
GateView: A Multi-Omics Platform for Gene Feature Analysis of Virus Receptors within Human Normal Tissues and Tumors

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Data Processing, Statistics and Web Implementation
2.2.1. Analysis of Receptor Expression Levels
2.2.2. Single-Cell Sequencing Data Analysis
2.2.3. Evolutionary Conservation Analysis and Phylogenetic Analyses
2.2.4. Cancer Data Analysis
2.2.5. Database Architecture
3. Results
3.1. The Main Features of GateView: Using ACE2 as an Example
3.1.1. GateView Demostrates the Correlation between Viruses and Receptor Molecules
3.1.2. Multiple Types of DNA Variations in ACE2 Gene across Different Cancer Types
3.1.3. ACE2 Expression Exhibits Gender or Age Differences in Specific Tissues
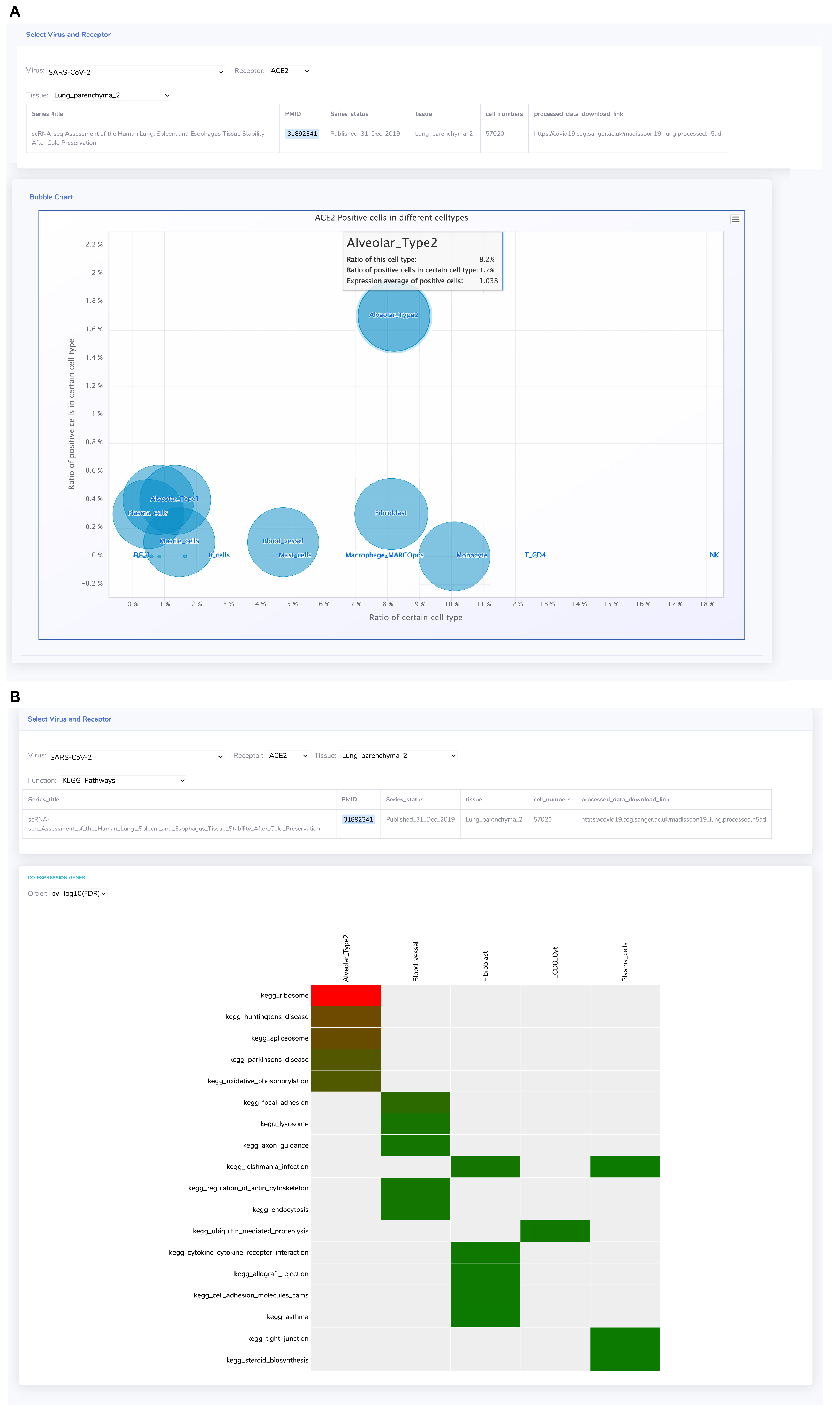
3.1.4. ACE2 Displays Distinct Co-Expression Patterns in Diverse Cell Types
3.2. Comprehensive Analysis of Diverse Human Virus Receptor Molecules
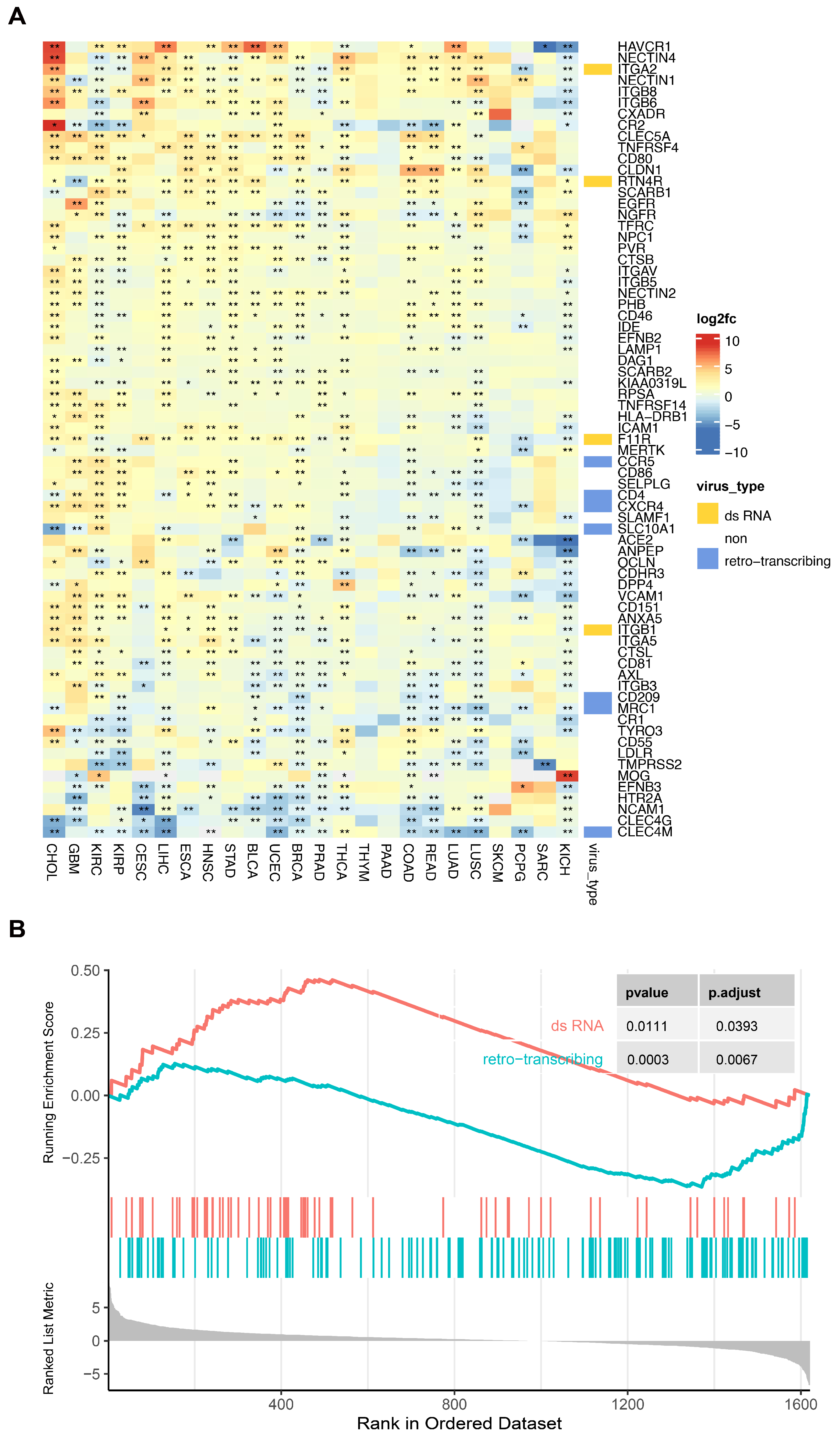
3.2.1. Comparative Analysis of Virus Receptors and Other Membrane Proteins
3.2.2. Receptor Molecules Demonstrate Pronounced Tissue Specificity
3.2.3. Receptor Molecule Expression Levels Differ among Populations
3.2.4. Receptor Molecule Expression Levels Are Generally Dysregulated in Cancer
4. Discussion
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mei, M.; Ye, J.; Qin, A.; Wang, L.; Hu, X.; Qian, K.; Shao, H. Identification of novel viral receptors with cell line expressing viral receptor-binding protein. Sci. Rep. 2015, 5, 7935. [Google Scholar] [CrossRef]
- Claireaux, M.; Robinot, R.; Kervevan, J.; Patgaonkar, M.; Staropoli, I.; Brelot, A.; Nouël, A.; Gellenoncourt, S.; Tang, X.; Héry, M.; et al. Low CCR5 expression protects HIV-specific CD4+ T cells of elite controllers from viral entry. Nat. Commun. 2022, 13, 521. [Google Scholar] [CrossRef]
- Freire, J.M.; Santos, N.C.; Veiga, A.S.; Da Poian, A.T.; Castanho, M.A. Rethinking the capsid proteins of enveloped viruses: Multifunctionality from genome packaging to genome transfection. FEBS J. 2015, 282, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Louten, J. Virus Structure and Classification. Essent. Human. Virol. 2016, 19–29. [Google Scholar] [CrossRef]
- Zhuang, M.W.; Cheng, Y.; Zhang, J.; Jiang, X.M.; Wang, L.; Deng, J.; Wang, P.A.-O. Increasing host cellular receptor-angiotensin-converting enzyme 2 expression by coronavirus may facilitate 2019-nCoV (or SARS-CoV-2) infection. J. Med. Virol. 2020, 92, 2693–2701. [Google Scholar] [CrossRef]
- Ying, K.A.-O.; Zhai, R.A.-O.; Pyrkov, T.A.-O.; Shindyapina, A.V.; Mariotti, M.; Fedichev, P.O.; Shen, X.A.-O.; Gladyshev, V.A.-O. Genetic and phenotypic analysis of the causal relationship between aging and COVID-19. Commun. Med. 2021, 1, 35. [Google Scholar] [CrossRef]
- Yang, Z.A.-O.; Macdonald-Dunlop, E.; Chen, J.A.-O.; Zhai, R.A.-O.; Li, T.; Richmond, A.; Klarić, L.A.-O.; Pirastu, N.A.-O.; Ning, Z.A.-O.; Zheng, C.; et al. Genetic Landscape of the ACE2 Coronavirus Receptor. Circulation 2022, 145, 1398–1411. [Google Scholar] [CrossRef]
- Kousathanas, A.; Pairo-Castineira, E.; Rawlik, K.; Stuckey, A.; Odhams, C.A.; Walker, S.; Russell, C.D.; Malinauskas, T.; Wu, Y.; Millar, J.; et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 2022, 607, 97–103. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Xu, D.; Xiao, L.; Qin, W.; Liu, B.; Yuan, X. Characterization of the SARS-CoV-2 co-receptor NRP1 expression profiles in healthy people and cancer patients: Implication for susceptibility to COVID-19 disease and potential therapeutic strategy. Front. Genet. 2022, 13, 995736. [Google Scholar] [CrossRef]
- Muus, C.A.-O.; Luecken, M.A.-O.; Eraslan, G.; Sikkema, L.A.-O.; Waghray, A.; Heimberg, G.; Kobayashi, Y.; Vaishnav, E.A.-O.; Subramanian, A.A.-O.; Smillie, C.; et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 2021, 27, 546–559. [Google Scholar] [CrossRef]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, Z.; Chen, W.; Cai, Z.; Xu, B.; Tan, Z.; Wu, A.; Ge, X.; Guo, X.; Tan, Z.; et al. Cell membrane proteins with high N-glycosylation, high expression and multiple interaction partners are preferred by mammalian viruses as receptors. Bioinformatics 2019, 35, 723–728. [Google Scholar] [CrossRef]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Rockweiler, N.B.; Ramu, A.; Nagirnaja, L.; Wong, W.H.; Noordam, M.J.; Drubin, C.W.; Huang, N.; Miller, B.; Todres, E.Z.; Vigh-Conrad, K.A.; et al. The origins and functional effects of postzygotic mutations throughout the human life span. Science 2023, 380, eabn7113. [Google Scholar] [CrossRef]
- Gruber, M.P.; Coldren, C.D.; Woolum, M.D.; Cosgrove, G.P.; Zeng, C.; Baron, A.E.; Moore, M.D.; Cool, C.D.; Worthen, G.S.; Brown, K.K.; et al. Human lung project: Evaluating variance of gene expression in the human lung. Am. J. Respir. Cell Mol. Biol. 2006, 35, 65–71. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Mikheev, A.M.; Nabekura, T.; Kaddoumi, A.; Bammler, T.K.; Govindarajan, R.; Hebert, M.F.; Unadkat, J.D. Profiling gene expression in human placentae of different gestational ages: An OPRU Network and UW SCOR Study. Reprod. Sci. 2008, 15, 866–877. [Google Scholar] [CrossRef]
- Uuskula, L.; Mannik, J.; Rull, K.; Minajeva, A.; Koks, S.; Vaas, P.; Teesalu, P.; Reimand, J.; Laan, M. Mid-gestational gene expression profile in placenta and link to pregnancy complications. PLoS ONE 2012, 7, e49248. [Google Scholar] [CrossRef]
- Rull, K.; Tomberg, K.; Koks, S.; Mannik, J.; Mols, M.; Sirotkina, M.; Varv, S.; Laan, M. Increased placental expression and maternal serum levels of apoptosis-inducing TRAIL in recurrent miscarriage. Placenta 2013, 34, 141–148. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Xuan, J.J.; Sun, W.J.; Lin, P.H.; Zhou, K.R.; Liu, S.; Zheng, L.L.; Qu, L.H.; Yang, J.H. RMBase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018, 46, D327–D334. [Google Scholar] [CrossRef]
- Monnerat, G.; Maior, A.S.; Tannure, M.; Back, L.; Santos, C.G.M. Single-Nucleotide-Polymorphism-Panel Population-Genetics Approach Based on the 1000 Genomes Database and Elite Soccer Players. Int. J. Sports Physiol. Perform. 2019, 14, 711–717. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.H.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell. 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, Y.; Tian, X.; Bai, Z.; Gong, Y.; Qi, J.; Liu, D.; Liu, W.; Li, J. Analysis of ACE2 Gene-Encoded Proteins Across Mammalian Species. Front. Vet. Sci. 2020, 7, 457. [Google Scholar] [CrossRef]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef]
- Madissoon, E.; Wilbrey-Clark, A.; Miragaia, R.J.; Saeb-Parsy, K.; Mahbubani, K.T.; Georgakopoulos, N.; Harding, P.; Polanski, K.; Huang, N.; Nowicki-Osuch, K.; et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019, 21, 1. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef]
- Edenfield, R.C.; Easley, C.A. Implications of testicular ACE2 and the renin–angiotensin system for SARS-CoV-2 on testis function. Nat. Rev. Urol. 2022, 19, 116–127. [Google Scholar] [CrossRef]
- Canas, L.S.; Sudre, C.H.; Capdevila Pujol, J.; Polidori, L.; Murray, B.; Molteni, E.; Graham, M.S.; Klaser, K.; Antonelli, M.; Berry, S.; et al. Early detection of COVID-19 in the UK using self-reported symptoms: A large-scale, prospective, epidemiological surveillance study. Lancet Digit. Health 2021, 3, e587–e598. [Google Scholar] [CrossRef]
- Xie, X.; Chen, J.; Wang, X.; Zhang, F.; Liu, Y. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006, 78, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 2020, 19, e13168. [Google Scholar] [CrossRef]
- Goldstein, E.; Lipsitch, M.; Cevik, M. On the Effect of Age on the Transmission of SARS-CoV-2 in Households, Schools, and the Community. J. Infect. Dis. 2021, 223, 362–369. [Google Scholar] [CrossRef]
- Demogines, A.; Abraham, J.; Choe, H.; Farzan, M.; Sawyer, S.L. Dual host-virus arms races shape an essential housekeeping protein. PLoS Biol. 2013, 11, e1001571. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. Virions at the gates: Receptors and the host-virus arms race. PLoS Biol. 2013, 11, e1001574. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, H.; Han, G.Z. Host-Virus Arms Races Drive Elevated Adaptive Evolution in Viral Receptors. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Jiang, B.; Qiu, M.; Qin, L.; Tang, J.; Zhan, S.; Lin, Q.; Wei, J.; Liu, Y.; Zhou, Z.; Liang, X.; et al. Associations between genetic variants in sphingolipid metabolism pathway genes and hepatitis B virus-related hepatocellular carcinoma survival. Front. Oncol. 2023, 13, 1252158. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Montemurro, N.; Pahwa, B.; Tayal, A.; Shukla, A.; De Jesus Encarnacion, M.; Ramirez, I.; Nurmukhametov, R.; Chavda, V.; De Carlo, A. Macrophages in Recurrent Glioblastoma as a Prognostic Factor in the Synergistic System of the Tumor Microenvironment. Neurol. Int. 2023, 15, 595–608. [Google Scholar] [CrossRef]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, G. Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun. 2021, 27, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood—Brain Barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cai, Z.; Mao, L.; Ge, X.-Y.; Peng, Y. EVIHVR: A platform for analysis of expression, variation and identification of human virus receptors. Infect. Med. 2022, 1, 59–62. [Google Scholar] [CrossRef]
- Chen, D.; Tan, C.; Ding, P.; Luo, L.; Zhu, J.; Jiang, X.; Ou, Z.; Ding, X.; Lan, T.; Zhu, Y.; et al. VThunter: A database for single-cell screening of virus target cells in the animal kingdom. Nucleic Acids Res. 2022, 50, D934–D942. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Huang, Z.-L.; Chen, W.-X.; Zhang, Y.-F.; Lei, H.-T.; Huang, Q.-J.; Lun, Z.-R.; Qu, L.-H.; Zheng, L.-L. GateView: A Multi-Omics Platform for Gene Feature Analysis of Virus Receptors within Human Normal Tissues and Tumors. Biomolecules 2024, 14, 516. https://doi.org/10.3390/biom14050516
Sun Y, Huang Z-L, Chen W-X, Zhang Y-F, Lei H-T, Huang Q-J, Lun Z-R, Qu L-H, Zheng L-L. GateView: A Multi-Omics Platform for Gene Feature Analysis of Virus Receptors within Human Normal Tissues and Tumors. Biomolecules. 2024; 14(5):516. https://doi.org/10.3390/biom14050516
Chicago/Turabian StyleSun, Yang, Zi-Liang Huang, Wen-Xin Chen, Yi-Feng Zhang, Hao-Tian Lei, Qiao-Juan Huang, Zhao-Rong Lun, Liang-Hu Qu, and Ling-Ling Zheng. 2024. "GateView: A Multi-Omics Platform for Gene Feature Analysis of Virus Receptors within Human Normal Tissues and Tumors" Biomolecules 14, no. 5: 516. https://doi.org/10.3390/biom14050516
APA StyleSun, Y., Huang, Z.-L., Chen, W.-X., Zhang, Y.-F., Lei, H.-T., Huang, Q.-J., Lun, Z.-R., Qu, L.-H., & Zheng, L.-L. (2024). GateView: A Multi-Omics Platform for Gene Feature Analysis of Virus Receptors within Human Normal Tissues and Tumors. Biomolecules, 14(5), 516. https://doi.org/10.3390/biom14050516