Cell Cultures upon Hyperglycemic Conditions. Antioxidants 2023, 12, 1459. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxid. Med. Cell Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef]
- Kong, J.Y.; Rabkin, S.W. Palmitate-induced apoptosis in cardiomyocytes is mediated through alterations in mitochondria: Prevention by cyclosporin A. Biochim. Biophys. Acta 2000, 1485, 45–55. [Google Scholar] [CrossRef]
- Ortiz-Rodriguez, A.; Acaz-Fonseca, E.; Boya, P.; Arevalo, M.A.; Garcia-Segura, L.M. Lipotoxic Effects of Palmitic Acid on Astrocytes Are Associated with Autophagy Impairment. Mol. Neurobiol. 2019, 56, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.O.; Blanco, A.; Lima, S.V.; Mancini, G.; Mendes, N.F.; Latini, A.; Gaspar, J.M. Palmitate Compromises C6 Astrocytic Cell Viability and Mitochondrial Function. Metabolites 2024, 14, 161. [Google Scholar] [CrossRef]
- Sparagna, G.C.; Hickson-Bick, D.L.; Buja, L.M.; McMillin, J.B. A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2124–H2132. [Google Scholar] [CrossRef]
- Hardy, S.; El-Assaad, W.; Przybytkowski, E.; Joly, E.; Prentki, M.; Langelier, Y. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. A role for cardiolipin. J. Biol. Chem. 2003, 278, 31861–31870. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, E.P.; Laker, R.C.; Breen, D.S.; Akhtar, Y.N.; Kenwood, B.M.; Liao, J.A.; Zhang, M.; Fazakerley, D.J.; Tomsig, J.L.; Harris, T.E.; et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2013, 3, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Bozi, L.H.M.; Campos, J.C.; Zambelli, V.O.; Ferreira, N.D.; Ferreira, J.C.B. Mitochondrially-targeted treatment strategies. Mol. Aspects Med. 2020, 71, 100836. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in β cells. Cell Metab. 2019, 29, 64–77. [Google Scholar] [CrossRef]
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Cell Physiol. 2012, 303, C1055–C1060. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Y.; Ge, W.; Zhou, X.; Pan, M. High glucose induces the apoptosis of HUVECs in mitochondria dependent manner by enhancing VDAC1 expression. Pharmazie 2018, 73, 725–728. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Dubinin, M.V.; Belosludtsev, K.N. Pore-Forming VDAC Proteins of the Outer Mitochondrial Membrane: Regulation and Pathophysiological Role. Biochemistry (Moscow) 2024, 89, 1061–1078. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D.; Keinan, N. Oligomerization of the mitochondrial protein VDAC1: From structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 2013, 117, 303–334. [Google Scholar] [CrossRef]
- Heslop, K.A.; Milesi, V.; Maldonado, E.N. VDAC Modulation of Cancer Metabolism: Advances and Therapeutic Challenges. Front. Physiol. 2021, 12, 742839. [Google Scholar] [CrossRef]
- Anwar, M.; Kasper, A.; Steck, A.R.; Schier, J.G. Bongkrekic Acid-a Review of a Lesser-Known Mitochondrial Toxin. J. Med. Toxicol. 2017, 13, 173–179. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Amplicon Length, bp | Tannealing, °C |
|---|---|---|---|
| SLC25A5-gRNA1 | TGCAAAGTAGAGCCAAAACT | - | - |
| SLC25A5-gRNA2 | TGGCATCGGGTGGTGCCGCA | ||
| SLC25A5-test-f | AGGGTCTGAAGGTCACACGGGT | 822 | 58 |
| SLC25A5-test-r | GGACTGTTAGGTTGGTTGGTACAATGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtseva, N.V.; Ilzorkina, A.I.; Serov, D.A.; Dubinin, M.V.; Talanov, E.Y.; Karagyaur, M.N.; Primak, A.L.; Liu, J.; Belosludtsev, K.N. ANT-Mediated Inhibition of the Permeability Transition Pore Alleviates Palmitate-Induced Mitochondrial Dysfunction and Lipotoxicity. Biomolecules 2024, 14, 1159. https://doi.org/10.3390/biom14091159
Belosludtseva NV, Ilzorkina AI, Serov DA, Dubinin MV, Talanov EY, Karagyaur MN, Primak AL, Liu J, Belosludtsev KN. ANT-Mediated Inhibition of the Permeability Transition Pore Alleviates Palmitate-Induced Mitochondrial Dysfunction and Lipotoxicity. Biomolecules. 2024; 14(9):1159. https://doi.org/10.3390/biom14091159
Chicago/Turabian StyleBelosludtseva, Natalia V., Anna I. Ilzorkina, Dmitriy A. Serov, Mikhail V. Dubinin, Eugeny Yu. Talanov, Maxim N. Karagyaur, Alexandra L. Primak, Jiankang Liu, and Konstantin N. Belosludtsev. 2024. "ANT-Mediated Inhibition of the Permeability Transition Pore Alleviates Palmitate-Induced Mitochondrial Dysfunction and Lipotoxicity" Biomolecules 14, no. 9: 1159. https://doi.org/10.3390/biom14091159
APA StyleBelosludtseva, N. V., Ilzorkina, A. I., Serov, D. A., Dubinin, M. V., Talanov, E. Y., Karagyaur, M. N., Primak, A. L., Liu, J., & Belosludtsev, K. N. (2024). ANT-Mediated Inhibition of the Permeability Transition Pore Alleviates Palmitate-Induced Mitochondrial Dysfunction and Lipotoxicity. Biomolecules, 14(9), 1159. https://doi.org/10.3390/biom14091159