
Mitochondrial Transplantation Therapy Ameliorates Muscular Dystrophy in mdx Mouse Model

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
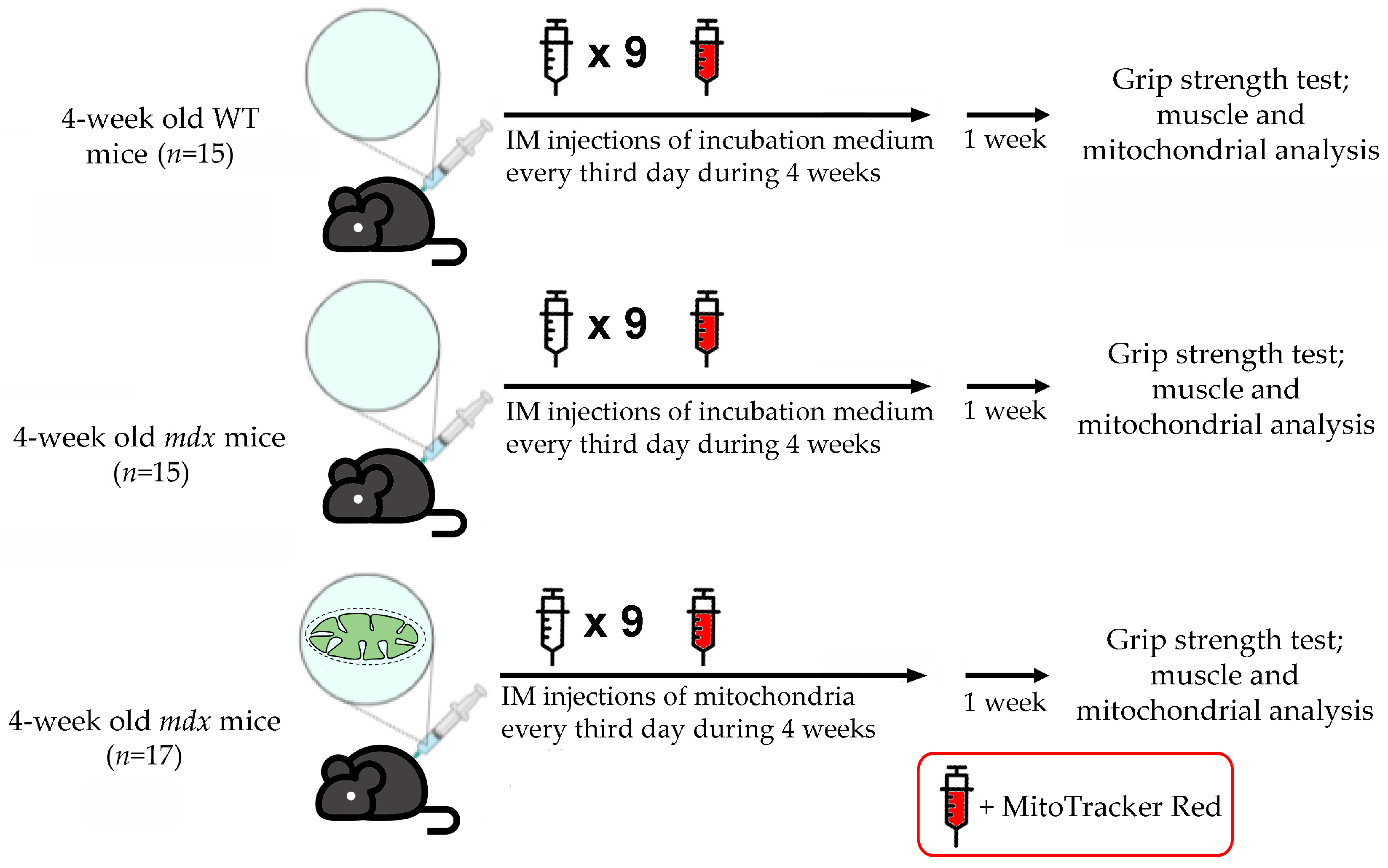
2.1. Animals
2.2. Isolation of Mitochondria from Skeletal Muscles of Mice and Functional Studies
2.3. Administration of WT Mouse Skeletal Muscle Mitochondria to mdx Animals and Labeling Them with MitoTracker Red Chloromethyl-X-Rosamine (CMXRos)
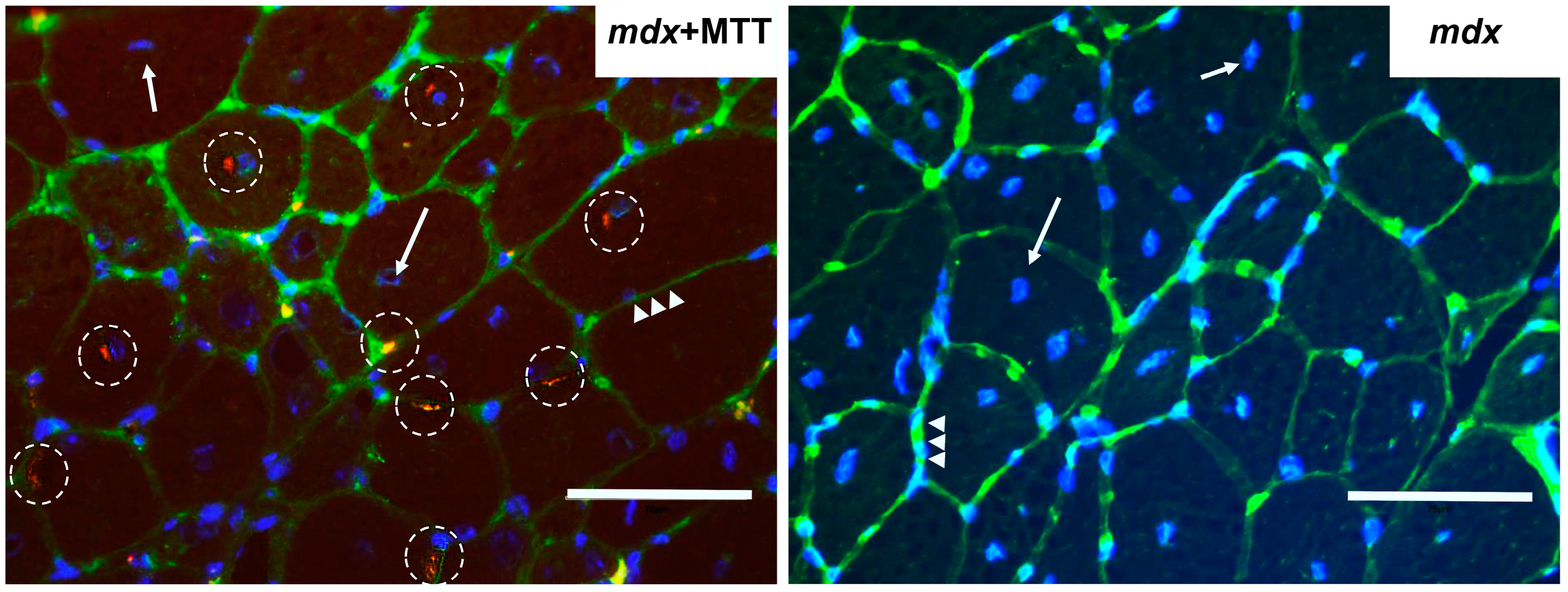
2.4. Imaging of Mitochondrial Transplantation
2.5. Grip Strength Test and Wheel-Running Activity
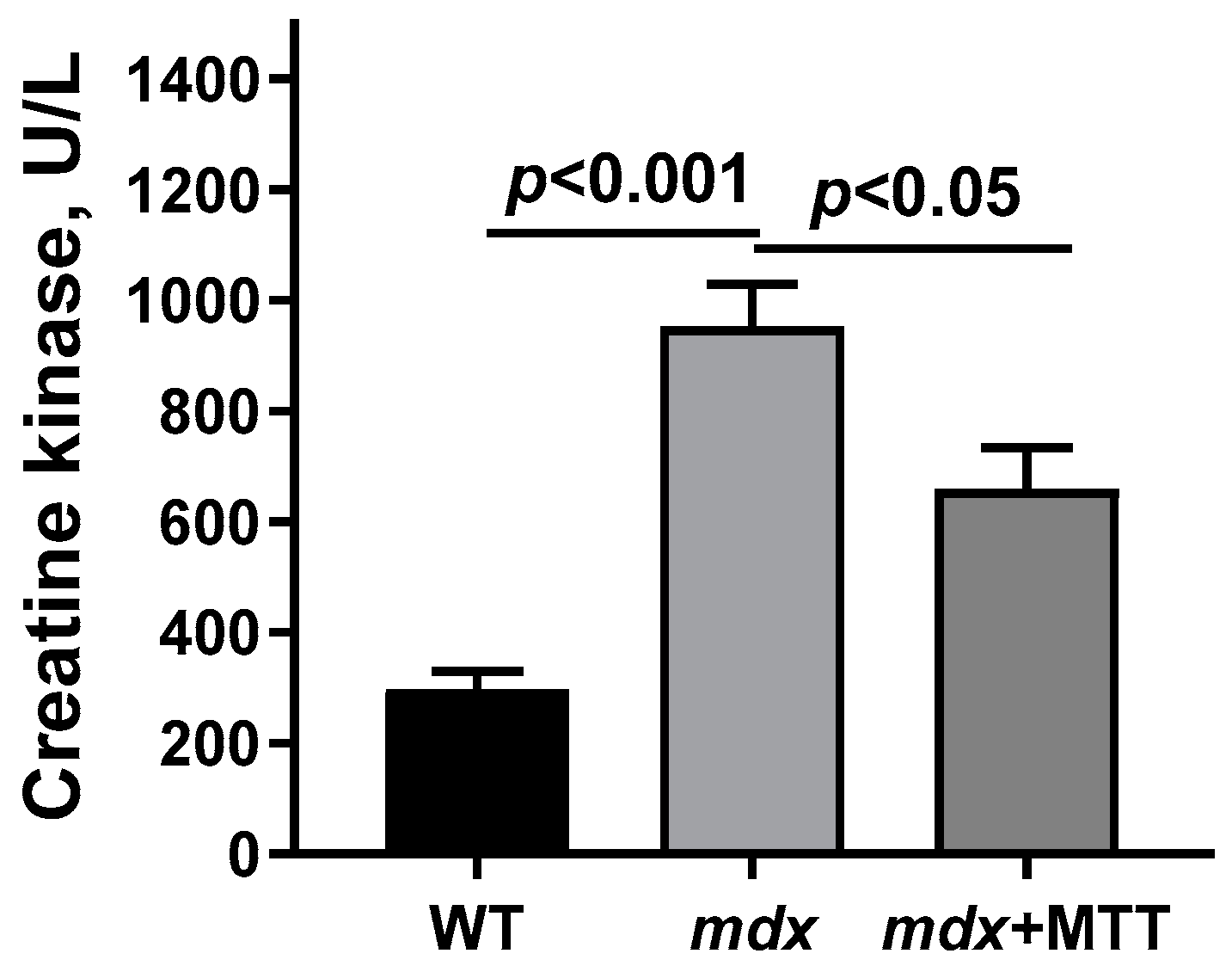
2.6. Creatine Kinase Analysis
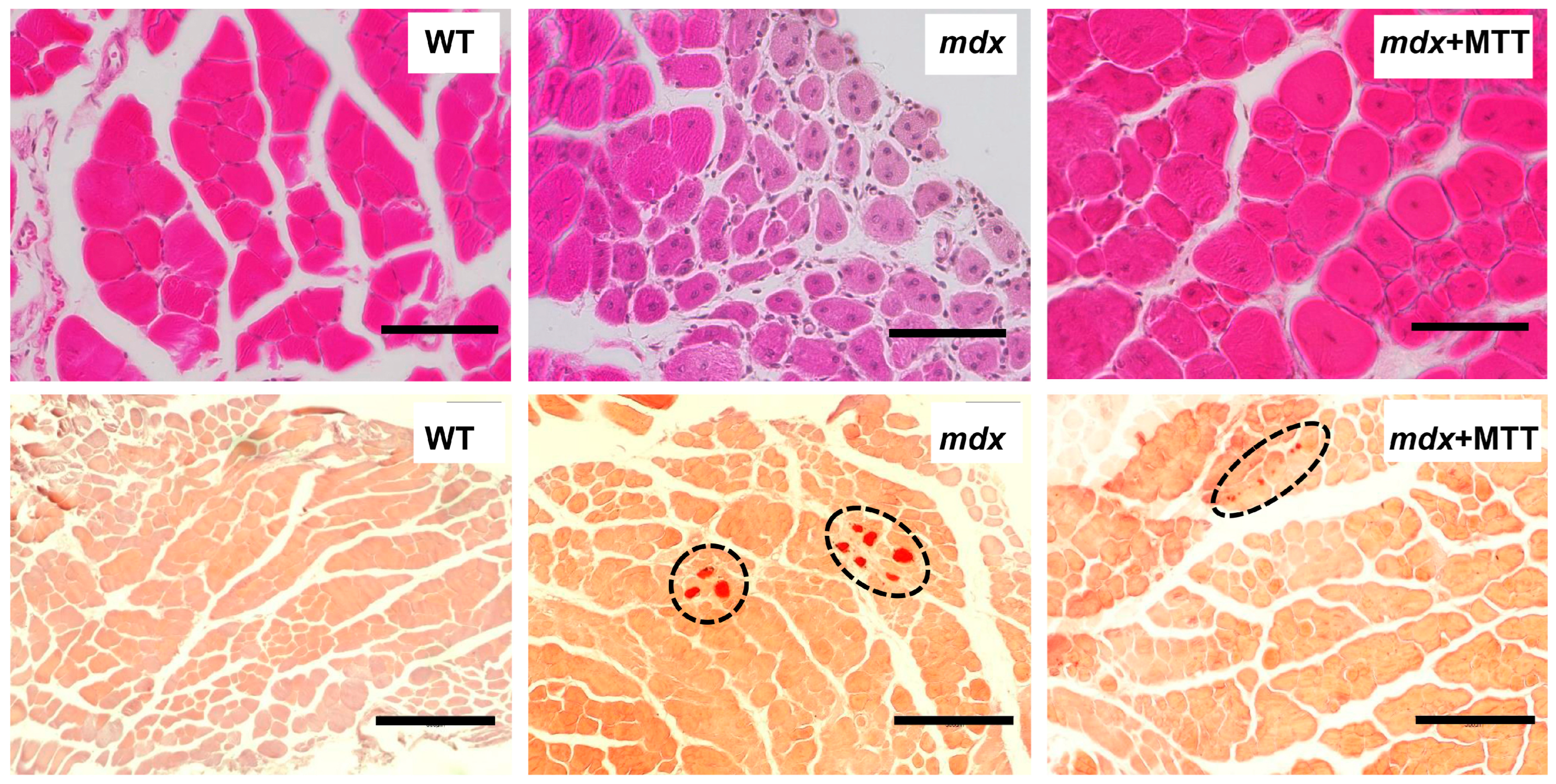
2.7. Histological Studies
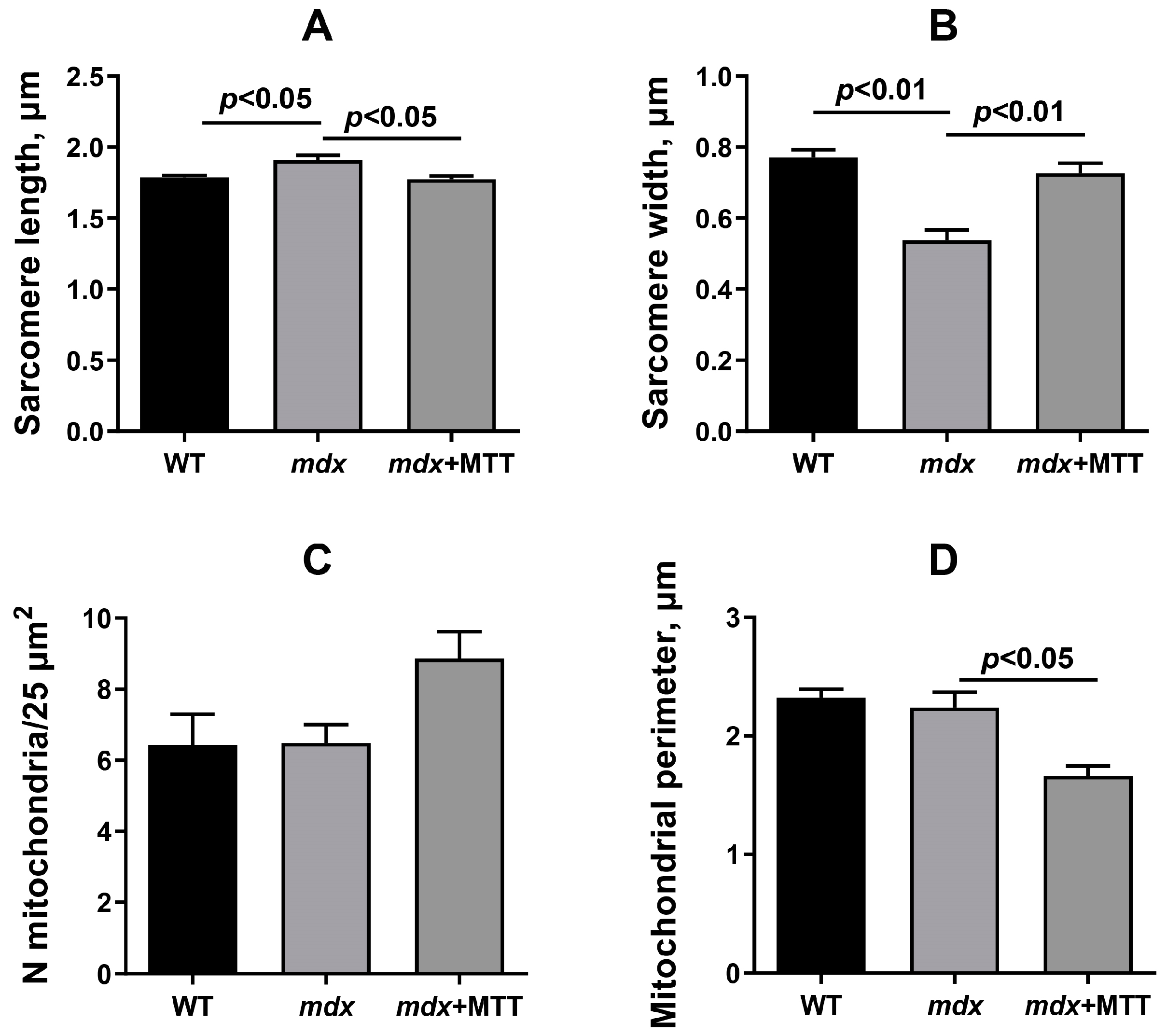
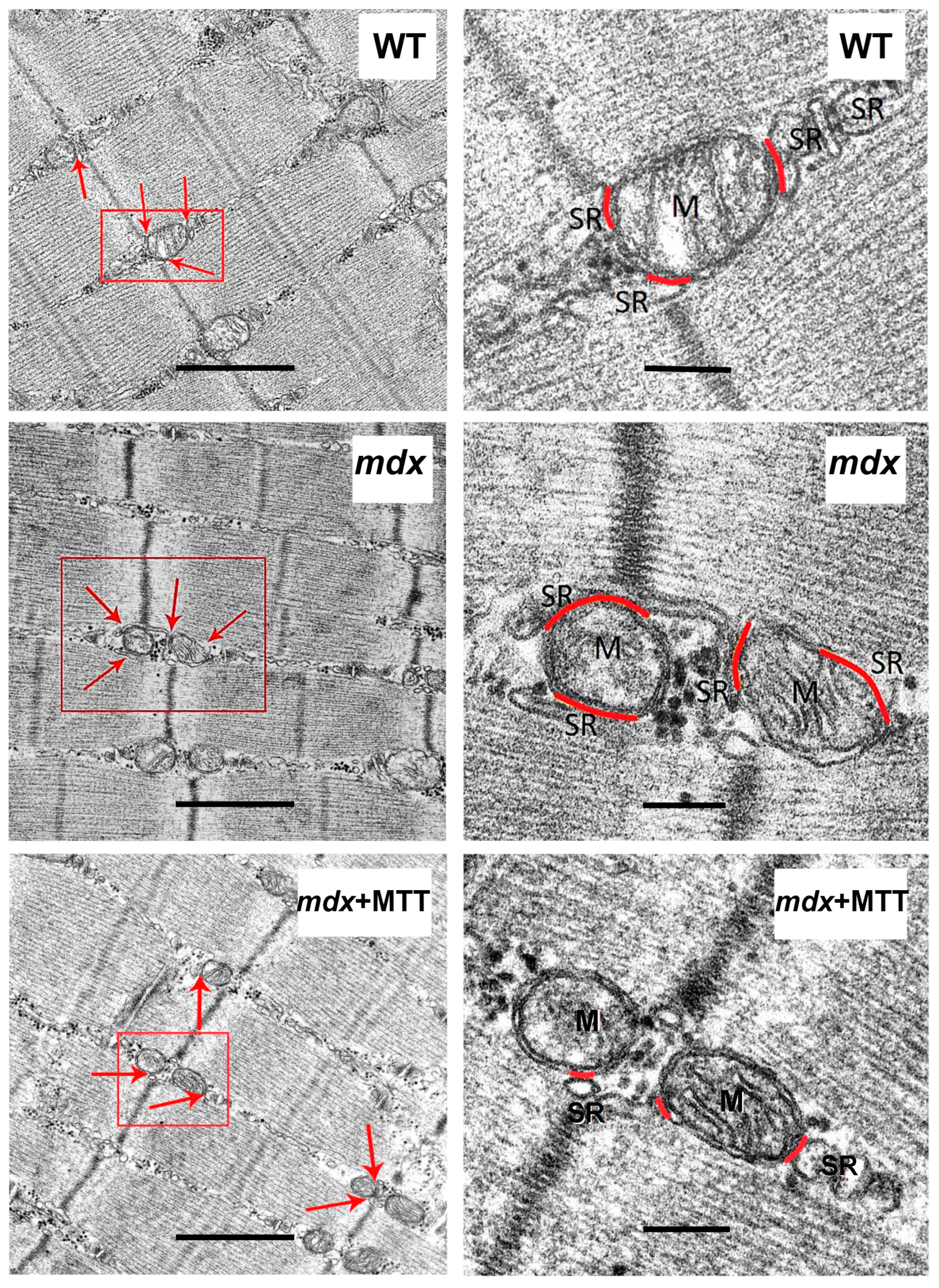
2.8. Transmission Electron Microscopy
2.9. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
2.10. Mitochondrial DNA Quantification
2.11. Statistical Analysis
3. Results
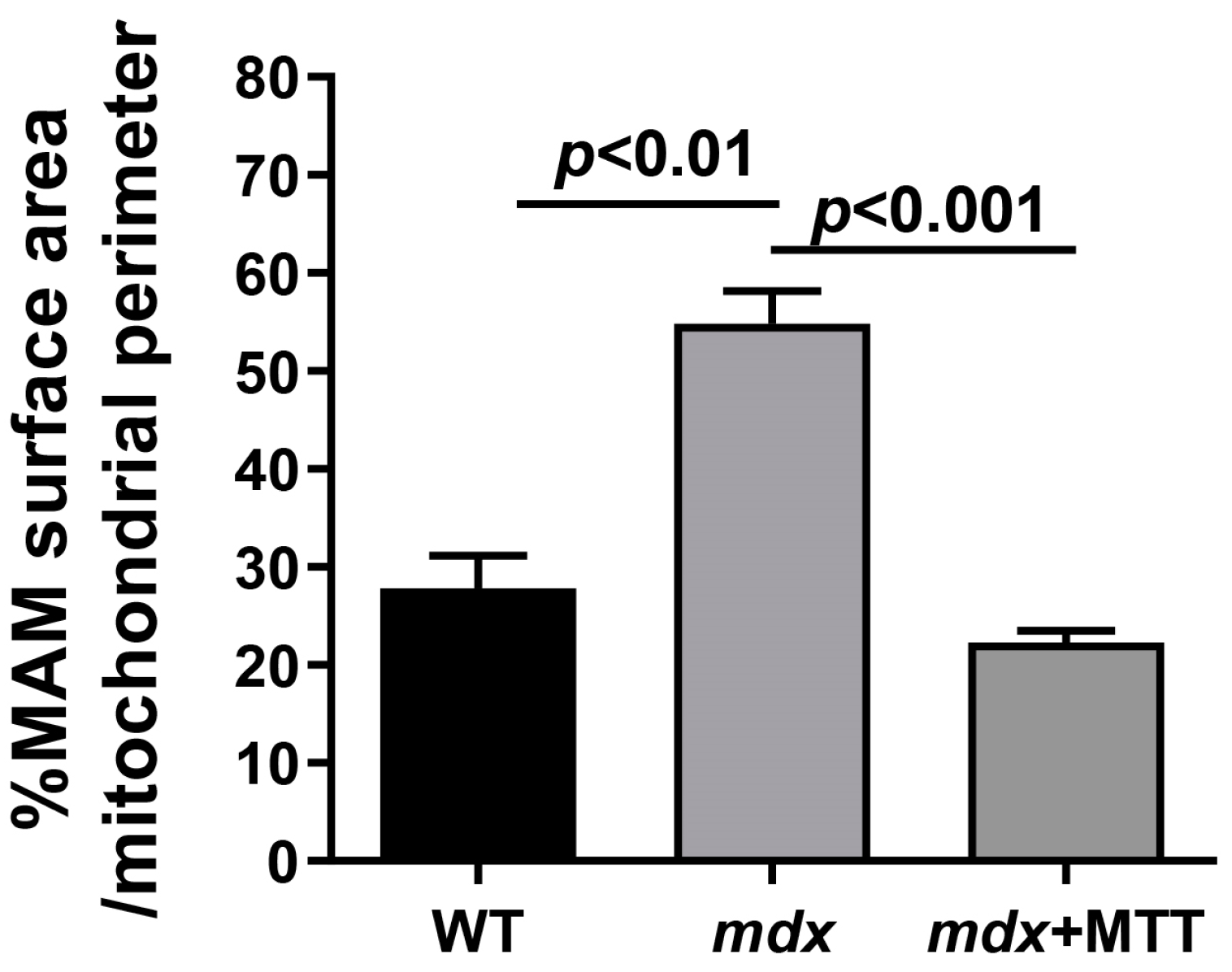
3.1. Visualization of MTT and Its Effect on the Skeletal Muscle Ultrastructure of mdx Mice
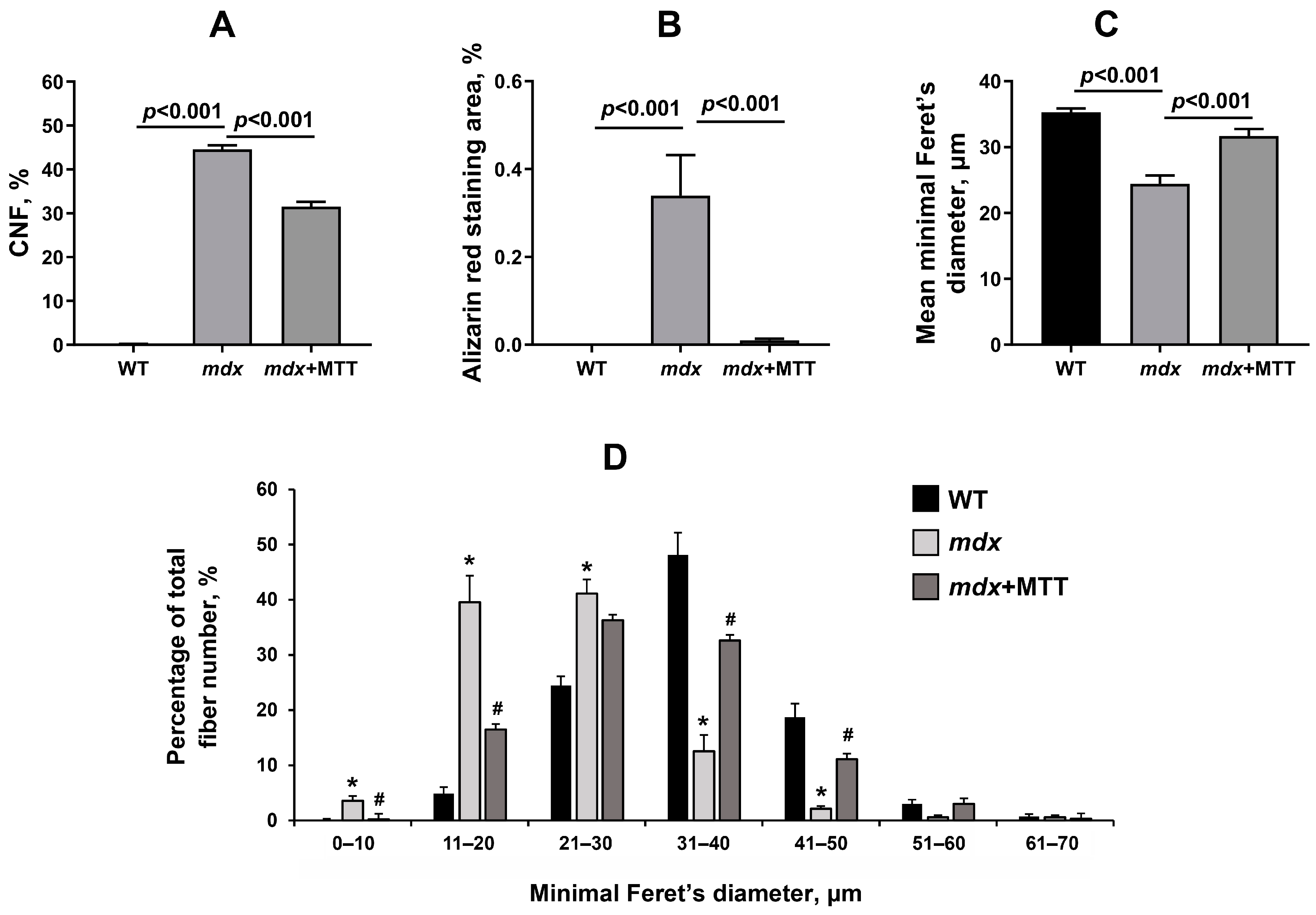
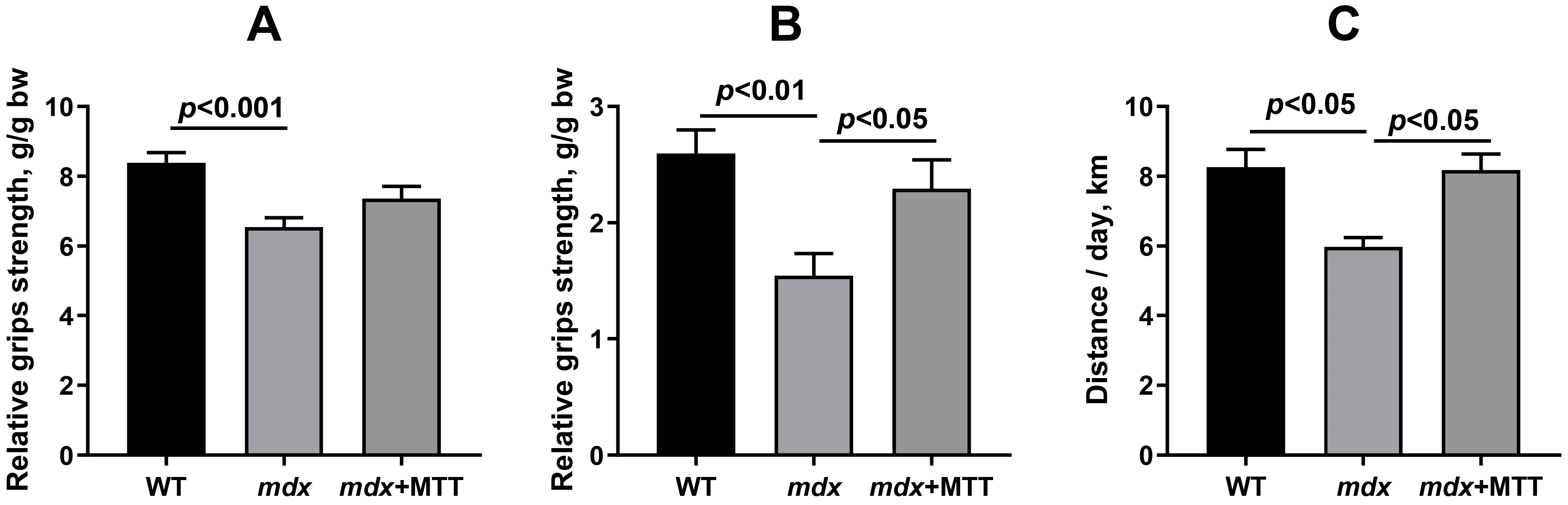
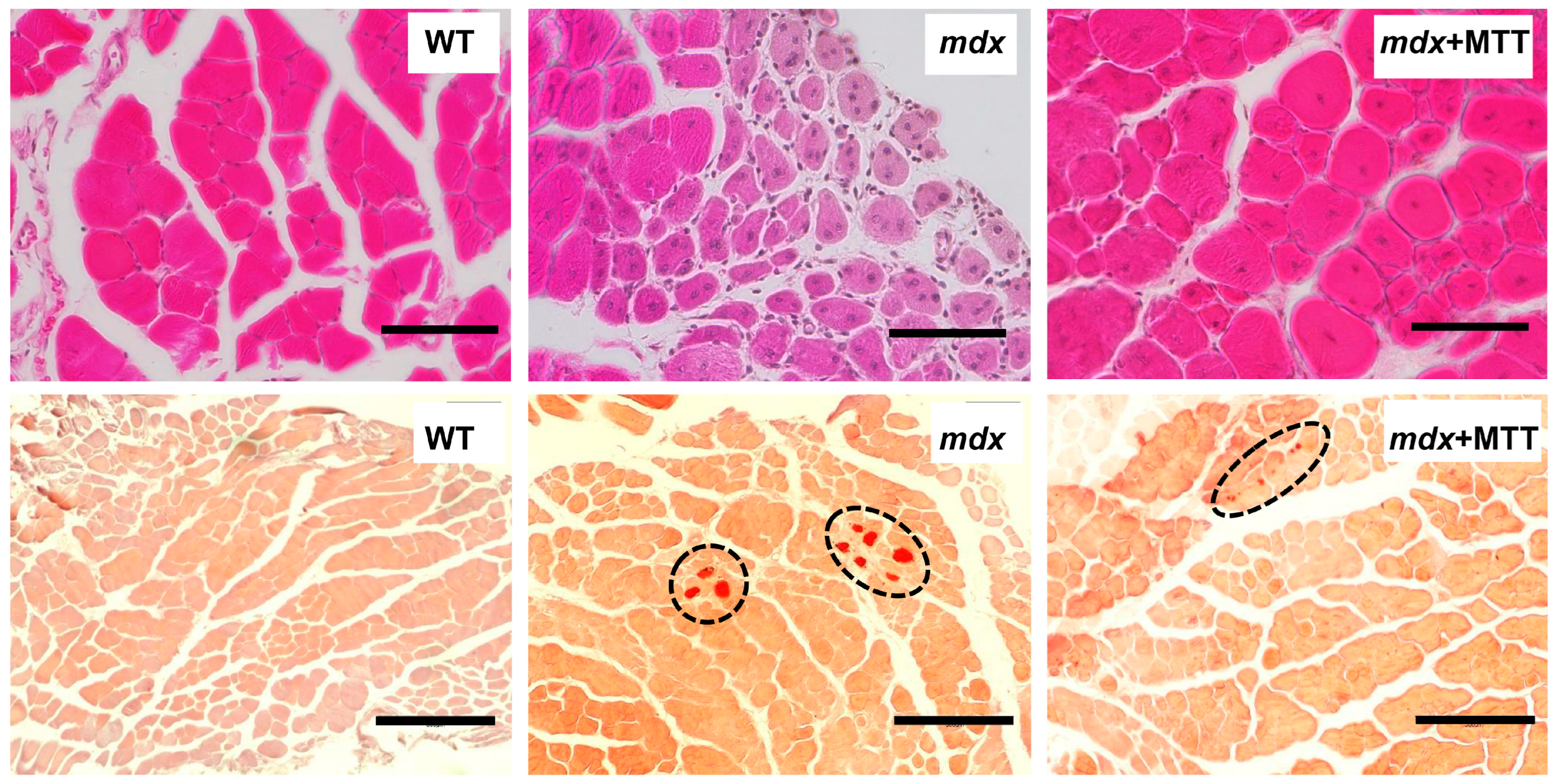
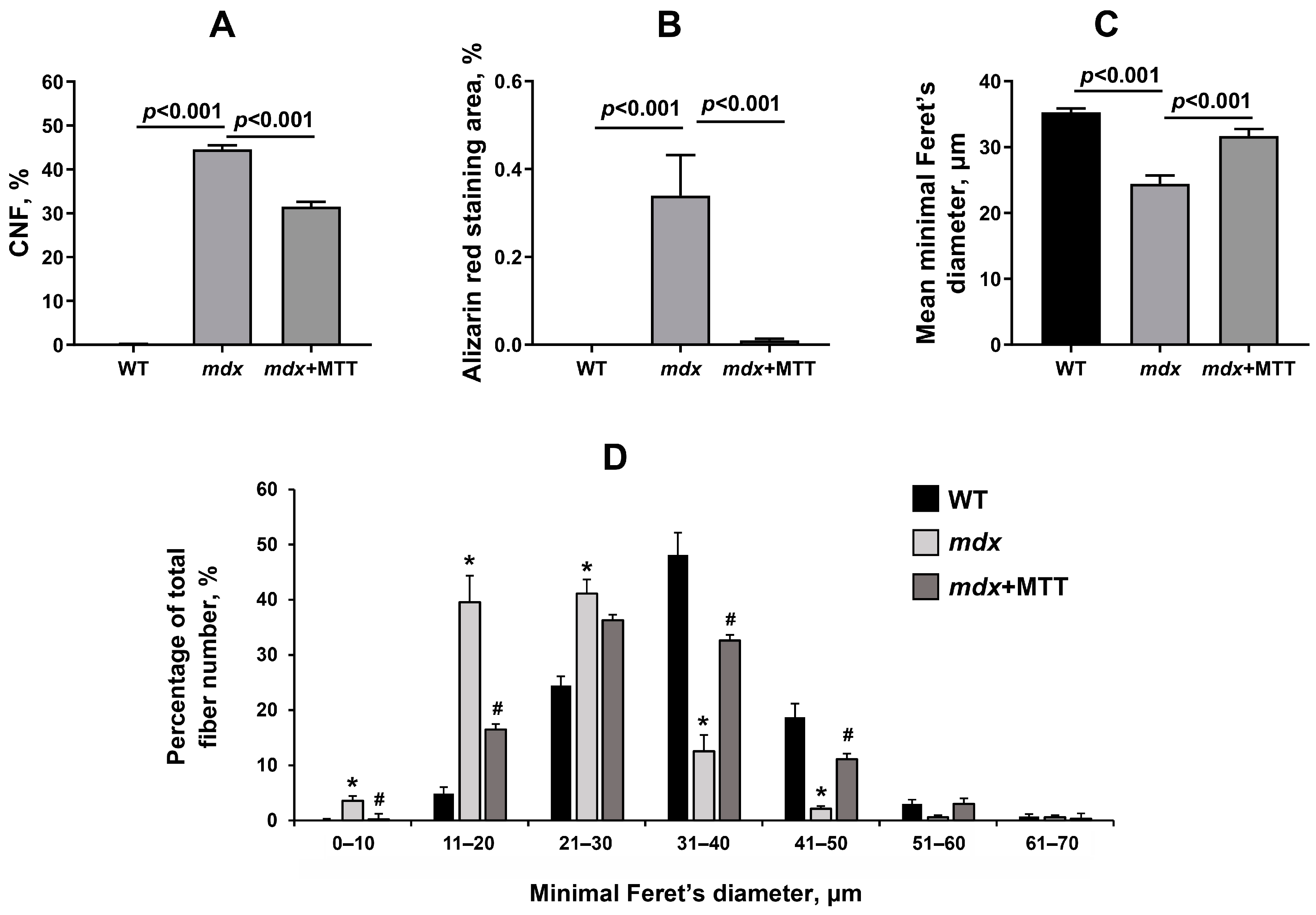
3.2. Effect of MTT on Skeletal Muscle Degeneration and Muscle Strength in mdx Mice
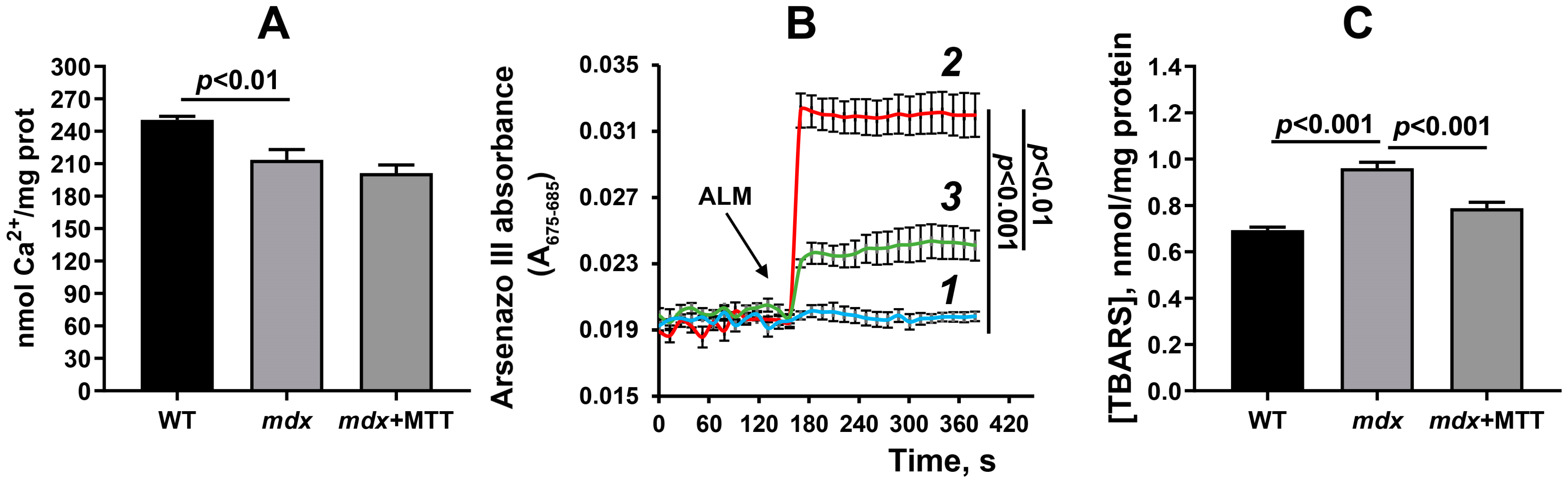
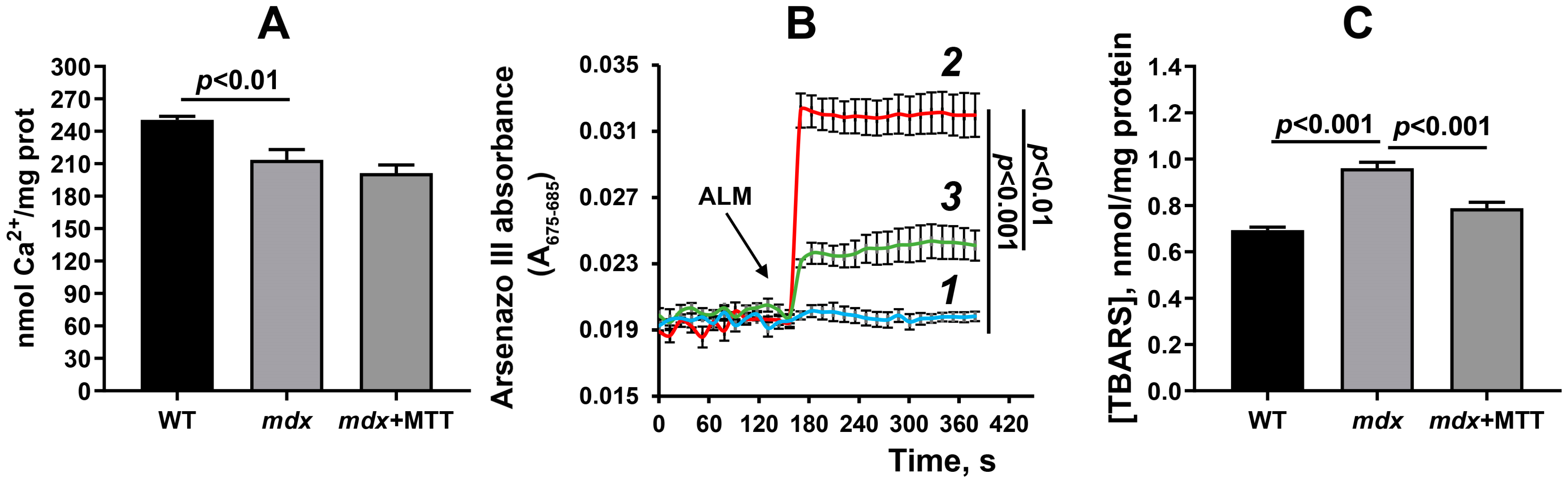
3.3. Effect of MTT on the Functioning of Skeletal Muscle Mitochondria in mdx Mice
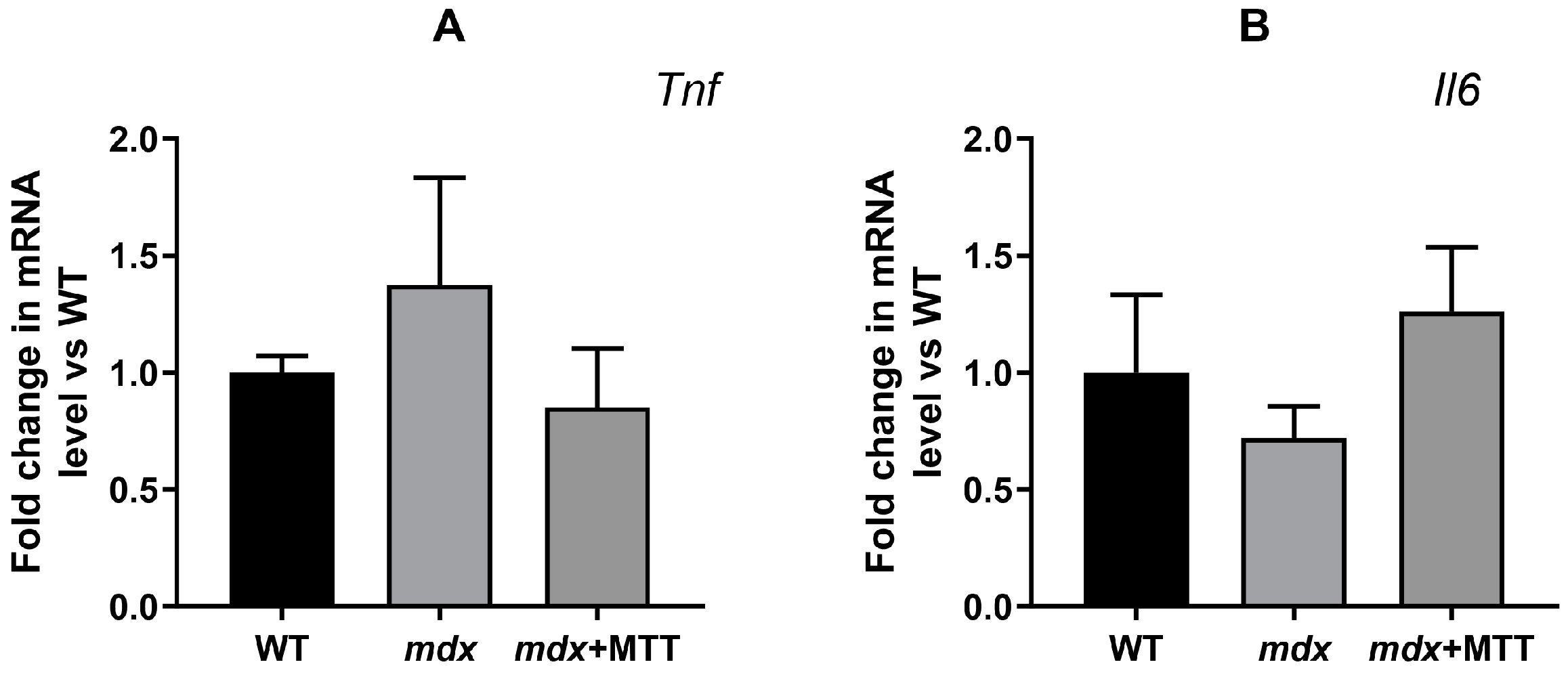
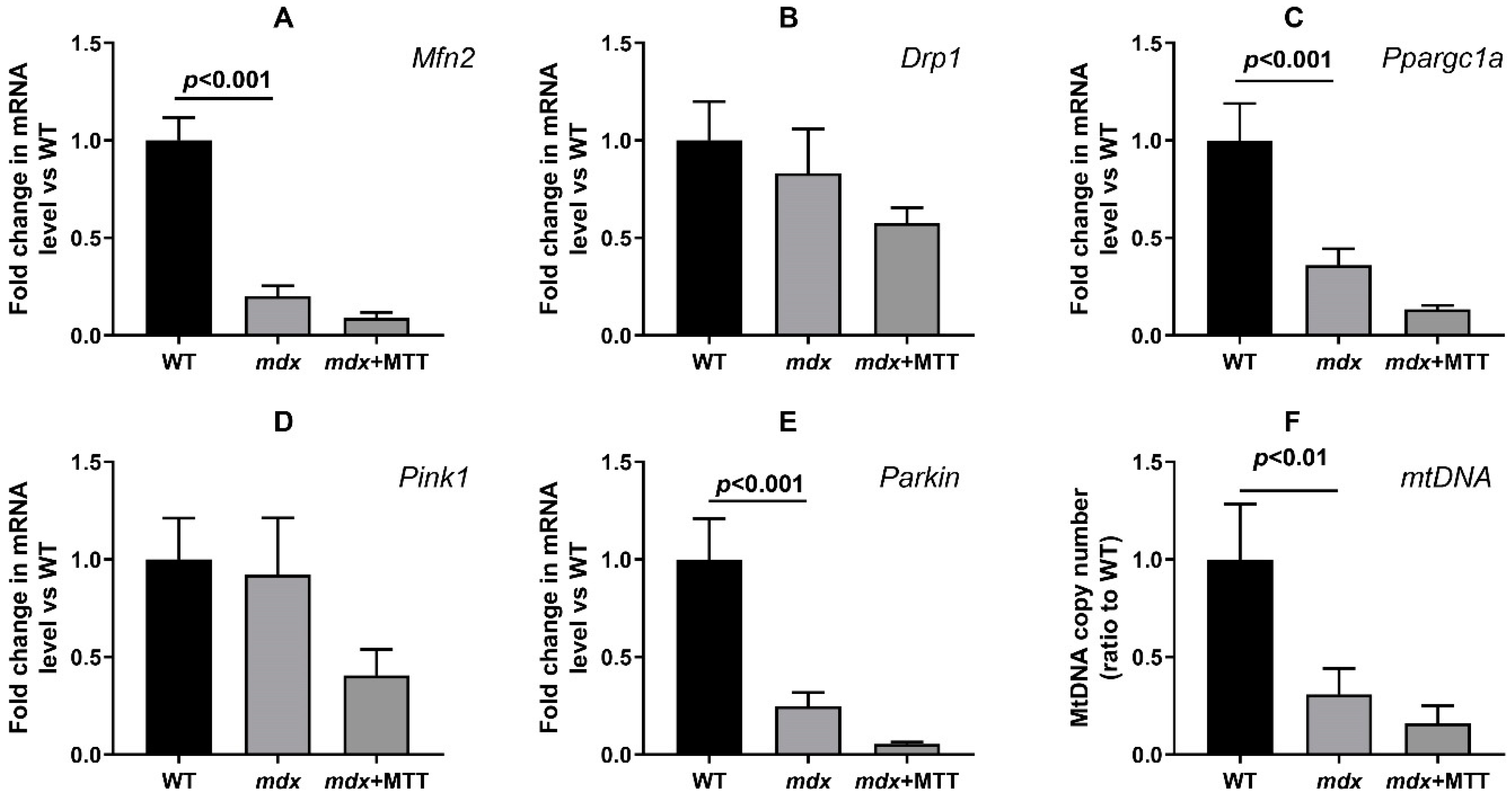
3.4. Effect of MTT on Expression of Mitochondrial Signature Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alway, S.E.; Paez, H.G.; Pitzer, C.R. The Role of Mitochondria in Mediation of Skeletal Muscle Repair. Muscles 2023, 2, 119–163. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Phys. Heart Circ. Phys. 2009, 296, 94–110. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously-derived mitochondria protects the heart from ischemia/reperfusion injury. Am. J. Phys. Heart Circ. Physiol. 2013, 304, 966–982. [Google Scholar] [CrossRef]
- Bobkova, N.V.; Zhdanova, D.Y.; Belosludtseva, N.V.; Penkov, N.V.; Mironova, G.D. Intranasal administration of mitochondria improves spatial memory in olfactory bulbectomized mice. Exp. Biol. Med. 2022, 247, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Paez, H.G.; Pitzer, C.R.; Ferrandi, P.J.; Khan, M.M.; Mohamed, J.S.; Carson, J.A.; Deschenes, M.R. Mitochondria transplant therapy improves regeneration and restoration of injured skeletal muscle. J. Cachexia Sarcopenia Muscle 2023, 14, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Liu, S.Y.; Lai, H.S.; Lai, I.R. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats. Shock 2013, 39, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Kitani, T.; Kami, D.; Matoba, S.; Gojo, S. Internalization of isolated functional mitochondria: Involvement of macropinocytosis. J. Cell Mol. Med. 2014, 18, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- Orfany, A.; Arriola, C.G.; Doulamis, I.P.; Guariento, A.; Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Rogers, C.; Del Nido, P.J.; et al. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 2020, 71, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Voglhuber, J.; Holzer, M.; Radulovic, S.; Thai, P.N.; Djalinac, N.; Matzer, I.; Wallner, M.; Bugger, H.; Zirlik, A.; Leitinger, G.; et al. Functional remodelling of perinuclear mitochondria alters nucleoplasmic Ca2+ signalling in heart failure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022, 377, 20210320. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Igoshkina, A.D.; Vafina, A.B.; Vedernikov, A.A.; Belosludtsev, K.N. BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy. Pharmaceutics 2022, 14, 2336. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, C.A.; Delfinis, L.J.; Hughes, M.C.; Turnbull, P.C.; Gandhi, S.; DiBenedetto, S.N.; Rahman, F.A.; Tadi, P.; Amaral, C.A.; Dehghani, A.; et al. Mitochondrial creatine sensitivity is lost in the D2.mdx model of Duchenne muscular dystrophy and rescued by the mitochondrial-enhancing compound Olesoxime. Am. J. Physiol. Cell Physiol. 2023, 324, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 1955, 217, 383–393. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Kumar, A.; Accorsi, A.; Rhee, Y.; Girgenrath, M. Do’s and don’ts in the preparation of muscle cryosections for histological analysis. J. Vis. Exp. 2015, 99, e52793. [Google Scholar] [CrossRef]
- Thoudam, T.; Ha, C.M.; Leem, J.; Chanda, D.; Park, J.S.; Kim, H.J.; Jeon, J.H.; Choi, Y.K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA ratio in mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Panza, E.; Vellecco, V.; Iannotti, F.A.; Paris, D.; Manzo, O.L.; Smimmo, M.; Mitilini, N.; Boscaino, A.; de Dominicis, G.; Bucci, M.; et al. Duchenne’s muscular dystrophy involves a defective transsulfuration pathway activity. Redox Biol. 2021, 45, 102040. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; Yackly, A.; et al. Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Ponzetti, M.; Ucci, A.; Maurizi, A.; Giacchi, L.; Teti, A.; Rucci, N. Lipocalin 2 influences bone and muscle phenotype in the mdx mouse model of Duchenne muscular dystrophy. Int. J. Mol. Sci. 2022, 23, 958. [Google Scholar] [CrossRef]
- Wu, K.K. Extracellular succinate: A physiological messenger and a pathological trigger. Int. J. Mol. Sci. 2023, 24, 11165. [Google Scholar] [CrossRef]
- Koriem, K.M.M.; Tharwat, H.A.K. Malic acid improves behavioral, biochemical, and molecular disturbances in the hypothalamus of stressed rats. J. Integr. Neurosci. 2023, 22, 98. [Google Scholar] [CrossRef]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef]
- Bround, M.J.; Havens, J.R.; York, A.J.; Sargent, M.A.; Karch, J.; Molkentin, J.D. ANT-dependent MPTP underlies necrotic myofiber death in muscular dystrophy. Sci. Adv. 2023, 9, eadi2767. [Google Scholar] [CrossRef]
- Doulamis, I.P.; Guariento, A.; Duignan, T.; Kido, T.; Orfany, A.; Saeed, M.Y.; Weixler, V.H.; Blitzer, D.; Shin, B.; Snay, E.R.; et al. Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 319, F403–F413. [Google Scholar] [CrossRef]
- Mokhtari, B.; Hamidi, M.; Badalzadeh, R.; Mahmoodpoor, A. Mitochondrial transplantation protects against sepsis-induced myocardial dysfunction by modulating mitochondrial biogenesis and fission/fusion and inflammatory response. Mol. Biol. Rep. 2023, 50, 2147–2158. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Schips, T.G.; Kwong, J.Q.; Davis, J.; Tjondrokoesoemo, A.; Brody, M.J.; Sargent, M.A.; Kanisicak, O.; Yi, H.; Gao, Q.Q.; et al. Thrombospondin expression in myofibers stabilizes muscle membranes. Elife 2016, 5, e17589. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Babenko, V.A.; Silachev, D.N.; Zorova, L.D.; Khryapenkova, T.G.; Savchenko, E.S.; Pevzner, I.B.; Zorov, D.B. Intercellular Transfer of Mitochondria. Biochemistry 2015, 80, 542–548. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. 2008, 586, 2003–2014. [Google Scholar] [CrossRef]
- Pinniger, G.J.; Terrill, J.R.; Assan, E.B.; Grounds, M.D.; Arthur, P.G. Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol. 2017, 595, 7093–7107. [Google Scholar] [CrossRef]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Jankauskas, S.S.; Zinovkin, R.A.; Zorova, L.D.; Zorov, S.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, D.B. A Combination of Kidney Ischemia and Injection of Isolated Mitochondria Leads to Activation of Inflammation and Increase in Mortality Rate in Rats. Bull. Exp. Biol. Med. 2020, 169, 213–217. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Genes of inflammatory response | ||
| Tnf (Tumor necrosis factor-α) | CCACGTCGTAGCAAACCACC | ACAAGGTACAACCCATCGGC |
| Il6 (interleukin-6) | GGGACTGATGCTGGTGACAA | TGCCATTGCACAACTCTTTTC |
| Genes for mitochondrial dynamics | ||
| Drp1 (Dynamin-related protein 1) | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 (Mitofusin 2) | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Gene for mitochondrial biogenesis | ||
| Ppargc1a (Peroxisome proliferator-activated receptor gamma coactivator 1-α) | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Genes of mitophagy | ||
| Pink1 (PTEN-induced kinase 1) | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Parkin | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Genes used to estimate relative mtDNA levels | ||
| Nd4 (NADH-ubiquinone oxidoreductase chain 4, mitochondrial) | ATTATTATTACCCGATGAGGGAACC | ATTAAGATGAGGGCAATTAGCAGT |
| Gapdh (Glyceraldehyde 3-phosphate dehydrogenase, nuclear) | GTGAGGGAGATGCYCAGTGT | CTGGCATTGCTCTCAATGAC |
| Housekeeping gene | ||
| Rplp2 (Ribosomal protein lateral stalk subunit P2) | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
| Group | Respiration Rate, nmol O2/min per 1 mg of Protein | RCR | |||
|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | ||
| WT | 19.7 ± 0.9 | 133.7 ± 2.8 | 32.3 ± 2.6 | 210.4 ± 8.3 | 4.2 ± 0.2 |
| mdx | 17.7 ± 0.8 | 121.6 ± 1.3 * | 33.4 ± 1.8 | 178.6 ± 1.9 * | 3.6 ± 0.1 * |
| Mdx + MTT | 17.2 ± 0.9 | 105.2 ± 1.9 *# | 31.4 ± 1.4 | 157.3 ± 1.8 *# | 3.3 ± 0.1 *# |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Mikheeva, I.B.; Stepanova, A.E.; Igoshkina, A.D.; Cherepanova, A.A.; Semenova, A.A.; Sharapov, V.A.; Kireev, I.I.; Belosludtsev, K.N. Mitochondrial Transplantation Therapy Ameliorates Muscular Dystrophy in mdx Mouse Model. Biomolecules 2024, 14, 316. https://doi.org/10.3390/biom14030316
Dubinin MV, Mikheeva IB, Stepanova AE, Igoshkina AD, Cherepanova AA, Semenova AA, Sharapov VA, Kireev II, Belosludtsev KN. Mitochondrial Transplantation Therapy Ameliorates Muscular Dystrophy in mdx Mouse Model. Biomolecules. 2024; 14(3):316. https://doi.org/10.3390/biom14030316
Chicago/Turabian StyleDubinin, Mikhail V., Irina B. Mikheeva, Anastasia E. Stepanova, Anastasia D. Igoshkina, Alena A. Cherepanova, Alena A. Semenova, Vyacheslav A. Sharapov, Igor I. Kireev, and Konstantin N. Belosludtsev. 2024. "Mitochondrial Transplantation Therapy Ameliorates Muscular Dystrophy in mdx Mouse Model" Biomolecules 14, no. 3: 316. https://doi.org/10.3390/biom14030316
APA StyleDubinin, M. V., Mikheeva, I. B., Stepanova, A. E., Igoshkina, A. D., Cherepanova, A. A., Semenova, A. A., Sharapov, V. A., Kireev, I. I., & Belosludtsev, K. N. (2024). Mitochondrial Transplantation Therapy Ameliorates Muscular Dystrophy in mdx Mouse Model. Biomolecules, 14(3), 316. https://doi.org/10.3390/biom14030316