
Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities

 ,
,  , , ,
, , ,  ,
,  ,
, 

 , and
, and
Abstract
1. Introduction to Pathological Calcification
2. Cellular Players in Vascular Calcification
2.1. Endothelial Cells
2.2. Vascular Smooth Muscle Cells
2.3. Macrophages
2.4. Pericytes and Fibroblasts
3. Molecular Networking in Vascular Calcification
3.1. Signaling Pathways in Vascular Calcification
3.2. Molecular Mediators of Vascular Calcification
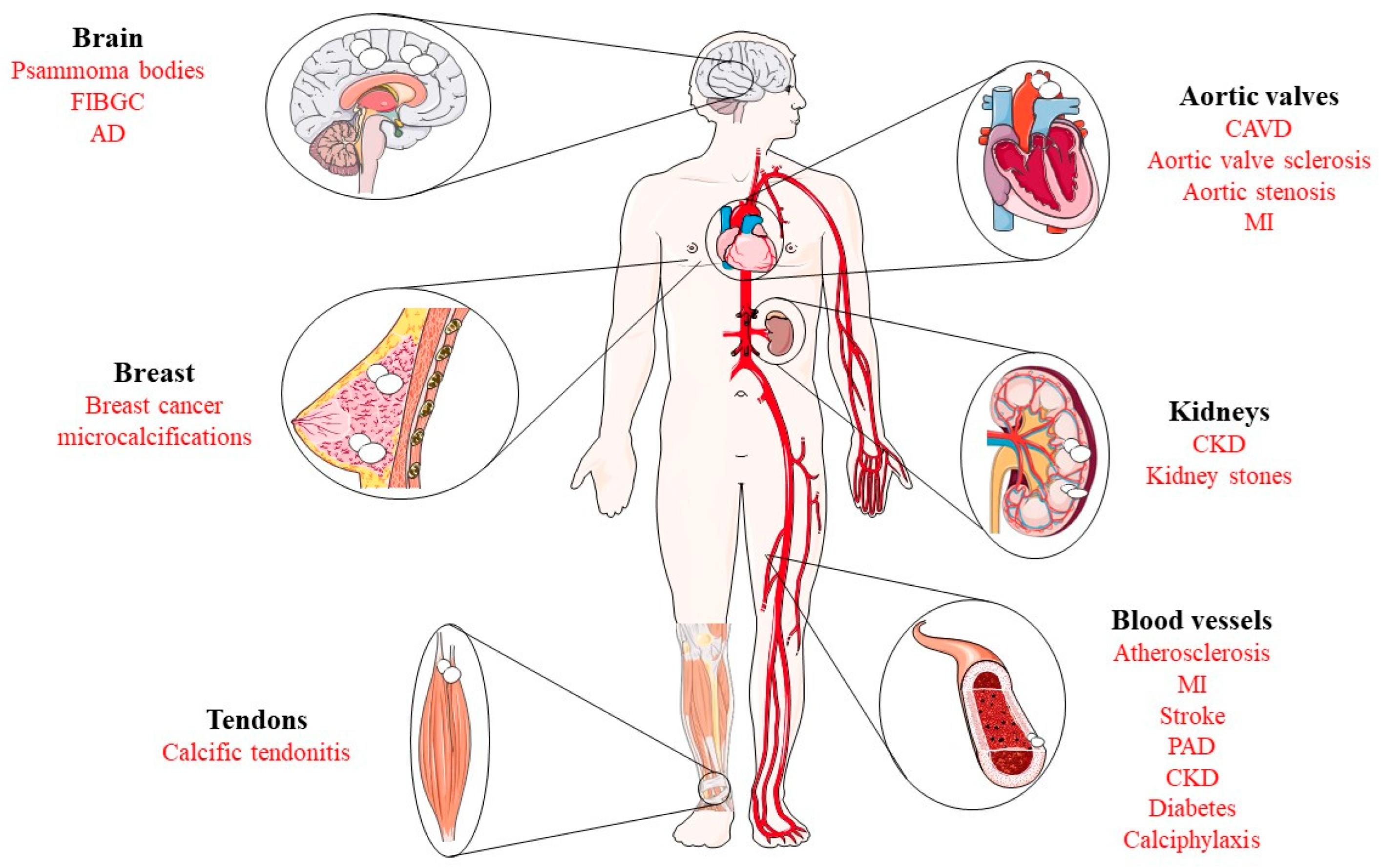
4. Pathological Implications
5. Translational Opportunities
5.1. Biomarkers of Vascular Calcification
5.2. Therapeutic Approaches

{kind=link}
| Compound | Target | Mechanism of Action | References |
|---|---|---|---|
| Sevelamer | Phosphate | Phosphate binder | [210,211] |
| Ppi analogs | Hydroxyapatite | Disruption of hydroxyapatite nucleation | [212,213] |
| Vitamin K2 | MGP | Activation of MGP | [214] |
| NaTS | Calcium | Chelation of calcium | [216] |
| Calcimimetics | Calcium-sensing receptors | Increase the sensitivity of calcium-sensing receptors | [219] |
| IP6 | Hydroxyapatite | Disruption of hydroxyapatite nucleation | [220] |
| Denosumab | RANKL | Inhibition of RANKL signaling | [223] |
6. Conclusions
Funding
Conflicts of Interest
References
- Proudfoot, D. Calcium Signaling and Tissue Calcification. Cold Spring Harb. Perspect. Biol. 2019, 11, a035303. [Google Scholar] [CrossRef]
- De Leon-Oliva, D.; Barrena-Blázquez, S.; Jiménez-Álvarez, L.; Fraile-Martinez, O.; García-Montero, C.; López-González, L.; Torres-carranza, D.; García-Puente, L.M.; Carranza, S.T.; Álvarez-Mon, M.Á.; et al. The RANK–RANKL–OPG System: A Multifaceted Regulator of Homeostasis, Immunity, and Cancer. Medicina 2023, 59, 1752. [Google Scholar] [CrossRef]
- Murshed, M. Mechanism of Bone Mineralization. Cold Spring Harb. Perspect. Med. 2018, 8, a031229. [Google Scholar] [CrossRef]
- De Leon-Oliva, D.; Boaru, D.L.; Perez-Exposito, R.E.; Fraile-Martinez, O.; García-Montero, C.; Díaz, R.; García-Honduvilla, N.; Lopez-Gonzalez, L.; Alvarez-Mon, M.; Saz, J.V.; et al. Advanced Hydrogel-Based Strategies for Enhanced Bone and Cartilage Regeneration: A Comprehensive Review. Gels 2023, 9, 885. [Google Scholar] [CrossRef]
- Fraile-Martínez, O.; García-Montero, C.; Coca, A.; Álvarez-Mon, M.A.; Monserrat, J.; Gómez-lahoz, A.M.; Coca, S.; Álvarez-mon, M.; Acero, J.; Bujan, J.; et al. Applications of Polymeric Composites in Bone Tissue Engineering and Jawbone Regeneration. Polymers 2021, 13, 3429. [Google Scholar] [CrossRef]
- Sutton, N.R.; Malhotra, R.; Hilaire, C.S.; Aikawa, E.; Blumenthal, R.S.; Gackenbach, G.; Goyal, P.; Johnson, A.; Nigwekar, S.U.; Shanahan, C.M.; et al. Molecular Mechanisms of Vascular Health: Insights From Vascular Aging and Calcification. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Vidavsky, N.; Kunitake, J.A.M.R.; Estroff, L.A. Multiple Pathways for Pathological Calcification in the Human Body. Adv. Healthc. Mater. 2021, 10, e2001271. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Inflammatory, Metabolic, and Genetic Mechanisms of Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Ortolani, F.; Rigonat, L.; Bonetti, A.; Contin, M.; Tubaro, F.; Rattazzi, M.; Marchini, M. Pro-Calcific Responses by Aortic Valve Interstitial Cells in a Novel in Vitro Model Simulating Dystrophic Calcification. Ital. J. Anat. Embryol. 2010, 115, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Senba, M.; Kawai, K.; Mori, N. Pathogenesis of Metastatic Calcification and Acute Pancreatitis in Adult T-Cell Leukemia under Hypercalcemic State. Leuk. Res. Treat. 2012, 2012, 128617. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Park, J.H.; Lee, J.B.; Kim, S.J. A Case of Dystrophic Calcification in the Masseter Muscle. Maxillofac. Plast. Reconstr. Surg. 2017, 39, 31. [Google Scholar] [CrossRef]
- Bonetti, A.; Della Mora, A.; Contin, M.; Tubaro, F.; Marchini, M.; Ortolani, F. Ultrastructural and Spectrophotometric Study on the Effects of Putative Triggers on Aortic Valve Interstitial Cells in In Vitro Models Simulating Metastatic Calcification. Anat. Rec. 2012, 295, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, A.; Chung, L. Calcinosis: Pathophysiology and Management. Curr. Opin. Rheumatol. 2015, 27, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Sawke, G.K.; Rai, T.; Sawke, N. Iatrogenic Calcinosis Cutis: A Rare Cytological Diagnosis. J. Cytol. 2016, 33, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Ibragimova, A.G.; Stanishevskiy, Y.M.; Plakkhin, A.M.; Zubko, A.V.; Darvish, N.A.; Koassary, A.K.; Shindyapina, A.V. Comparative Analysis of Calcified Soft Tissues Revealed Shared Deregulated Pathways. Front. Aging Neurosci. 2023, 15, 1131548. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Saez, M.Á.; Asúnsolo, Á.; Romero, B.; Bravo, C.; Coca, S.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Upregulation of VEGF and PEDF in Placentas of Women with Lower Extremity Venous Insufficiency during Pregnancy and Its Implication in Villous Calcification. BioMed Res. Int. 2019, 2019, 5320902. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Tran, T.X.M.; Song, H.; Park, B. Microcalcifications, Mammographic Breast Density, and Risk of Breast Cancer: A Cohort Study. Breast Cancer Res. 2022, 24, 96. [Google Scholar] [CrossRef]
- Monfrini, E.; Arienti, F.; Rinchetti, P.; Lotti, F.; Riboldi, G.M. Brain Calcifications: Genetic, Molecular, and Clinical Aspects. Int. J. Mol. Sci. 2023, 24, 8995. [Google Scholar] [CrossRef]
- Wyatt, C.M.; Drueke, T.B. Vascular Calcification in Chronic Kidney Disease: Here to Stay? Kidney Int. 2017, 92, 276–278. [Google Scholar] [CrossRef]
- Kosuru, V.; Mohammed, A.; Kapoor, R.; Jhaveri, K.; Medepalli, V.; Mulloy, L.; Padala, S.A. Metastatic Calcinosis of Gastric Mucosa. J. Investig. Med. High Impact Case Rep. 2020, 8, 2324709620940482. [Google Scholar] [CrossRef]
- Kim, M.-S.; Kim, I.-W.; Lee, S.; Shin, S.-J. Diagnosis and Treatment of Calcific Tendinitis of the Shoulder. Clin. Shoulder Elb. 2020, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Jarjou’i, A.; Bogot, N.; Kalak, G.; Chen-Shuali, C.; Rokach, A.; Izbicki, G.; Arish, N. Diffuse Pulmonary Calcifications: A Case Series and Review of Literature. Respirol. Case Rep. 2021, 9, e0839. [Google Scholar] [CrossRef] [PubMed]
- Poloni, L.N.; Ward, M.D. The Materials Science of Pathological Crystals. Chem. Mater. 2014, 26, 477–495. [Google Scholar] [CrossRef]
- Bazin, D.; Daudon, M.; Combes, C.; Rey, C. Characterization and Some Physicochemical Aspects of Pathological Microcalcifications. Chem. Rev. 2012, 112, 5092–5120. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, A.; Contin, M.; Marchini, M.; Ortolani, F. Ultrastructural and Immunohistochemical Detection of Hydroxyapatite Nucleating Role by RRNA and Nuclear Chromatin Derivatives in Aortic Valve Calcification: In Vitro and In Vivo Pro-Calcific Animal Models and Actual Calcific Disease in Humans. Int. J. Mol. Sci. 2023, 24, 266. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular Calcification: Pathobiology of a Multifaceted Disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, A.; Marchini, M.; Ortolani, F. Ectopic Mineralization in Heart Valves: New Insights from in Vivo and in Vitro Procalcific Models and Promising Perspectives on Noncalcifiable Bioengineered Valves. J. Thorac. Dis. 2019, 11, 2126–2143. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Kuczumow, A.; Gorzelak, M.; Kosiński, J.; Lasota, A.; Blicharski, T.; Gągała, J.; Nowak, J.; Jarzębski, M.; Jabłoński, M. Hierarchy of Bioapatites. Int. J. Mol. Sci. 2022, 23, 9537. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Arterial Stiffness: A Focus on Vascular Calcification and Its Link to Bone Mineralization. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef]
- You, A.Y.F.; Bergholt, M.S.; St-Pierre, J.P.; Kit-Anan, W.; Pence, I.J.; Chester, A.H.; Yacoub, M.H.; Bertazzo, S.; Stevens, M.M. Raman Spectroscopy Imaging Reveals Interplay between Atherosclerosis and Medial Calcification in the Human Aorta. Sci. Adv. 2017, 3, e1701156. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Hannan, F.M.; Lanzer, J.D.; Janzen, J.; Raggi, P.; Furniss, D.; Schuchardt, M.; Thakker, R.; Fok, P.W.; Saez-Rodriguez, J.; et al. Medial Arterial Calcification: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1145–1165. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.I.; Guzman, R.J. Medial Artery Calcification in Peripheral Artery Disease. Front. Cardiovasc. Med. 2023, 10, 1093355. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Mackenzie, N.C.W.; Farquharson, C.; MacRae, V.E. Mechanisms and Clinical Consequences of Vascular Calcification. Front. Endocrinol. 2012, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019, 25, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Lerman, D.A.; Prasad, S.; Alotti, N. Calcific Aortic Valve Disease: Molecular Mechanisms and Therapeutic Approaches. Eur. Cardiol. 2015, 10, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Moncla, L.H.M.; Briend, M.; Bossé, Y.; Mathieu, P. Calcific Aortic Valve Disease: Mechanisms, Prevention and Treatment. Nat. Rev. Cardiol. 2023, 20, 546–559. [Google Scholar] [CrossRef]
- Kraler, S.; Blaser, M.C.; Aikawa, E.; Camici, G.G.; Lüscher, T.F. Calcific Aortic Valve Disease: From Molecular and Cellularmechanisms to Medical Therapy. Eur. Heart J. 2022, 43, 683–697. [Google Scholar] [CrossRef]
- Pawade, T.; Sheth, T.; Guzzetti, E.; Dweck, M.R.; Clavel, M.A. Why and How to Measure Aortic Valve Calcification in Patients With Aortic Stenosis. JACC Cardiovasc. Imaging 2019, 12, 1835–1848. [Google Scholar] [CrossRef]
- Chen, H.Y.; Engert, J.C.; Thanassoulis, G. Risk Factors for Valvular Calcification. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 96–102. [Google Scholar] [CrossRef]
- Ahn, B.Y.; Jeong, Y.; Kim, S.; Zhang, Y.; Kim, S.W.; Leem, Y.E.; Kang, J.S. Cdon Suppresses Vascular Smooth Muscle Calcification via Repression of the Wnt/Runx2 Axis. Exp. Mol. Med. 2023, 55, 120–131. [Google Scholar] [CrossRef]
- Yang, P.; Troncone, L.; Augur, Z.M.; Kim, S.S.J.; McNeil, M.E.; Yu, P.B. The Role of Bone Morphogenetic Protein Signaling in Vascular Calcification. Bone 2020, 141, 115542. [Google Scholar] [CrossRef]
- Semenova, D.; Kostina, A.; Irtyuga, O.; Moiseeva, O.; Malashicheva, A. The Mechanisms of Notch-Dependent Aortic Valve Calcification. Struct. Heart 2019, 3, 171. [Google Scholar] [CrossRef]
- Pustlauk, W.; Westhoff, T.H.; Claeys, L.; Roch, T.; Geißler, S.; Babel, N. Induced Osteogenic Differentiation of Human Smooth Muscle Cells as a Model of Vascular Calcification. Sci. Rep. 2020, 10, 5951. [Google Scholar] [CrossRef]
- Cui, L.; Houston, D.A.; Farquharson, C.; MacRae, V.E. Characterisation of Matrix Vesicles in Skeletal and Soft Tissue Mineralisation. Bone 2016, 87, 147–158. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Z.; Li, Y.; Chen, C.; Yang, H.; Lin, Q.; Hu, M.; Qin, X. The Cell Origin and Role of Osteoclastogenesis and Osteoblastogenesis in Vascular Calcification. Front. Cardiovasc. Med. 2021, 8, 639740. [Google Scholar] [CrossRef]
- Yap, C.; Mieremet, A.; De Vries, C.J.M.; Micha, D.; De Waard, V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef] [PubMed]
- Tintut, Y.; Honda, H.M.; Demer, L.L. Biomolecules Orchestrating Cardiovascular Calcification. Biomolecules 2021, 11, 1482. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, L.; Zhang, L.; Zang, G.; Sun, Z.; Wang, Z. Role of Endothelial Cells in Vascular Calcification. Front. Cardiovasc. Med. 2022, 9, 895005. [Google Scholar] [CrossRef] [PubMed]
- Wylie-Sears, J.; Aikawa, E.; Levine, R.A.; Yang, J.H.; Bischoff, J. Mitral Valve Endothelial Cells with Osteogenic Differentiation Potential. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Wang, H.; Zhu, Y.Z. The Drug Developments of Hydrogen Sulfide on Cardiovascular Disease. Oxid. Med. Cell. Longev. 2018, 1, 4010395. [Google Scholar] [CrossRef]
- Kim, S.H.L.; Lee, S.S.; Kim, I.; Kwon, J.; Kwon, S.; Bae, T.; Hur, J.; Lee, H.; Hwang, N.S. Ectopic Transient Overexpression of OCT-4 Facilitates BMP4-Induced Osteogenic Transdifferentiation of Human Umbilical Vein Endothelial Cells. J. Tissue Eng. 2020, 11, 2041731420909208. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Lang, F.; Eckardt, K.U.; Amann, K.; Kuro-o, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling Pathways Involved in Vascular Smooth Muscle Cell Calcification during Hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of Mouse Aortic Smooth Muscle Cells to a Macrophage-like State after Cholesterol Loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Shan, S.K.; Xu, F.; Zhong, J.Y.; Wu, F.; Duan, J.Y.; Guo, B.; Li, F.X.Z.; Wang, Y.; Zheng, M.H.; et al. The Crosstalk between Endothelial Cells and Vascular Smooth Muscle Cells Aggravates High Phosphorus-Induced Arterial Calcification. Cell Death Dis. 2022, 13, 650. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.C.; Nesmith, A.P.; Parker, K.K. The Role of Mechanotransduction on Vascular Smooth Muscle Myocytes Cytoskeleton and Contractile Function. Anat. Rec. 2014, 297, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Gomel, M.A.; Lee, R.; Grande-Allen, K.J. Comparing the Role of Mechanical Forces in Vascular and Valvular Calcification Progression. Front. Cardiovasc. Med. 2019, 5, 197. [Google Scholar] [CrossRef] [PubMed]
- Balogh, E.; Tóth, A.; Méhes, G.; Trencsényi, G.; Paragh, G.; Jeney, V. Hypoxia Triggers Osteochondrogenic Differentiation of Vascular Smooth Muscle Cells in an HIF-1 (Hypoxia-Inducible Factor 1)-Dependent and Reactive Oxygen Species-Dependent Manner. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1088–1099. [Google Scholar] [CrossRef]
- Csiki, D.M.; Ababneh, H.; Tóth, A.; Lente, G.; Szöőr, Á.; Tóth, A.; Fillér, C.; Juhász, T.; Nagy, B.; Balogh, E.; et al. Hypoxia-Inducible Factor Activation Promotes Osteogenic Transition of Valve Interstitial Cells and Accelerates Aortic Valve Calcification in a Mice Model of Chronic Kidney Disease. Front. Cardiovasc. Med. 2023, 10, 1168339. [Google Scholar] [CrossRef]
- Huang, X.; Akgün, E.E.; Mehmood, K.; Zhang, H.; Tang, Z.; Li, Y. Mechanism of Hypoxia-Mediated Smooth Muscle Cell Proliferation Leading to Vascular Remodeling. BioMed Res. Int. 2022, 24, 3959845. [Google Scholar] [CrossRef] [PubMed]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Metzinger-Le Meuth, V.; Hénaut, L.; Djelouat, M.S.E.I.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic Phosphate Accelerates the Migration of Vascular Smooth Muscle Cells: Evidence for the Involvement of MiR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef] [PubMed]
- Shimokado, A.; Sun, Y.; Nakanishi, M.; Sato, F.; Oikawa, K.; Akasaka, T.; Muragaki, Y. Smad3 Plays an Inhibitory Role in Phosphate-Induced Vascular Smooth Muscle Cell Calcification. Exp. Mol. Pathol. 2014, 97, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Metzinger-Le Meuth, V.; Metzinger, L. MiR-223 and Other MiRNA’s Evaluation in Chronic Kidney Disease: Innovative Biomarkers and Therapeutic Tools. Non-Coding RNA Res. 2019, 4, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of Vascular Smooth Muscle Cells Is Induced by Secondary Calciprotein Particles and Enhanced by Tumor Necrosis Factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.D.; Estepa, M.; Boehme, B.; Pieske, B.; Lang, F.; Eckardt, K.U.; Voelkl, J.; Alesutan, I. Inhibition of Vascular Smooth Muscle Cell Calcification by Vasorin through Interference with TGFβ1 Signaling. Cell. Signal. 2019, 64, 109414. [Google Scholar] [CrossRef]
- Cao, J.; Chen, L.; Zhong, X.; Shen, Y.; Gao, Y.; Chen, Q.; Zu, X.; Liu, J. MiR32-5p Promoted Vascular Smooth Muscle Cell Calcification by Upregulating TNFα in the Microenvironment. BMC Immunol. 2020, 21, 3. [Google Scholar] [CrossRef]
- Song, X.; Song, Y.; Ma, Q.; Fang, K.; Chang, X. M1-Type Macrophages Secrete TNF-α to Stimulate Vascular Calcification by Upregulating CA1 and CA2 Expression in VSMCs. J. Inflamm. Res. 2023, 16, 3019–3032. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Z.; Zhang, L.; Yan, J.; Shao, C.; Jing, L.; Li, L.; Wang, Z. Role of Macrophages in the Progression and Regression of Vascular Calcification. Front. Pharmacol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Waring, O.J.; Skenteris, N.T.; Biessen, E.A.L.; Donners, M.M.P.C. Two-Faced Janus: The Dual Role of Macrophages in Atherosclerotic Calcification. Cardiovasc. Res. 2022, 118, 2768–2777. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage Subsets in Atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed Cell Death in Atherosclerosis and Vascular Calcification. Cell Death Dis. 2022, 13, 467. [Google Scholar] [CrossRef]
- Hénaut, L.; Candellier, A.; Boudot, C.; Grissi, M.; Mentaverri, R.; Choukroun, G.; Brazier, M.; Kamel, S.; Massy, Z.A. New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease. Toxins 2019, 11, 529. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification in Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Farzaneh-Far, A.; Shanahan, C.M.; Weissberg, P.L. The Role of Apoptosis in the Initiation of Vascular Calcification. Z. Kardiol. 2001, 90, 43–46. [Google Scholar] [CrossRef]
- Doherty, M.J.; Ashton, B.A.; Walsh, S.; Beresford, J.N.; Grant, M.E.; Canfield, A.E. Vascular Pericytes Express Osteogenic Potential in Vitro and in Vivo. J. Bone Miner. Res. 1998, 13, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Canfield, A.E.; Doherty, M.J.; Wood, A.C.; Farrington, C.; Ashton, B.; Begum, N.; Harvey, B.; Poole, A.; Grant, M.E.; Boot-Handford, R.P. Role of Pericytes in Vascular Calcification: A Review. Z. Kardiol. 2000, 89, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Davaine, J.M.; Quillard, T.; Brion, R.; Lapérine, O.; Guyomarch, B.; Merlini, T.; Chatelais, M.; Guilbaud, F.; Brennan, M.Á.; Charrier, C.; et al. Osteoprotegerin, Pericytes and Bone-like Vascular Calcification Are Associated with Carotid Plaque Stability. PLoS ONE 2014, 9, e107642. [Google Scholar] [CrossRef] [PubMed]
- Hortells, L.; Sur, S.; Hilaire, C.S. Cell Phenotype Transitions in Cardiovascular Calcification. Front. Cardiovasc. Med. 2018, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, A.; Simionescu, D.T.; Vyavahare, N.R. Osteogenic Responses in Fibroblasts Activated by Elastin Degradation Products and Transforming Growth Factor-Β1: Role of Myofibroblasts in Vascular Calcification. Am. J. Pathol. 2007, 171, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Su, S.; Chen, J.; Ma, H.; Xiang, M. Emerging Roles of Fibroblasts in Cardiovascular Calcification. J. Cell. Mol. Med. 2021, 25, 1808–1816. [Google Scholar] [CrossRef]
- Bundy, K.; Boone, J.; Simpson, C.L.S. Wnt Signaling in Vascular Calcification. Front. Cardiovasc. Med. 2021, 8, 3–8. [Google Scholar] [CrossRef]
- Shimizu, T.; Tanaka, T.; Iso, T.; Doi, H.; Sato, H.; Kawai-Kowase, K.; Arai, M.; Kurabayashi, M. Notch Signaling Induces Osteogenic Differentiation and Mineralization of Vascular Smooth Muscle Cells: Role of Msx2 Gene Induction via Notch-RBP-Jk Signaling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1104–1111. [Google Scholar] [CrossRef]
- Zhao, X.-K.; Zhu, M.-M.; Wang, S.-N.; Zhang, T.-T.; Wei, X.-N.; Wang, C.-Y.; Zheng, J.; Zhu, W.-Y.; Jiang, M.-X.; Xu, S.-W.; et al. Transcription Factor 21 Accelerates Vascular Calcification in Mice by Activating the IL-6/STAT3 Signaling Pathway and the Interplay between VSMCs and ECs. Acta Pharmacol. Sin. 2023, 44, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Martín-Núñez, E.; Hernández-Carballo, C.; Ferri, C.; Tagua, V.G.; Delgado-Molinos, A.; López-Castillo, Á.; Rodríguez-Ramos, S.; Cerro-López, P.; López-Tarruella, V.C.; et al. Fibroblast Growth Factor 23 Expression in Human Calcified Vascular Tissues. Aging 2019, 11, 7899–7913. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix Vesicles Induce Calcification of Recipient Vascular Smooth Muscle Cells through Multiple Signaling Pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed]
- Issa, H.; Hénaut, L.; Abdallah, J.B.; Boudot, C.; Lenglet, G.; Avondo, C.; Ibrik, A.; Caus, T.; Brazier, M.; Mentaverri, R.; et al. Activation of the Calcium-Sensing Receptor in Human Valvular Interstitial Cells Promotes Calcification. J. Mol. Cell. Cardiol. 2019, 129, 2–12. [Google Scholar] [CrossRef]
- Molostvov, G.; Hiemstra, T.F.; Fletcher, S.; Bland, R.; Zehnder, D. Arterial Expression of the Calcium-Sensing Receptor Is Maintained by Physiological Pulsation and Protects against Calcification. PLoS ONE 2015, 10, e0138833. [Google Scholar] [CrossRef]
- Ma, C.; Gu, R.; Wang, X.; He, S.; Bai, J.; Zhang, L.; Zhang, J.; Li, Q.; Qu, L.; Xin, W.; et al. CircRNA CDR1as Promotes Pulmonary Artery Smooth Muscle Cell Calcification by Upregulating CAMK2D and CNN3 via Sponging MiR-7-5p. Mol. Ther. Nucleic Acids 2020, 22, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Li, S.J.; Kao, Y.H.; Chung, C.C.; Cheng, W.L.; Lin, Y.K.; Chen, Y.J. Vascular Endothelial Growth Factor on Runt-Related Transcript Factor-2 in Aortic Valve Cells. Eur. J. Clin. Investig. 2021, 51, e13470. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, A.; Allegri, L.; Baldan, F.; Contin, M.; Battistella, C.; Damante, G.; Marchini, M.; Ortolani, F. Critical Involvement of Calcium-Dependent Cytosolic Phospholipase A2α in Aortic Valve Interstitial Cell Calcification. Int. J. Mol. Sci. 2020, 21, 6398. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-Catenin Signalling: Function, Biological Mechanisms, and Therapeutic Opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kawano, T.; Murata, M.; Toita, R. Vascular Calcification and Cellular Signaling Pathways as Potential Therapeutic Targets. Life Sci. 2024, 336, 122309. [Google Scholar] [CrossRef] [PubMed]
- Albanese, I.; Khan, K.; Barratt, B.; Al-Kindi, H.; Schwertani, A. Atherosclerotic Calcification: Wnt Is the Hint. J. Am. Heart Assoc. 2018, 7, e007356. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Albanese, I.; Yu, B.; Al-Kindi, H.; Barratt, B.; Ott, L.; Al-Refai, M.; De Varennes, B.; Shum-Tim, D.; Cerruti, M.; Gourgas, O.; et al. Role of Noncanonical Wnt Signaling Pathway in Human Aortic Valve Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 543–552. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; ten Dijke, P. Bone Morphogenetic Protein Receptors: Structure, Function and Targeting by Selective Small Molecule Kinase Inhibitors. Bone 2020, 138, 115472. [Google Scholar] [CrossRef]
- Niu, Z.; Su, G.; Li, T.; Yu, H.; Shen, Y.; Zhang, D.; Liu, X. Vascular Calcification: New Insights Into BMP Type I Receptor, A. Front. Pharmacol. 2022, 13, 887253. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; Ten Dijke, P.; Goumans, M.J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP Signalling in Cardiovascular Disease and Anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef]
- Cai, J.; Pardali, E.; Sánchez-Duffhues, G.; Ten Dijke, P. BMP Signaling in Vascular Diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef]
- Ortega, M.A.; Asúnsolo, Á.; Pekarek, L.; Alvarez-Mon, M.A.; Delforge, A.; Sáez, M.A.; Coca, S.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; et al. Histopathological Study of JNK in Venous Wall of Patients with Chronic Venous Insufficiency Related to Osteogenesis Process. Int. J. Med. Sci. 2021, 18, 1921–1934. [Google Scholar] [CrossRef]
- Ye, D.; Liu, Y.; Pan, H.; Feng, Y.; Lu, X.; Gan, L.; Wan, J.; Ye, J. Insights into Bone Morphogenetic Proteins in Cardiovascular Diseases. Front. Pharmacol. 2023, 14, 1125642. [Google Scholar] [CrossRef]
- Zhang, M.; Sara, J.D.; Wang, F.L.; Liu, L.P.; Su, L.X.; Zhe, J.; Wu, X.; Liu, J.H. Increased Plasma BMP-2 Levels Are Associated with Atherosclerosis Burden and Coronary Calcification in Type 2 Diabetic Patients. Cardiovasc. Diabetol. 2015, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; López-Ongil, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL Increases Vascular Smooth Muscle Cell Calcification through a Rank-Bmp4-Dependent Pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef]
- Huang, J.; Pu, Y.; Zhang, H.; Xie, L.; He, L.; Zhang, C.L.; Cheng, C.K.; Huo, Y.; Wan, S.; Chen, S.; et al. KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling. Circ. Res. 2021, 129, E87–E100. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.M.; Sánchez-Duffhues, G.; Ten Dijke, P.; Yu, P.B. Bone Morphogenetic Protein 6 and Oxidized Low-Density Lipoprotein Synergistically Recruit Osteogenic Differentiation in Endothelial Cells. Cardiovasc. Res. 2015, 108, 278–287. [Google Scholar] [CrossRef]
- Davies, M.R.; Lund, R.J.; Hruska, K.A. BMP-7 Is an Efficacious Treatment of Vascular Calcification in a Murine Model of Atherosclerosis and Chronic Renal Failure. J. Am. Soc. Nephrol. 2003, 14, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Kuo, W.H.; Tain, Y.L.; Wang, Y.; Lee, W.C. Exogenous BMP7 Administration Attenuated Vascular Calcification and Improved Bone Disorders in Chronic Uremic Rats. Biochem. Biophys. Res. Commun. 2022, 621, 8–13. [Google Scholar] [CrossRef]
- Rusanescu, G.; Weissleder, R.; Aikawa, E. Notch Signaling in Cardiovascular Disease and Calcification. Curr. Cardiol. Rev. 2008, 4, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, Y.; Lu, P.; Wu, B.; Zhou, B. NOTCH Signaling in Aortic Valve Development and Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2021, 8, 682298. [Google Scholar] [CrossRef]
- Majumdar, U.; Manivannan, S.; Basu, M.; Ueyama, Y.; Blaser, M.C.; Cameron, E.; McDermott, M.R.; Lincoln, J.; Cole, S.E.; Wood, S.; et al. Nitric Oxide Prevents Aortic Valve Calcification by S-Nitrosylation of USP9X to Activate NOTCH Signaling. Sci. Adv. 2021, 7, eabe3706. [Google Scholar] [CrossRef]
- Beazley, K.E.; Nurminsky, D.; Lima, F.; Gandhi, C.; Nurminskaya, M.V. Wnt16 Attenuates TGFβ-Induced Chondrogenic Transformation in Vascular Smooth Muscle. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 573–579. [Google Scholar] [CrossRef]
- Lin, X.; Li, S.; Wang, Y.J.; Wang, Y.; Zhong, J.Y.; He, J.Y.; Cui, X.J.; Zhan, J.K.; Liu, Y.S. Exosomal Notch3 from High Glucose-Stimulated Endothelial Cells Regulates Vascular Smooth Muscle Cells Calcification/Aging. Life Sci. 2019, 232, 116582. [Google Scholar] [CrossRef] [PubMed]
- White, M.P.; Theodoris, C.V.; Liu, L.; Collins, W.J.; Blue, K.W.; Lee, J.H.; Meng, X.; Robbins, R.C.; Ivey, K.N.; Srivastava, D. NOTCH1 Regulates Matrix Gla Protein and Calcification Gene Networks in Human Valve Endothelium. J. Mol. Cell. Cardiol. 2015, 84, 13–23. [Google Scholar] [CrossRef]
- Ouyang, L.; Zhang, K.; Chen, J.; Wang, J.; Huang, H. Roles of Platelet-Derived Growth Factor in Vascular Calcification. J. Cell. Physiol. 2018, 233, 2804–2814. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, S.; Peng, H.; Lv, Y.; Li, W.; Cheng, S.; Liu, J. Fibroblast Growth Factor 21 Attenuates Vascular Calcification by Alleviating Endoplasmic Reticulum Stress Mediated Apoptosis in Rats. Int. J. Biol. Sci. 2019, 15, 138–147. [Google Scholar] [CrossRef]
- Olapoju, S.O.; Adejobi, O.I.; Le Thi, X. Fibroblast Growth Factor 21; Review on Its Participation in Vascular Calcification Pathology. Vascul. Pharmacol. 2020, 125–126, 106636. [Google Scholar] [CrossRef]
- Sung, D.C.; Bowen, C.J.; Vaidya, K.A.; Zhou, J.; Chapurin, N.; Recknagel, A.; Zhou, B.; Chen, J.; Kotlikoff, M.; Butcher, J.T. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1627–1637. [Google Scholar] [CrossRef]
- Vaidya, K.A.; Donnelly, M.P.; Mahmut, A.; Jang, J.W.; Gee, T.W.; Aibo, M.A.I.; Bossong, R.; Hall, C.; Samb, S.; Chen, J.; et al. Rac1 Mediates Cadherin-11 Induced Cellular Pathogenic Processes in Aortic Valve Calcification. Cardiovasc. Pathol. 2022, 58, 107414. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Chen, J.; Sewell-Loftin, M.K.; Ryzhova, L.M.; Fisher, C.I.; Su, Y.R.; David Merryman, W. Cadherin-11 Regulates Cell-Cell Tension Necessary for Calcific Nodule Formation by Valvular Myofibroblasts. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Bowler, M.A.; Bersi, M.R.; Ryzhova, L.M.; Jerrell, R.J.; Parekh, A.; Merryman, W.D. Cadherin-11 as a Regulator of Valve Myofibroblast Mechanobiology. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1614–H1626. [Google Scholar] [CrossRef]
- Radvar, E.; Griffanti, G.; Tsolaki, E.; Bertazzo, S.; Nazhat, S.N.; Addison, O.; Mata, A.; Shanahan, C.M.; Elsharkawy, S. Engineered In Vitro Models for Pathological Calcification: Routes Toward Mechanistic Understanding. Adv. NanoBiomed Res. 2021, 1, 2100042. [Google Scholar] [CrossRef]
- Bjørklund, G.; Svanberg, E.; Dadar, M.; Card, D.J.; Chirumbolo, S.; Harrington, D.J.; Aaseth, J. The Role of Matrix Gla Protein (MGP) in Vascular Calcification. Curr. Med. Chem. 2020, 27, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M. Matrix Gla-Protein (MGP) Not Only Inhibits Calcification in Large Arteries But Also May Be Renoprotective: Connecting the Dots. EBioMedicine 2016, 4, 16–17. [Google Scholar] [CrossRef][Green Version]
- Jaminon, A.M.G.; Dai, L.; Qureshi, A.R.; Evenepoel, P.; Ripsweden, J.; Söderberg, M.; Witasp, A.; Olauson, H.; Schurgers, L.J.; Stenvinkel, P. Matrix Gla Protein Is an Independent Predictor of Both Intimal and Medial Vascular Calcification in Chronic Kidney Disease. Sci. Rep. 2020, 10, 6586. [Google Scholar] [CrossRef]
- Barrett, H.; O’Keeffe, M.; Kavanagh, E.; Walsh, M.; O’Connor, E.M. Is Matrix Gla Protein Associated with Vascular Calcification? A Systematic Review. Nutrients 2018, 10, 415. [Google Scholar] [CrossRef]
- Ge, Q.; Ruan, C.C.; Ma, Y.; Tang, X.F.; Wu, Q.H.; Wang, J.G.; Zhu, D.L.; Gao, P.J. Osteopontin Regulates Macrophage Activation and Osteoclast Formation in Hypertensive Patients with Vascular Calcification. Sci. Rep. 2017, 7, 40253. [Google Scholar] [CrossRef]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef]
- Van Campenhout, A.; Golledge, J. Osteoprotegerin, Vascular Calcification and Atherosclerosis. Atherosclerosis 2009, 204, 321–329. [Google Scholar] [CrossRef]
- Zhou, S.; Fang, X.; Xin, H.; Li, W.; Qiu, H.; Guan, S. Osteoprotegerin Inhibits Calcification of Vascular Smooth Muscle Cell via Down Regulation of the Notch1-RBP-Jκ/Msx2 Signaling Pathway. PLoS ONE 2013, 8, e68987. [Google Scholar] [CrossRef]
- Dekker, M.; Waissi, F.; Silvis, M.J.M.; Bennekom, J.V.; Schoneveld, A.H.; de Winter, R.J.; Isgum, I.; Lessmann, N.; Velthuis, B.K.; Pasterkamp, G.; et al. High Levels of Osteoprotegerin Are Associated with Coronary Artery Calcification in Patients Suspected of a Chronic Coronary Syndrome. Sci. Rep. 2021, 11, 18946. [Google Scholar] [CrossRef]
- Qin, Z.; Liao, R.; Xiong, Y.; Jiang, L.; Li, J.; Wang, L.; Han, M.; Sun, S.; Geng, J.; Yang, Q.; et al. A Narrative Review of Exosomes in Vascular Calcification. Ann. Transl. Med. 2021, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Zazzeroni, L.; Faggioli, G.; Pasquinelli, G. Mechanisms of Arterial Calcification: The Role of Matrix Vesicles. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yu, H.; Zhang, D.; Feng, T.; Miao, M.; Li, J.; Liu, X. Matrix Vesicles as a Therapeutic Target for Vascular Calcification. Front. Cell Dev. Biol. 2022, 10, 825622. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, A.L.; Moysés, R.M.A.; Canziani, M.E. Vascular Calcification in Peritoneal Dialysis Patients. Perit. Dial. Int. J. Int. Soc. Perit. Dial. 2008, 28, 20–25. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Vascular Calcification: Key Roles of Phosphate and Pyrophosphate. Int. J. Mol. Sci. 2021, 22, 13536. [Google Scholar] [CrossRef] [PubMed]
- Kenkre, J.S.; Bassett, J.H.D. The Bone Remodelling Cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, C.; Iwata, H.; Aikawa, E. Parathyroid Hormone. Critical Bridge Between Bone Metabolism and Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1333–1335. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Phosphonoformic Acid Prevents Vascular Smooth Muscle Cell Calcification by Inhibiting Calcium-Phosphate Deposition. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 761–766. [Google Scholar] [CrossRef]
- Chen, J.J.; Zhang, J.; Cai, Y.; Zhou, Y.B.; Wen, G.B.; Tang, C.S.; Qi, Y.F.; Jiang, Z.S. C-Type Natriuretic Peptide Inhibiting Vascular Calcification Might Involve Decreasing Bone Morphogenic Protein 2 and Osteopontin Levels. Mol. Cell. Biochem. 2014, 392, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.B.; Gao, Q.; Li, P.; Han, Y.; Zhang, F.; Qi, Y.F.; Tang, C.S.; Gao, X.Y.; Zhu, G.Q. Adrenomedullin Attenuates Vascular Calcification in Fructose-Induced Insulin Resistance Rats. Acta Physiol. 2013, 207, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Jin, J.S.; Yi, D.W.; Son, S.M. Bone Morphogenetic Protein-7 Inhibits Vascular Calcification Induced by High Vitamin D in Mice. Tohoku J. Exp. Med. 2010, 221, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Shanahan, C.M.; De Baaij, J.H.F. Magnesium Counteracts Vascular Calcification: Passive Interference or Active Modulation? Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar] [CrossRef]
- Ciceri, P.; Elli, F.; Cappelletti, L.; Tosi, D.; Savi, F.; Bulfamante, G.; Cozzolino, M. Osteonectin (SPARC) Expression in Vascular Calcification: In Vitro and Ex Vivo Studies. Calcif. Tissue Int. 2016, 99, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Morioka, T.; Shoji, T.; Emoto, M. The Inhibitory Roles of Vitamin k in Progression of Vascular Calcification. Nutrients 2020, 12, 583. [Google Scholar] [CrossRef] [PubMed]
- Abedi, F.; Sadeghi, M.; Omidkhoda, N.; Kelesidis, T.; Ramezani, J.; Samadi, S.; Mohammadpour, A.H. HDL-Cholesterol Concentration and Its Association with Coronary Artery Calcification: A Systematic Review and Meta-Analysis. Lipids Health Dis. 2023, 22, 60. [Google Scholar] [CrossRef] [PubMed]
- Nanao-Hamai, M.; Son, B.K.; Hashizume, T.; Ogawa, S.; Akishita, M. Protective Effects of Estrogen against Vascular Calcification via Estrogen Receptor α-Dependent Growth Arrest-Specific Gene 6 Transactivation. Biochem. Biophys. Res. Commun. 2016, 480, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tao, S.; Zhang, D.; Xiao, J.; Wang, X.; Yuan, L.; Pan, H.; Wang, D. Association between Fibrinogen/Albumin Ratio and Severity of Coronary Artery Calcification in Patients with Chronic Kidney Disease: A Retrospective Study. PeerJ 2022, 10, e13550. [Google Scholar] [CrossRef]
- Cheng, Z.Y.; Ye, T.; Ling, Q.Y.; Wu, T.; Wu, G.Y.; Zong, G.J. Parathyroid Hormone Promotes Osteoblastic Differentiation of Endothelial Cells via the Extracellular Signal-Regulated Protein Kinase 1/2 and Nuclear Factor-ΚB Signaling Pathways. Exp. Ther. Med. 2018, 15, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.B.; Chen, Y.; Xin, S.X.; Yuan, N.; Liu, Y.F.; Sun, J.B.; Meng, X.Y.; Qi, Y.F. Positive Association of Leptin and Artery Calcification of Lower Extremity in Patients With Type 2 Diabetes Mellitus: A Pilot Study. Front. Endocrinol. 2021, 12, 583575. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced Glycation End Products Induce Calcification of Vascular Smooth Muscle Cells through RAGE/P38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Rashdan, N.A.; Chapman, K.E.; Hadoke, P.W.; MacRae, V.E. A Novel Role for the Mineralocorticoid Receptor in Glucocorticoid Driven Vascular Calcification. Vascul. Pharmacol. 2016, 86, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, Z.; Li, L.; Yuan, W.; Wang, Z. Role of Collagen in Vascular Calcification. J. Cardiovasc. Pharmacol. 2022, 80, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.T.; Wang, C.G.; Zhang, T.L.; Wang, K. Fibronectin Enhances in Vitro Vascular Calcification by Promoting Osteoblastic Differentiation of Vascular Smooth Muscle Cells via ERK Pathway. J. Cell. Biochem. 2006, 99, 1343–1352. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, Y.; Liu, W.; Liu, X.; Chen, A.; Yang, X.; Li, Y.; Wang, S.; Fu, M.; Ou, J.S.; et al. 25-Hydroxycholesterol Promotes Vascular Calcification via Activation of Endoplasmic Reticulum Stress. Eur. J. Pharmacol. 2020, 880, 173165. [Google Scholar] [CrossRef]
- Peng, Y.Q.; Xiong, D.; Lin, X.; Cui, R.R.; Xu, F.; Zhong, J.Y.; Zhu, T.; Wu, F.; Mao, M.Z.; Liao, X.B.; et al. Oestrogen Inhibits Arterial Calcification by Promoting Autophagy. Sci. Rep. 2017, 7, 3549. [Google Scholar] [CrossRef]
- Chao, C.-T.; Lin, S.H. Uremic Vascular Calcification: The Pathogenic Roles and Gastrointestinal Decontamination of Uremic Toxins. Toxins 2020, 12, 812. [Google Scholar] [CrossRef]
- Stubbs, J.R.; Liu, S.; Tang, W.; Zhou, J.; Wang, Y.; Yao, X.; Quarles, L.D. Role of Hyperphosphatemia and 1,25-Dihydroxyvitamin D in Vascular Calcification and Mortality in Fibroblastic Growth Factor 23 Null Mice. J. Am. Soc. Nephrol. 2007, 18, 2116–2124. [Google Scholar] [CrossRef]
- Prosdocimo, D.A.; Wyler, S.C.; Romani, A.M.; O’Neill, W.C.; Dubyak, G.R. Regulation of Vascular Smooth Muscle Cell Calcification by Extracellular Pyrophosphate Homeostasis: Synergistic Modulation by Cyclic AMP and Hyperphosphatemia. Am. J. Physiol. Cell Physiol. 2010, 298, C702–C713. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Pan, Y.; Yang, C.; Zhang, L.; Zhao, Z.; Ye, K.; Li, L.; Xia, S.; Lu, X.; Shi, H.; et al. TNFα Activation and TGFβ Blockage Act Synergistically for Smooth Muscle Cell Calcification in Patients with Venous Thrombosis via TGFβ/ERK Pathway. J. Cell. Mol. Med. 2022, 26, 4479–4491. [Google Scholar] [CrossRef] [PubMed]
- Tóth, A.; Balogh, E.; Jeney, V. Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants 2020, 9, 963. [Google Scholar] [CrossRef]
- Liu, X.; Cao, F.; Liu, S.; Mi, Y.; Liu, J. BMP2/Smad Signaling Pathway Is Involved in the Inhibition Function of Fibroblast Growth Factor 21 on Vascular Calcification. Biochem. Biophys. Res. Commun. 2018, 503, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.F.; Yu, L.X.; Yin, X.Y.; Ye, J.M.; Li, S.S. Correlation Between Soluble Klotho and Vascular Calcification in Chronic Kidney Disease: A Meta-Analysis and Systematic Review. Front. Physiol. 2021, 12, 711904. [Google Scholar] [CrossRef] [PubMed]
- Shobeiri, N.; Bendeck, M.P. Interleukin-1β Is a Key Biomarker and Mediator of Inflammatory Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 179–180. [Google Scholar] [CrossRef]
- Henaut, L.; Massy, Z.A. New Insights into the Key Role of Interleukin 6 in Vascular Calcification of Chronic Kidney Disease. Nephrol. Dial. Transplant. 2018, 33, 543–548. [Google Scholar] [CrossRef]
- Kozakova, M.; Morizzo, C.; Jamagidze, G.; Della Latta, D.; Chiappino, S.; Chiappino, D.; Palombo, C. Association between Low-Density Lipoprotein Cholesterol and Vascular Biomarkers in Primary Prevention. Biomedicines 2023, 11, 1753. [Google Scholar] [CrossRef]
- Henze, L.A.; Luong, T.T.D.; Boehme, B.; Masyout, J.; Schneider, M.P.; Brachs, S.; Lang, F.; Pieske, B.; Pasch, A.; Eckardt, K.U.; et al. Impact of C-Reactive Protein on Osteo-/Chondrogenic Transdifferentiation and Calcification of Vascular Smooth Muscle Cells. Aging 2019, 11, 5445–5462. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Yu, C.M.; Ji, Q.W.; Cai, M.; Zhao, Y.X.; Zhou, Y.J. Current Understanding of Coronary Artery Calcification. J. Geriatr. Cardiol. 2015, 12, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Chang, H.J.; Sung, J.M.; Park, H.B.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, Y.J.; et al. Effects of Statins on Coronary Atherosclerotic Plaques: The PARADIGM Study. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, Y.; Inoue, I.; Inoue, K.; Shinoda, Y.; Iida, S.; Goto, S.; Nakano, T.; Shimada, A.; Noda, M. The Annual Rate of Coronary Artery Calcification with Combination Therapy with a PCSK9 Inhibitor and a Statin Is Lower than That with Statin Monotherapy. npj Aging Mech. Dis. 2018, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Généreux, P.; Madhavan, M.V.; Mintz, G.S.; Maehara, A.; Palmerini, T.; Lasalle, L.; Xu, K.; McAndrew, T.; Kirtane, A.; Lansky, A.J.; et al. Ischemic Outcomes after Coronary Intervention of Calcified Vessels in Acute Coronary Syndromes: Pooled Analysis from the HORIZONS-AMI (Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction) and ACUITY (Acute Catheterization. J. Am. Coll. Cardiol. 2014, 63, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Du, H.; Li, J.; Chen, X. Intracranial Artery Calcification as an Independent Predictor of Ischemic Stroke: A Systematic Review and a Meta-Analysis. BMC Neurol. 2023, 23, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Chen, Z.; Zhang, M. Arterial Calcification and Its Association With Stroke: Implication of Risk, Prognosis, Treatment Response, and Prevention. Front. Cell. Neurosci. 2022, 16, 845215. [Google Scholar] [CrossRef] [PubMed]
- Fote, G.M.; Raefsky, S.; Mock, K.; Chaudhari, A.; Shafie, M.; Yu, W. Intracranial Arterial Calcifications: Potential Biomarkers of Stroke Risk and Outcome. Front. Neurol. 2022, 13, 900579. [Google Scholar] [CrossRef] [PubMed]
- Palit, S.; Kendrick, J. Vascular Calcification in Chronic Kidney Disease: Role of Disordered Mineral Metabolism. Curr. Pharm. Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef]
- Dube, P.; DeRiso, A.; Patel, M.; Battepati, D.; Khatib-Shahidi, B.; Sharma, H.; Gupta, R.; Malhotra, D.; Dworkin, L.; Haller, S.; et al. Vascular Calcification in Chronic Kidney Disease: Diversity in the Vesselwall. Biomedicines 2021, 9, 404. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Dounousi, E.; Salmas, M.; Eleftheriadis, T.; Liakopoulos, V. Vascular Calcification in Chronic Kidney Disease: The Role of Vitamin K- Dependent Matrix Gla Protein. Front. Med. 2020, 7, 154. [Google Scholar] [CrossRef]
- Stabley, J.N.; Towler, D.A. Arterial Calcification in Diabetes Mellitus: Preclinical Models and Translational Implications. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 205–217. [Google Scholar] [CrossRef]
- Harper, E.; Forde, H.; Davenport, C.; Rochfort, K.D.; Smith, D.; Cummins, P.M. Vascular Calcification in Type-2 Diabetes and Cardiovascular Disease: Integrative Roles for OPG, RANKL and TRAIL. Vascul. Pharmacol. 2016, 82, 30–40. [Google Scholar] [CrossRef]
- Ghosh, S.; Luo, D.; He, W.; Chen, J.; Su, X.; Huang, H. Diabetes and Calcification: The Potential Role of Anti-Diabetic Drugs on Vascular Calcification Regression. Pharmacol. Res. 2020, 158, 104861. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Bourron, O.; Vemeer, C.; Theuwissen, E.; Magdeleyns, E.; Aubert, C.E.; Brazier, M.; Mentaverri, R.; Hartemann, A.; Massy, Z.A. Erratum to Vascular Calcification in Patients with Type 2 Diabetes: The Involvement of Matrix Gla Protein. Cardiovasc. Diabetol. 2015, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.; Hung, J.; Bermejo, J.; Chambers, J.B.; Evangelista, A.; Griffin, B.P.; Iung, B.; Otto, C.M.; Pellikka, P.A.; Quiñones, M. Echocardiographic Assessment of Valve Stenosis: EAE/ASE Recommendations for Clinical Practice. Eur. J. Echocardiogr. 2009, 10, 1–25. [Google Scholar] [CrossRef]
- Gaisne, R.; Péré, M.; Menoyo, V.; Hourmant, M.; Larmet-Burgeot, D. Calciphylaxis Epidemiology, Risk Factors, Treatment and Survival among French Chronic Kidney Disease Patients: A Case-Control Study. BMC Nephrol. 2020, 21, 63. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Zhao, S.; Wenger, J.; Hymes, J.L.; Maddux, F.W.; Thadhani, R.I.; Chan, K.E. A Nationally Representative Study of Calcific Uremic Arteriolopathy Risk Factors. J. Am. Soc. Nephrol. 2016, 27, 3421–3429. [Google Scholar] [CrossRef]
- Chang, J.J. Calciphylaxis: Diagnosis, Pathogenesis, and Treatment. Adv. Ski. Wound Care 2019, 32, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Olaoye, O.A.; Koratala, A. Calcific Uremic Arteriolopathy. Oxf. Med. Case Rep. 2017, 2017, 197–198. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Biomarkers of Vascular Calcification in Serum. Adv. Clin. Chem. 2020, 98, 91–147. [Google Scholar] [CrossRef]
- Clemente, A.; Traghella, I.; Mazzone, A.; Sbrana, S.; Vassalle, C. Vascular and Valvular Calcification Biomarkers, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 95, ISBN 9780128211656. [Google Scholar]
- Jahnen-Dechent, W.; Heiss, A.; Schäfer, C.; Ketteler, M. Fetuin-A Regulation of Calcified Matrix Metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar] [CrossRef]
- Hamano, T.; Matsui, I.; Mikami, S.; Tomida, K.; Fujii, N.; Imai, E.; Rakugi, H.; Isaka, Y. Fetuin-Mineral Complex Reflects Extraosseous Calcification Stress in CKD. J. Am. Soc. Nephrol. 2010, 21, 1998–2007. [Google Scholar] [CrossRef]
- Cranenburg, E.C.M.; Vermeer, C.; Koos, R.; Boumans, M.L.; Hackeng, T.M.; Bouwman, F.G.; Kwaijtaal, M.; Brandenburg, V.M.; Ketteler, M.; Schurgers, L.J. The Circulating Inactive Form of Matrix Gla Protein (UcMGP) as a Biomarker for Cardiovascular Calcification. J. Vasc. Res. 2008, 45, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E. TGFβ Signaling and Cardiovascular Diseases. Int. J. Biol. Sci. 2012, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Burgess, K.A.; Herrick, A.L.; Watson, R.E.B. Systemic Sclerosis Skin Is a Primed Microenvironment for Soft Tissue Calcification—A Hypothesis. Rheumatology 2021, 60, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.E.H.; Duca, L. Elastic Fibers: Formation, Function, and Fate during Aging and Disease. FEBS J. 2022, 289, 3704–3730. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Brandenburg, V.; Kalantar-Zadeh, K.; Stenvinkel, P.; Magnusson, P. Alkaline Phosphatase: A Novel Treatment Target for Cardiovascular Disease in CKD. Nat. Rev. Nephrol. 2017, 13, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Leonard, O.; Spaak, J.; Goldsmith, D. Regression of Vascular Calcification in Chronic Kidney Disease—Feasible or Fantasy? A Review of the Clinical Evidence. Br. J. Clin. Pharmacol. 2013, 76, 560–572. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.C.; Riser, B.L. Treatment with Pyrophosphate Inhibits Uremic Vascular Calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Jüppner, H. Phosphate and FGF-23. Kidney Int. 2011, 79, 25–28. [Google Scholar] [CrossRef]
- Hortells, L.; Sosa, C.; Guillén, N.; Lucea, S.; Millán, Á.; Sorribas, V. Identifying Early Pathogenic Events during Vascular Calcification in Uremic Rats. Kidney Int. 2017, 92, 1384–1394. [Google Scholar] [CrossRef]
- Barinda, A.J.; Ikeda, K.; Hirata, K.I.; Emoto, N. Macrophages Highly Express Carbonic Anhydrase 2 and Play a Significant Role in Demineralization of the Ectopic Calcification. Kobe J. Med. Sci. 2017, 63, E45–E50. [Google Scholar]
- Adeva-Andany, M.M.; Fernández-Fernández, C.; Sánchez-Bello, R.; Donapetry-García, C.; Martínez-Rodríguez, J. The Role of Carbonic Anhydrase in the Pathogenesis of Vascular Calcification in Humans. Atherosclerosis 2015, 241, 183–191. [Google Scholar] [CrossRef]
- Silaghi, C.N.; Ilyés, T.; Van Ballegooijen, A.J.; Crăciun, A.M. Calciprotein Particles and Serum Calcification Propensity: Hallmarks of Vascular Calcifications in Patients with Chronic Kidney Disease. J. Clin. Med. 2020, 9, 1287. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Feenstra, L.; Kostyunin, A.E.; Yuzhalin, A.E.; Hillebrands, J.L.; Krenning, G. Calciprotein Particles: Balancing Mineral Homeostasis and Vascular Pathology. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1607–1624. [Google Scholar] [CrossRef]
- Bundy, J.D.; Cai, X.; Mehta, R.C.; Scialla, J.J.; de Boer, I.H.; Hsu, C.Y.; Go, A.S.; Dobre, M.A.; Chen, J.; Rao, P.S.; et al. Serum Calcification Propensity and Clinical Events in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, M.; Kamel, S.; Choukroun, G.; Liabeuf, S.; Laville, S.M. Serum Calcification Propensity Represents a Good Biomarker of Vascular Calcification: A Systematic Review. Toxins 2022, 14, 637. [Google Scholar] [CrossRef]
- Hyafil, F.; Messika-Zeitoun, D.; Burg, S.; Rouzet, F.; Benali, K.; Iung, B.; Vahanian, A.; Le Guludec, D. Detection of 18fluoride Sodium Accumulation by Positron Emission Tomography in Calcified Stenotic Aortic Valves. Am. J. Cardiol. 2012, 109, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, W.S.A.; Vesey, A.T.; Shah, A.S.V.; Pawade, T.A.; Chin, C.W.L.; White, A.C.; Fletcher, A.; Cartlidge, T.R.G.; Mitchell, A.J.; Pringle, M.A.H.; et al. Valvular 18F-Fluoride and 18F-Fluorodeoxyglucose Uptake Predict Disease Progression and Clinical Outcome in Patients with Aortic Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1200–1201. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanihaghjo, A.; Argani, H.; Golmohamadi, Z.; Rashtchizadeh, N.; Abbasi, M.M.; Bargahi, N.; Vatankhah, A.M.; Sanajou, D. Linkage of Fibroblast Growth Factor 23 and Phosphate in Serum: Phosphate and Fibroblast Growth Factor 23 Reduction by Increasing Dose of Sevelamer. J. Bone Metab. 2018, 25, 153. [Google Scholar] [CrossRef]
- Adema, A.Y.; De Jong, M.A.; De Borst, M.H.; Ter Wee, P.M.; Vervloet, M.G. Phosphate Binding Therapy to Lower Serum Fibroblast-Growth-Factor-23 Concentrations in Chronic Kidney Disease: Rationale and Study Design of the Sevelamer on FGF23 Trial (SoFT). Nephron 2016, 134, 215–220. [Google Scholar] [CrossRef]
- Giger, E.V.; Castagner, B.; Leroux, J.C. Biomedical Applications of Bisphosphonates. J. Control. Release 2013, 167, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.E.; Gottesman, G.S.; McAlister, W.H.; Mumm, S.; Madson, K.L.; Kiffer-Moreira, T.; Sheen, C.; Millán, J.L.; Ericson, K.L.; Whyte, M.P. Severe Skeletal Toxicity from Protracted Etidronate Therapy for Generalized Arterial Calcification of Infancy. J. Bone Miner. Res. 2013, 28, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Oikonomaki, T.; Papasotiriou, M.; Ntrinias, T.; Kalogeropoulou, C.; Zabakis, P.; Kalavrizioti, D.; Papadakis, I.; Goumenos, D.S.; Papachristou, E. The Effect of Vitamin K2 Supplementation on Vascular Calcification in Haemodialysis Patients: A 1-Year Follow-up Randomized Trial. Int. Urol. Nephrol. 2019, 51, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- de Vriese, A.S.; Caluwé, R.; Pyfferoen, L.; de Bacquer, D.; de Boeck, K.; Delanote, J.; de Surgeloose, D.; van Hoenacker, P.; van Vlem, B.; Verbeke, F. Multicenter Randomized Controlled Trial of Vitamin K Antagonist Replacement by Rivaroxaban with or without Vitamin K2 in Hemodialysis Patients with Atrial Fibrillation: The Valkyrie Study. J. Am. Soc. Nephrol. 2020, 31, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Portales-Castillo, I.; Seethapathy, R.; Durant, O.; Mengesha, B.; Krinsky, S.; Kroshinsky, D.; Kalim, S.; Goverman, J.; Nazarian, R.M.; et al. Intravenous Sodium Thiosulphate for Calciphylaxis of Chronic Kidney Disease: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2023, 6, E2310068. [Google Scholar] [CrossRef] [PubMed]
- Djuric, P.; Dimkovic, N.; Schlieper, G.; Djuric, Z.; Pantelic, M.; Mitrovic, M.; Jankovic, A.; Milanov, M.; Kuzmanovic Pficer, J.; Floege, J. Sodium Thiosulphate and Progression of Vascular Calcification in End-Stage Renal Disease Patients: A Double-Blind, Randomized, Placebo-Controlled Study. Nephrol. Dial. Transplant. 2020, 35, 162–169. [Google Scholar] [CrossRef]
- Zu, Y.; Lu, X.; Song, J.; Yu, L.; Li, H.; Wang, S. Cinacalcet Treatment Significantly Improves All-Cause and Cardiovascular Survival in Dialysis Patients: Results from a Meta-Analysis. Kidney Blood Press. Res. 2019, 44, 1327–1338. [Google Scholar] [CrossRef]
- Torres, P.A.U.; De Broe, M. Calcium-Sensing Receptor, Calcimimetics, and Cardiovascular Calcifications in Chronic Kidney Disease. Kidney Int. 2012, 82, 19–25. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Ketteler, M.; Tur, F.; Tur, E.; Isern, B.; Salcedo, C.; Joubert, P.H.; Behets, G.J.; Neven, E.; D’Haese, P.C.; et al. Characterization of SNF472 Pharmacokinetics and Efficacy in Uremic and Non-Uremic Rats Models of Cardiovascular Calcification. PLoS ONE 2018, 13, e0197061. [Google Scholar] [CrossRef]
- Hedayati, S.S. A Novel Treatment for Vascular Calcification in Patients with Dialysis-Dependent Chronic Kidney Disease: Are We There Yet? Circulation 2020, 141, 740–742. [Google Scholar] [CrossRef]
- Singh, A.; Tandon, S.; Tandon, C. An Update on Vascular Calcification and Potential Therapeutics. Mol. Biol. Rep. 2021, 48, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Helas, S.; Goettsch, C.; Schoppet, M.; Zeitz, U.; Hempel, U.; Morawietz, H.; Kostenuik, P.J.; Erben, R.G.; Hofbauer, L.C. Inhibition of Receptor Activator of NF-κB Ligand by Denosumab Attenuates Vascular Calcium Deposition in Mice. Am. J. Pathol. 2009, 175, 473–478. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, M.A.; De Leon-Oliva, D.; Gimeno-Longas, M.J.; Boaru, D.L.; Fraile-Martinez, O.; García-Montero, C.; de Castro, A.V.; Barrena-Blázquez, S.; López-González, L.; Amor, S.; et al. Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities. Biomolecules 2024, 14, 275. https://doi.org/10.3390/biom14030275
Ortega MA, De Leon-Oliva D, Gimeno-Longas MJ, Boaru DL, Fraile-Martinez O, García-Montero C, de Castro AV, Barrena-Blázquez S, López-González L, Amor S, et al. Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities. Biomolecules. 2024; 14(3):275. https://doi.org/10.3390/biom14030275
Chicago/Turabian StyleOrtega, Miguel A., Diego De Leon-Oliva, Maria José Gimeno-Longas, Diego Liviu Boaru, Oscar Fraile-Martinez, Cielo García-Montero, Amador Velazquez de Castro, Silvestra Barrena-Blázquez, Laura López-González, Silvia Amor, and et al. 2024. "Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities" Biomolecules 14, no. 3: 275. https://doi.org/10.3390/biom14030275
APA StyleOrtega, M. A., De Leon-Oliva, D., Gimeno-Longas, M. J., Boaru, D. L., Fraile-Martinez, O., García-Montero, C., de Castro, A. V., Barrena-Blázquez, S., López-González, L., Amor, S., García-Honduvilla, N., Buján, J., Guijarro, L. G., Castillo-Ruiz, E., Álvarez-Mon, M. Á., Albillos, A., Álvarez-Mon, M., Diaz, R., & Saez, M. A. (2024). Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities. Biomolecules, 14(3), 275. https://doi.org/10.3390/biom14030275