
Guardians of the Genome: How the Single-Stranded DNA-Binding Proteins RPA and CST Facilitate Telomere Replication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
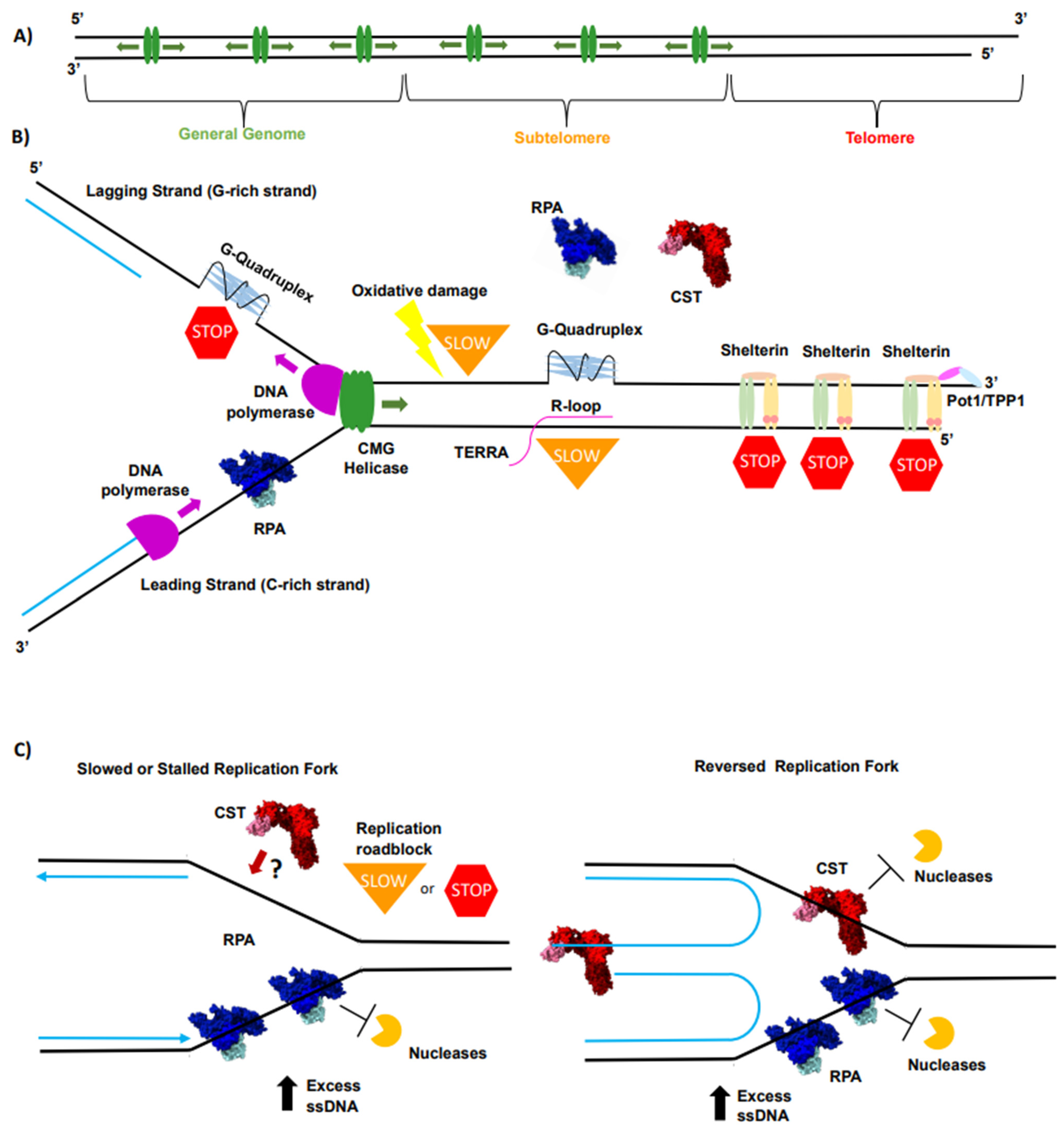
2. Telomeres Are the “Problem Child” for Conventional Replication Machinery
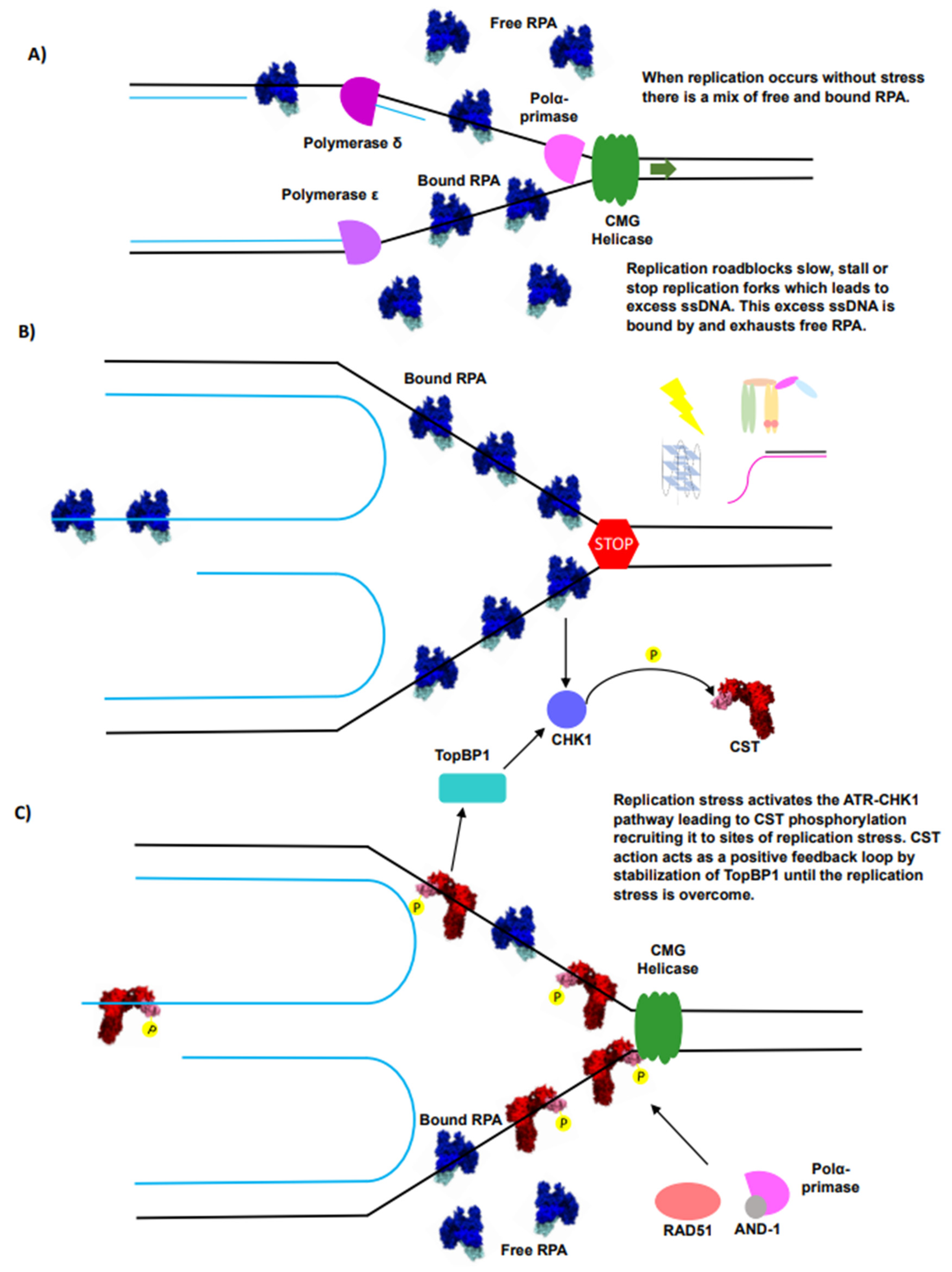
3. RPA Is the ssDNA Guardian of the Genome
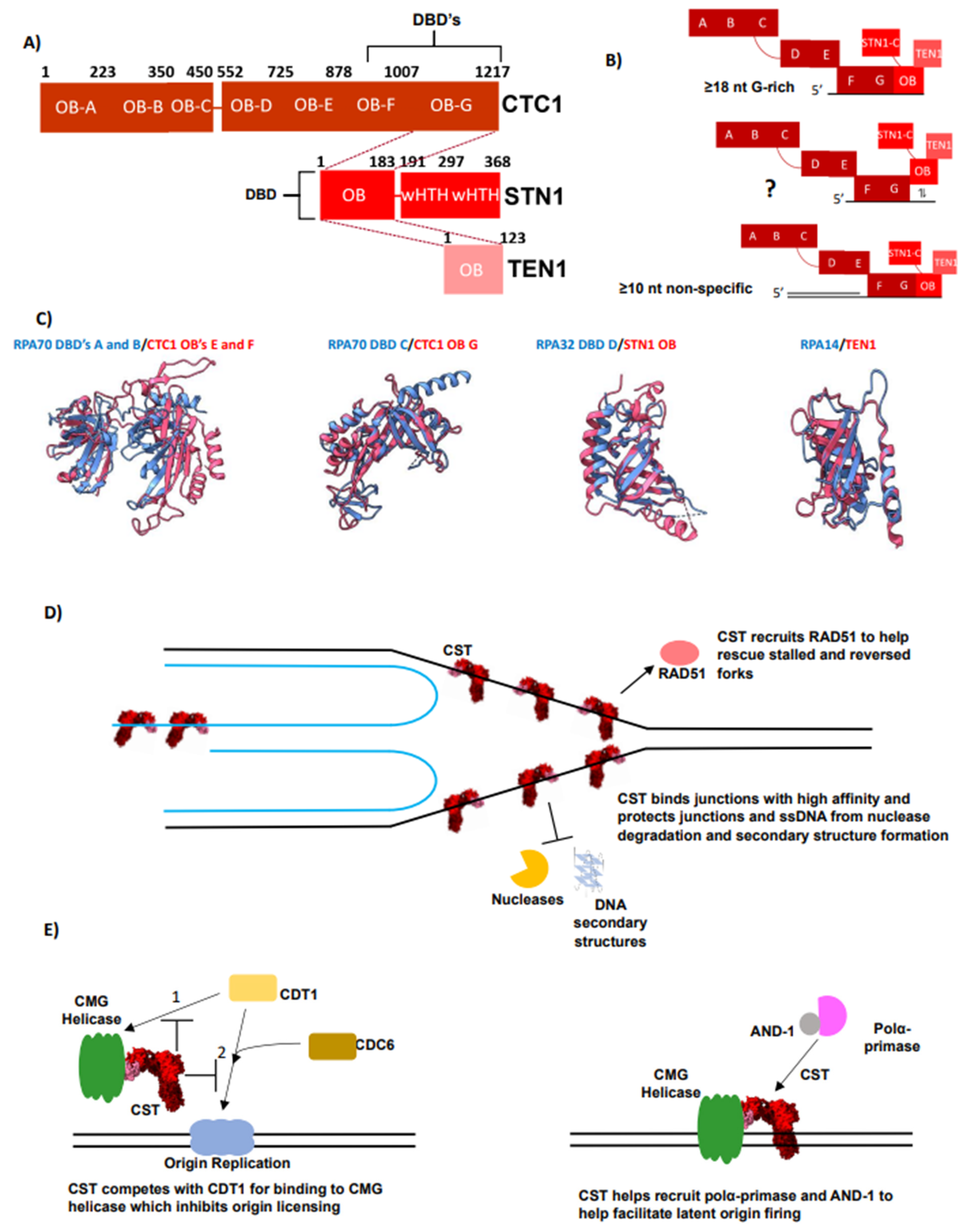
4. CST Is an RPA-Like Protein Essential for Telomere Overhang Replication
5. The Interplay of RPA and CST Supports Efficient Telomere Replication
6. CST Functions Outside of Telomeres
7. Conclusions and Outstanding Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olovnikov, A.M. A Theory of Marginotomy. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Watson, J. Origin of Concatemeric T7DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.V.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere Length Predicts Replicative Capacity of Human Fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10114–10118. [Google Scholar] [CrossRef]
- Lim, C.J.; Cech, T.R. Shaping Human Telomeres: From Shelterin and CST Complexes to Telomeric Chromatin Organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Moyzis, R.K.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.-R. A Highly Conserved Repetitive DNA Sequence, (TTAGGG)., Present at the Telomeres of Human Chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef]
- Makarov, V.L.; Hirose, Y.; Langmore, J.P. Long G Tails at Both Ends of Human Chromosomes Suggest a C Strand Degradation Mechanism for Telomere Shortening. Cell 1997, 88, 657–666. [Google Scholar] [CrossRef]
- Zhao, Y.; Hoshiyama, H.; Shay, J.W.; Wright, W.E. Quantitative Telomeric Overhang Determination Using a Double-Strand Specific Nuclease. Nucleic Acids Res. 2007, 36, e14. [Google Scholar] [CrossRef]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, Health, and Hallmarks of Aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef]
- Grill, S.; Nandakumar, J. Molecular Mechanisms of Telomere Biology Disorders. J. Biol. Chem. 2021, 296, 100064. [Google Scholar] [CrossRef]
- Maciejowski, J.; De Lange, T. Telomeres in Cancer: Tumour Suppression and Genome Instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Brenner, K.A.; Nandakumar, J. Consequences of Telomere Replication Failure: The Other End-Replication Problem. Trends Biochem. Sci. 2022, 47, 506–517. [Google Scholar] [CrossRef]
- Lu, R.; Pickett, H.A. Telomeric Replication Stress: The Beginning and the End for Alternative Lengthening of Telomeres Cancers. Open Biol. 2022, 12, 220011. [Google Scholar] [CrossRef]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human Telomeres Contain Two Distinct Myb–Related Proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–235. [Google Scholar] [CrossRef]
- Van Steensel, B.; de Lange, T. Control of Telomere Length by the Human Telomeric Protein TRF1. Nature 1997, 385, 740–743. [Google Scholar] [CrossRef]
- Bilaud, T.; Brun, C.; Ancelin, K.; Koering, C.E.; Laroche, T.; Gilson, E. Telomeric Localization of TRF2, a Novel Human Telobox Protein. Nat. Genet. 1997, 17, 236–239. [Google Scholar] [CrossRef]
- Bianchi, A. TRF1 Is a Dimer and Bends Telomeric DNA. EMBO J. 1997, 16, 1785–1794. [Google Scholar] [CrossRef]
- Ye, J.Z.-S.; Donigian, J.R.; van Overbeek, M.; Loayza, D.; Luo, Y.; Krutchinsky, A.N.; Chait, B.T.; de Lange, T. TIN2 Binds TRF1 and TRF2 Simultaneously and Stabilizes the TRF2 Complex on Telomeres. J. Biol. Chem. 2004, 279, 47264–47271. [Google Scholar] [CrossRef]
- Liu, D.; O’Connor, M.S.; Qin, J.; Songyang, Z. Telosome, a Mammalian Telomere-Associated Complex Formed by Multiple Telomeric Proteins. J. Biol. Chem. 2004, 279, 51338–51342. [Google Scholar] [CrossRef]
- O’Connor, M.S.; Safari, A.; Xin, H.; Liu, D.; Songyang, Z. A Critical Role for TPP1 and TIN2 Interaction in High-Order Telomeric Complex Assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11874–11879. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1–TPP1 Telomere Complex Is a Telomerase Processivity Factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef]
- Kibe, T.; Osawa, G.A.; Keegan, C.E.; De Lange, T. Telomere Protection by TPP1 Is Mediated by POT1a and POT1b. Mol. Cell. Biol. 2010, 30, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cech, T.R. Pot1, the Putative Telomere End-Binding Protein in Fission Yeast and Humans. Science 2001, 292, 1171–1175. [Google Scholar] [CrossRef]
- Zaug, A.J.; Podell, E.R.; Cech, T.R. Human POT1 Disrupts Telomeric G-Quadruplexes Allowing Telomerase Extension In Vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 10864–10869. [Google Scholar] [CrossRef] [PubMed]
- Latrick, C.M.; Cech, T.R. POT1–TPP1 Enhances Telomerase Processivity by Slowing Primer Dissociation and Aiding Translocation. EMBO J. 2010, 29, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Sekne, Z.; Ghanim, G.E.; Van Roon, A.-M.M.; Nguyen, T.H.D. Structural Basis of Human Telomerase Recruitment by TPP1-POT1. Science 2022, 375, 1173–1176. [Google Scholar] [CrossRef]
- Liu, B.; He, Y.; Wang, Y.; Song, H.; Zhou, Z.H.; Feigon, J. Structure of Active Human Telomerase with Telomere Shelterin Protein TPP1. Nature 2022, 604, 578–583. [Google Scholar] [CrossRef]
- Nandakumar, J.; Bell, C.F.; Weidenfeld, I.; Zaug, A.J.; Leinwand, L.A.; Cech, T.R. The TEL Patch of Telomere Protein TPP1 Mediates Telomerase Recruitment and Processivity. Nature 2012, 492, 285–289. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a Specific Telomere Terminal Transferase Activity in Tetrahymena Extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A Telomeric Sequence in the RNA of Tetrahymena Telomerase Required for Telomere Repeat Synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef]
- Lingner, J.; Hughes, T.R.; Shevchenko, A.; Mann, M.; Lundblad, V.; Cech, T.R. Reverse Transcriptase Motifs in the Catalytic Subunit of Telomerase. Science 1997, 276, 561–567. [Google Scholar] [CrossRef]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Roake, C.M.; Artandi, S.E. Regulation of Human Telomerase in Homeostasis and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Abreu, E.; Kim, J.; Stadler, G.; Eskiocak, U.; Terns, M.P.; Terns, R.M.; Shay, J.W.; Wright, W.E. Processive and Distributive Extension of Human Telomeres by Telomerase under Homeostatic and Nonequilibrium Conditions. Mol. Cell 2011, 42, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Y.; Redon, S.; Lingner, J. The Human CST Complex Is a Terminator of Telomerase Activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef]
- Zaug, A.J.; Lim, C.J.; Olson, C.L.; Carilli, M.T.; Goodrich, K.J.; Wuttke, D.S.; Cech, T.R. CST Does Not Evict Elongating Telomerase but Prevents Initiation by ssDNA Binding. Nucleic Acids Res. 2021, 49, 11653–11665. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST Has Independent Functions during Telomere Duplex Replication and C-Strand Fill-In. Cell Rep. 2012, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Lin, X.; Chavez, B.L.; Agrawal, S.; Lusk, B.L.; Lim, C.J. Structures of the human CST-Polα-primase complex bound to telomere templates. Nature 2022, 608, 826–832. [Google Scholar] [CrossRef]
- Olson, C.L.; Barbour, A.T.; Wuttke, D.S. Filling in the Blanks: How the C-Strand Catches up to the G-Strand at Replicating Telomeres Using CST. Nat. Struct. Mol. Biol. 2022, 29, 730–733. [Google Scholar] [CrossRef]
- Mason, J.M.; Frydrychova, R.C.; Biessmann, H. Drosophila Telomeres: An Exception Providing New Insights. BioEssays 2008, 30, 25–37. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zou, L. Alternative Lengthening of Telomeres_ from Molecular Mechanism to Therapeutic Outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Lenain, C.; Bauwens, S.; Amiard, S.; Brunori, M.; Giraud-Panis, M.-J.; Gilson, E. The Apollo 5′ Exonuclease Functions Together with TRF2 to Protect Telomeres from DNA Repair. Curr. Biol. 2006, 16, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.C.; Akhter, S.; Gu, P.; Ye, J.; Poulet, A.; Giraud-Panis, M.-J.; Bailey, S.M.; Gilson, E.; Legerski, R.J.; Chang, S. SNMIB/Apollo Protects Leading-Strand Telomeres against NHEJ-Mediated Repair. EMBO J. 2010, 29, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Takai, H.; de Lange, T. Telomeric 3′ Overhangs Derive from Resection by Exo1 and Apollo and Fill-In by POT1b-Associated CST. Cell 2012, 150, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Lingner, J.; Cooper, J.P.; Cech, T.R. Telomerase and DNA end replication: No longer a lagging strand problem? Science 1995, 269, 1533–1534. [Google Scholar] [CrossRef]
- Dai, X.; Huang, C.; Bhusari, A.; Sampathi, S.; Schubert, K.; Chai, W. Molecular Steps of G-Overhang Generation at Human Telomeres and Its Function in Chromosome End Protection. EMBO J. 2010, 29, 2788–2801. [Google Scholar] [CrossRef]
- Chow, T.T.; Zhao, Y.; Mak, S.S.; Shay, J.W.; Wright, W.E. Early and Late Steps in Telomere Overhang Processing in Normal Human Cells: The Position of the Final RNA Primer Drives Telomere Shortening. Genes Dev. 2012, 26, 1167–1178. [Google Scholar] [CrossRef]
- Flynn, R.L.; Centore, R.C.; O’Sullivan, R.J.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 Orchestrate an RPA-to-POT1 Switch on Telomeric Single-Stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef]
- Lin, C.-Y.G.; Näger, A.C.; Lunardi, T.; Vančevska, A.; Lossaint, G.; Lingner, J. The Human Telomeric Proteome during Telomere Replication. Nucleic Acids Res. 2021, 49, 12119–12135. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; De Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Timashev, L.A.; De Lange, T. Characterization of T-Loop Formation by TRF2. Nucleus 2020, 11, 164–177. [Google Scholar] [CrossRef]
- Bonnell, E.; Pasquier, E.; Wellinger, R.J. Telomere Replication: Solving Multiple End Replication Problems. Front. Cell Dev. Biol. 2021, 9, 668171. [Google Scholar] [CrossRef]
- Saxena, S.; Zou, L. Hallmarks of DNA Replication Stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Replication Protein A: A Multifunctional Protein with Roles in DNA Replication, Repair and Beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.A.; Wang, F.; Chaiken, M.F.; Kasbek, C.; Chastain, P.D.; Wright, W.E.; Price, C.M. Human CST Promotes Telomere Duplex Replication and General Replication Restart after Fork Stalling: CST Promotes Replication Restart. EMBO J. 2012, 31, 3537–3549. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, W.C.; Deng, Z.; Twayana, S.; Kosiyatrakul, S.T.; Vladimirova, O.; Lieberman, P.M.; Schildkraut, C.L. TRF2 Mediates Replication Initiation within Human Telomeres to Prevent Telomere Dysfunction. Cell Rep. 2020, 33, 108379. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, W.C.; Kosiyatrakul, S.T.; Yan, Z.; Calderano, S.G.; Schildkraut, C.L. Human Telomeres Replicate Using Chromosome-Specific, Rather than Universal, Replication Programs. J. Cell Biol. 2012, 197, 253–266. [Google Scholar] [CrossRef]
- Canceill, D.; Ehrlich, S.D. Copy-Choice Recombination Mediated by DNA Polymerase III Holoenzyme from Escherichia coli. Proc. Natl. Acad. Sci. USA 1996, 93, 6647–6652. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Ohshima, K.; Shimizu, M.; Amirhaeri, S.; Wells, R.D. Pausing of DNA Synthesis in Vitro at Specific Loci in CTG and CGG Triplet Repeats from Human Hereditary Disease Genes. J. Biol. Chem. 1995, 270, 27014–27021. [Google Scholar] [CrossRef]
- Hartenstine, M.J.; Goodman, M.F.; Petruska, J. Base Stacking and Even/Odd Behavior of Hairpin Loops in DNA Triplet Repeat Slippage and Expansion with DNA Polymerase. J. Biol. Chem. 2000, 275, 18382–18390. [Google Scholar] [CrossRef]
- Williamson, J.R.; Raghuraman, M.K.; Cech, T.R. Monovalent cation-induced structure of telomeric DNA: The G-quartet model. Cell 1989, 59, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The Regulation and Functions of DNA and RNA G-Quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M. G-Quadruplexes at Telomeres: Friend or Foe? Molecules 2020, 25, 3686. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The Impact of Replication Stress on Replication Dynamics and DNA Damage in Vertebrate Cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; Van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ Promotes DNA Synthesis through G-quadruplex Structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M. Mechanisms of DNA Replication and Repair: Insights from the Study of G-Quadruplexes. Molecules 2019, 24, 3439. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Shao, R.; Kwong Yung, P.Y.; Elsässer, S.J. Genome-Wide Mapping of G-Quadruplex Structures with CUT&Tag. Nucleic Acids Res. 2022, 50, e13. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative Visualization of DNA G-Quadruplex Structures in Human Cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-Quadruplex Structures Mark Human Regulatory Chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Spiegel, J.; Marsico, G.; Tannahill, D.; Balasubramanian, S. Genome-Wide Mapping of Endogenous G-Quadruplex DNA Structures by Chromatin Immunoprecipitation and High-Throughput Sequencing. Nat. Protoc. 2018, 13, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Vannier, J.-B.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.I.R.; Ding, H.; Boulton, S.J. RTEL1 Dismantles T Loops and Counteracts Telomeric G4-DNA to Maintain Telomere Integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, W.C.; Kosiyatrakul, S.T.; Schildkraut, C.L. BLM Helicase Facilitates Telomere Replication during Leading Strand Synthesis of Telomeres. J. Cell Biol. 2015, 210, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Nguyen, G.H.; Oshima, J.; Maizels, N.; Harris, C.C.; Monnat, R.J. The Werner Syndrome RECQ Helicase Targets G4 DNA in Human Cells to Modulate Transcription. Hum. Mol. Genet. 2016, 25, 2060–2069. [Google Scholar] [CrossRef]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric Repeat–Containing RNA and RNA Surveillance Factors at Mammalian Chromosome Ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Feuerhahn, S.; Reichenbach, P.; Lingner, J. Molecular Dissection of Telomeric Repeat-Containing RNA Biogenesis Unveils the Presence of Distinct and Multiple Regulatory Pathways. Mol. Cell. Biol. 2010, 30, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Bonetti, D.; Lockhart, A.; Serhal, K.; Kellner, V.; Maicher, A.; Jolivet, P.; Teixeira, M.T.; Luke, B. Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell 2017, 170, 72–85.e14. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; De Lange, T. ATRX Affects the Repair of Telomeric DSBs by Promoting Cohesion and a DAXX-Dependent Activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative Lengthening of Telomeres Renders Cancer Cells Hypersensitive to ATR Inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic Oxidative Stress Compromises Telomere Integrity and Accelerates the Onset of Senescence in Human Endothelial Cells. J. Cell Sci. 2004, 117, 2417–2426. [Google Scholar] [CrossRef]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The Impact of Oxidative DNA Damage and Stress on Telomere Homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef]
- Barnes, R.P.; De Rosa, M.; Thosar, S.A.; Detwiler, A.C.; Roginskaya, V.; Van Houten, B.; Bruchez, M.P.; Stewart-Ornstein, J.; Opresko, P.L. Telomeric 8-Oxo-Guanine Drives Rapid Premature Senescence in the Absence of Telomere Shortening. Nat. Struct. Mol. Biol. 2022, 29, 639–652. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Warraich, U.-A.; Hussain, F.; Kayani, H.U.R. Aging—Oxidative Stress, Antioxidants and Computational Modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef]
- Douglas, M.E.; Diffley, J.F.X. Budding Yeast Rap1, but Not Telomeric DNA, Is Inhibitory for Multiple Stages of DNA Replication in Vitro. Nucleic Acids Res. 2021, 49, 5671–5683. [Google Scholar] [CrossRef]
- Ohki, R. Telomere-Bound TRF1 and TRF2 Stall the Replication Fork at Telomeric Repeats. Nucleic Acids Res. 2004, 32, 1627–1637. [Google Scholar] [CrossRef]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; De Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef]
- Zimmermann, M.; Kibe, T.; Kabir, S.; De Lange, T. TRF1 Negotiates TTAGGG Repeat-Associated Replication Problems by Recruiting the BLM Helicase and the TPP1/POT1 Repressor of ATR Signaling. Genes Dev. 2014, 28, 2477–2491. [Google Scholar] [CrossRef]
- Martínez, P.; Thanasoula, M.; Muñoz, P.; Liao, C.; Tejera, A.; McNees, C.; Flores, J.M.; Fernández-Capetillo, O.; Tarsounas, M.; Blasco, M.A. Increased Telomere Fragility and Fusions Resulting from TRF1 Deficiency Lead to Degenerative Pathologies and Increased Cancer in Mice. Genes Dev. 2009, 23, 2060–2075. [Google Scholar] [CrossRef]
- Yang, Z.; Sharma, K.; De Lange, T. TRF1 Uses a Noncanonical Function of TFIIH to Promote Telomere Replication. Genes Dev. 2022, 36, 956–969. [Google Scholar] [CrossRef]
- Sarek, G.; Kotsantis, P.; Ruis, P.; Van Ly, D.; Margalef, P.; Borel, V.; Zheng, X.-F.; Flynn, H.R.; Snijders, A.P.; Chowdhury, D.; et al. CDK Phosphorylation of TRF2 Controls T-Loop Dynamics during the Cell Cycle. Nature 2019, 575, 523–527. [Google Scholar] [CrossRef]
- Stansel, R.M. T-Loop Assembly in Vitro Involves Binding of TRF2 near the 3′ Telomeric Overhang. EMBO J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef]
- Nera, B.; Huang, H.-S.; Lai, T.; Xu, L. Elevated Levels of TRF2 Induce Telomeric Ultrafine Anaphase Bridges and Rapid Telomere Deletions. Nat. Commun. 2015, 6, 10132. [Google Scholar] [CrossRef]
- Williams, R.M.; Zhang, X. Roles of ATM and ATR in DNA Double Strand Breaks and Replication Stress. Prog. Biophys. Mol. Biol. 2021, 161, 27–38. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork Reversal, Fork Protection, and Genome Stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef]
- Wang, F.; Stewart, J.; Price, C.M. Human CST Abundance Determines Recovery from Diverse Forms of DNA Damage and Replication Stress. Cell Cycle 2014, 13, 3488–3498. [Google Scholar] [CrossRef]
- Chastain, M.; Zhou, Q.; Shiva, O.; Fadri-Moskwik, M.; Whitmore, L.; Jia, P.; Dai, X.; Huang, C.; Ye, P.; Chai, W. Human CST Facilitates Genome-Wide RAD51 Recruitment to GC-Rich Repetitive Sequences in Response to Replication Stress. Cell Rep. 2016, 16, 1300–1314. [Google Scholar] [CrossRef]
- Lyu, X.; Sang, P.B.; Chai, W. CST in Maintaining Genome Stability: Beyond Telomeres. DNA Repair 2021, 102, 103104. [Google Scholar] [CrossRef]
- Wold, M.S. REPLICATION PROTEIN A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Litman Flynn, R.; Chang, S.; Zou, L. RPA and POT1: Friends or Foes at Telomeres? Cell Cycle 2012, 11, 652–657. [Google Scholar] [CrossRef]
- Fan, J.; Pavletich, N.P. Structure and Conformational Change of a Replication Protein A Heterotrimer Bound to ssDNA. Genes Dev. 2012, 26, 2337–2347. [Google Scholar] [CrossRef]
- Bochkarev, A.; Pfuetzner, R.A.; Edwards, A.M.; Frappier, L. Structure of the Single-Stranded-DNA-Binding Domain of Replication Protein A Bound to DNA. Nature 1997, 385, 176–181. [Google Scholar] [CrossRef]
- Pfuetzner, R.A.; Bochkarev, A.; Frappier, L.; Edwards, A.M. Replication Protein A. J. Biol. Chem. 1997, 272, 430–434. [Google Scholar] [CrossRef]
- Bochkareva, E. Structure of the RPA Trimerization Core and Its Role in the Multistep DNA-Binding Mechanism of RPA. EMBO J. 2002, 21, 1855–1863. [Google Scholar] [CrossRef]
- Dickey, T.H.; Altschuler, S.E.; Wuttke, D.S. Single-Stranded DNA-Binding Proteins: Multiple Domains for Multiple Functions. Structure 2013, 21, 1074–1084. [Google Scholar] [CrossRef]
- Nguyen, D.-D.; Kim, E.Y.; Sang, P.B.; Chai, W. Roles of OB-Fold Proteins in Replication Stress. Front. Cell Dev. Biol. 2020, 8, 574466. [Google Scholar] [CrossRef]
- Wu, Y.; Fu, W.; Zang, N.; Zhou, C. Structural Characterization of Human RPA70N Association with DNA Damage Response Proteins. eLife 2023, 12, e81639. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. RPA-Coated Single-Stranded DNA as a Platform for Post-Translational Modifications in the DNA Damage Response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef]
- Yates, L.A.; Aramayo, R.J.; Pokhrel, N.; Caldwell, C.C.; Kaplan, J.A.; Perera, R.L.; Spies, M.; Antony, E.; Zhang, X. A Structural and Dynamic Model for the Assembly of Replication Protein A on Single-Stranded DNA. Nat. Commun. 2018, 9, 5447. [Google Scholar] [CrossRef]
- Brosey, C.A.; Yan, C.; Tsutakawa, S.E.; Heller, W.T.; Rambo, R.P.; Tainer, J.A.; Ivanov, I.; Chazin, W.J. A New Structural Framework for Integrating Replication Protein A into DNA Processing Machinery. Nucleic Acids Res. 2013, 41, 2313–2327. [Google Scholar] [CrossRef] [PubMed]
- Wieser, T.A.; Wuttke, D.S. Replication Protein A Utilizes Differential Engagement of Its DNA-Binding Domains to Bind Biologically Relevant ssDNAs in Diverse Binding Modes. Biochemistry 2022, 61, 2592–2606. [Google Scholar] [CrossRef]
- Kim, C.; Wold, M.S. Recombinant Human Replication Protein A Binds to Polynucleotides with Low Cooperativity. Biochemistry 1995, 34, 2058–2064. [Google Scholar] [CrossRef]
- Bastin-Shanower, S.A.; Brill, S.J. Functional Analysis of the Four DNA Binding Domains of Replication Protein A. J. Biol. Chem. 2001, 276, 36446–36453. [Google Scholar] [CrossRef]
- Walther, A.P.; Gomes, X.V.; Lao, Y.; Lee, C.G.; Wold, M.S. Replication Protein A Interactions with DNA. 1. Functions of the DNA-Binding and Zinc-Finger Domains of the 70-kDa Subunit. Biochemistry 1999, 38, 3963–3973. [Google Scholar] [CrossRef]
- Iftode, C.; Borowiec, J.A. 5‘ → 3‘ Molecular Polarity of Human Replication Protein A (hRPA) Binding to Pseudo-Origin DNA Substrates. Biochemistry 2000, 39, 11970–11981. [Google Scholar] [CrossRef]
- Kolpashchikov, D.M. Polarity of Human Replication Protein A Binding to DNA. Nucleic Acids Res. 2001, 29, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E. A Dynamic Model for Replication Protein A (RPA) Function in DNA Processing Pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef]
- Pokhrel, N.; Caldwell, C.C.; Corless, E.I.; Tillison, E.A.; Tibbs, J.; Jocic, N.; Tabei, S.M.A.; Wold, M.S.; Spies, M.; Antony, E. Dynamics and Selective Remodeling of the DNA-Binding Domains of RPA. Nat. Struct. Mol. Biol. 2019, 26, 129–136. [Google Scholar] [CrossRef]
- Sangeeta; Bhattacherjee, A. Interdomain Dynamics in Human Replication Protein A Regulates Kinetics and Thermodynamics of Its Binding to ssDNA. PLoS ONE 2023, 18, e0278396. [Google Scholar] [CrossRef]
- Krasikova, Y.S.; Rechkunova, N.I.; Lavrik, O.I. Replication Protein A as a Major Eukaryotic Single-Stranded DNA-Binding Protein and Its Role in DNA Repair. Mol. Biol. 2016, 50, 649–662. [Google Scholar] [CrossRef]
- Kuppa, S.; Deveryshetty, J.; Chadda, R.; Mattice, J.R.; Pokhrel, N.; Kaushik, V.; Patterson, A.; Dhingra, N.; Pangeni, S.; Sadauskas, M.K.; et al. Rtt105 regulates RPA function by configurationally stapling the flexible domains. Nat. Commun. 2022, 13, 5152. [Google Scholar] [CrossRef]
- Arunkumar, A.I.; Stauffer, M.E.; Bochkareva, E.; Bochkarev, A.; Chazin, W.J. Independent and Coordinated Functions of Replication Protein A Tandem High Affinity Single-Stranded DNA Binding Domains. J. Biol. Chem. 2003, 278, 41077–41082. [Google Scholar] [CrossRef]
- Olson, C.L.; Barbour, A.T.; Wieser, T.A.; Wuttke, D.S. RPA Engages Telomeric G-Quadruplexes More Effectively than CST. Nucleic Acids Res. 2023, 51, 5073–5086. [Google Scholar] [CrossRef]
- Spegg, V.; Panagopoulos, A.; Stout, M.; Krishnan, A.; Reginato, G.; Imhof, R.; Roschitzki, B.; Cejka, P.; Altmeyer, M. Phase Separation Properties of RPA Combine High-Affinity ssDNA Binding with Dynamic Condensate Functions at Telomeres. Nat. Struct. Mol. Biol. 2023, 30, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.M.; Friend, C.M.; Bell, S.P. Distinct RPA Functions Promote Eukaryotic DNA Replication Initiation and Elongation. Nucleic Acids Res. 2023, 51, 10506–10518. [Google Scholar] [CrossRef]
- Braun, K.A.; Lao, Y.; He, Z.; Ingles, C.J.; Wold, M.S. Role of protein-protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase alpha by multiple mechanisms. Biochemistry 1997, 36, 8443–8454. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Stillman, B. Multiple replication factors augment DNA synthesis by the two eukaryotic DNA polymerases, alpha and delta. EMBO J. 1989, 8, 3883–3889. [Google Scholar] [CrossRef]
- Kenny, M.K.; Lee, S.-H.; Hurwitz, J. Multiple Functions of Human Single-Stranded-DNA Binding Protein in Simian Virus 40 DNA Replication: Single-Strand Stabilization and Stimulation of DNA Polymerases a and 6. Proc. Natl. Acad. Sci. USA 1989, 86, 9757–9761. [Google Scholar] [CrossRef]
- Waga, S.; Stillman, B. The DNA Replication Fork in Eukaryotic Cells. Annu. Rev. Biochem. 1998, 67, 721–751. [Google Scholar] [CrossRef]
- Ciccia, A.; Bredemeyer, A.L.; Sowa, M.E.; Terret, M.-E.; Jallepalli, P.V.; Harper, J.W.; Elledge, S.J. The SIOD Disorder Protein SMARCAL1 Is an RPA-Interacting Protein Involved in Replication Fork Restart. Genes Dev. 2009, 23, 2415–2425. [Google Scholar] [CrossRef]
- Bansbach, C.E.; Bétous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The Annealing Helicase SMARCAL1 Maintains Genome Integrity at Stalled Replication Forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Brissett, N.C.; Ehlinger, A.; Keen, B.A.; Kolesar, P.; Taylor, E.M.; Bailey, L.J.; Lindsay, H.D.; Chazin, W.J.; Doherty, A.J. Molecular Basis for PrimPol Recruitment to Replication Forks by RPA. Nat. Commun. 2017, 8, 15222. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Spies, M. Dynamic Elements of Replication Protein A at the Crossroads of DNA Replication, Recombination, and Repair. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 482–507. [Google Scholar] [CrossRef]
- Cai, S.W.; de Lange, T. CST–Polα/Primase: The Second Telomere Maintenance Machine. Genes Dev. 2023, 37, 555–569. [Google Scholar] [CrossRef]
- He, Q.; Lim, C.J. Models for human telomere C-strand fill-in by CST-Polα-primase. Trends Biochem. Sci. 2023, 48, 860–872. [Google Scholar] [CrossRef]
- Barbour, A.T.; Wuttke, D.S. RPA-like Single-Stranded DNA-Binding Protein Complexes Including CST Serve as Specialized Processivity Factors for Polymerases. Curr. Opin. Struct. Biol. 2023, 81, 102611. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Barbour, A.T.; Zaug, A.J.; Goodrich, K.J.; McKay, A.E.; Wuttke, D.S.; Cech, T.R. The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 2020, 368, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Hom, R.A.; Wuttke, D.S. Human CST Prefers G-Rich but Not Necessarily Telomeric Sequences. Biochemistry 2017, 56, 4210–4218. [Google Scholar] [CrossRef]
- Cai, S.W.; Zinder, J.C.; Svetlov, V.; Bush, M.W.; Nudler, E.; Walz, T.; de Lange, T. Cryo-EM Structure of the Human CST–Polα/Primase Complex in a Recruitment State. Nat. Struct. Mol. Biol. 2022, 29, 813–819. [Google Scholar] [CrossRef]
- Gu, P.; Jia, S.; Takasugi, T.; Smith, E.; Nandakumar, J.; Hendrickson, E.; Chang, S. CTC1-STN1 Coordinates G- and C-Strand Synthesis to Regulate Telomere Length. Aging Cell 2018, 17, e12783. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Hsu, S.-J.; Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. CTC1-STN1 Terminates Telomerase While STN1-TEN1 Enables C-Strand Synthesis during Telomere Replication in Colon Cancer Cells. Nat. Commun. 2018, 9, 2827. [Google Scholar] [CrossRef]
- Stewart, J.A.; Wang, Y.; Ackerson, S.M.; Schuck, P.L. Emerging roles of CST in maintaining genome stability and human disease. Front. Biosci. 2018, 23, 1564–1586. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. Dynamic DNA Binding, Junction Recognition and G4 Melting Activity Underlie the Telomeric and Genome-Wide Roles of Human CST. Nucleic Acids Res. 2017, 45, 12311–12324. [Google Scholar] [CrossRef]
- Kemmerich, F.E.; Daldrop, P.; Pinto, C.; Levikova, M.; Cejka, P.; Seidel, R. Force Regulated Dynamics of RPA on a DNA Fork. Nucleic Acids Res. 2016, 44, 5837–5848. [Google Scholar] [CrossRef]
- Chaires, J.B.; Gray, R.D.; Dean, W.L.; Monsen, R.; DeLeeuw, L.W.; Stribinskis, V.; Trent, J.O. Human POT1 Unfolds G-Quadruplexes by Conformational Selection. Nucleic Acids Res. 2020, 48, 4976–4991. [Google Scholar] [CrossRef]
- Feng, X.; Hsu, S.-J.; Kasbek, C.; Chaiken, M.; Price, C.M. CTC1-Mediated C-Strand Fill-in Is an Essential Step in Telomere Length Maintenance. Nucleic Acids Res. 2017, 45, 4281–4293. [Google Scholar] [CrossRef]
- Zaug, A.J.; Goodrich, K.J.; Song, J.J.; Sullivan, A.E.; Cech, T.R. Reconstitution of a Telomeric Replicon Organized by CST. Nature 2022, 608, 819–825. [Google Scholar] [CrossRef]
- Huang, C.; Dai, X.; Chai, W. Human Stn1 Protects Telomere Integrity by Promoting Efficient Lagging-Strand Synthesis at Telomeres and Mediating C-Strand Fill-In. Cell Res. 2012, 22, 1681–1695. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Guo, T.-T.; Zheng, Y.-T.; Lai, C.-W.; Sun, B.; Xi, X.-G.; Hou, X.-M. Replication Protein A Plays Multifaceted Roles Complementary to Specialized Helicases in Processing G-Quadruplex DNA. iScience 2021, 24, 102493. [Google Scholar] [CrossRef]
- Lei, K.-H.; Yang, H.-L.; Chang, H.-Y.; Yeh, H.-Y.; Nguyen, D.D.; Lee, T.-Y.; Lyu, X.; Chastain, M.; Chai, W.; Li, H.-W.; et al. Crosstalk between CST and RPA Regulates RAD51 Activity during Replication Stress. Nat. Commun. 2021, 12, 6412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chai, W. Pathogenic CTC1 Mutations Cause Global Genome Instabilities under Replication Stress. Nucleic Acids Res. 2018, 46, 3981–3992. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Lei, K.-H.; Chastain, M.; Wang, Y.; Shiva, O.; Li, S.; You, Z.; Chi, P.; Chai, W. CaMKK2 and CHK1 Phosphorylate Human STN1 in Response to Replication Stress to Protect Stalled Forks from Aberrant Resection. Nat. Commun. 2023, 14, 7882. [Google Scholar] [CrossRef]
- Schimmel, J.; Muñoz-Subirana, N.; Kool, H.; van Schendel, R.; Tijsterman, M. Small Tandem DNA Duplications Result from CST-Guided Pol α-Primase Action at DNA Break Termini. Nat. Commun. 2021, 12, 4843. [Google Scholar] [CrossRef]
- Primo, L.M.F.; Teixeira, L.K. DNA Replication Stress: Oncogenes in the Spotlight. Genet. Mol. Biol. 2020, 43, e20190138. [Google Scholar] [CrossRef]
- Ackerson, S.M.; Gable, C.I.; Stewart, J.A. Human CTC1 Promotes TopBP1 Stability and CHK1 Phosphorylation in Response to Telomere Dysfunction and Global Replication Stress. Cell Cycle 2020, 19, 3491–3507. [Google Scholar] [CrossRef]
- Wang, Y.; Brady, K.S.; Caiello, B.P.; Ackerson, S.M.; Stewart, J.A. Human CST Suppresses Origin Licensing and Promotes AND-1/Ctf4 Chromatin Association. Life Sci. Alliance 2019, 2, e201800270. [Google Scholar] [CrossRef] [PubMed]
- Kratz, K.; de Lange, T. Protection of Telomeres 1 Proteins POT1a and POT1b Can Repress ATR Signaling by RPA Exclusion, but Binding to CST Limits ATR Repression by POT1b. J. Biol. Chem. 2018, 293, 14384–14392. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.J.; Lev, A.; Zhang, Y.; Weiss, B.; Rylova, A.; Eyal, E.; Kol, N.; Barel, O.; Cesarkas, K.; Soudack, M.; et al. Mutations in STN1 Cause Coats plus Syndrome and Are Associated with Genomic and Telomere Defects. J. Exp. Med. 2016, 213, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Barazas, M.; Annunziato, S.; Pettitt, S.J.; de Krijger, I.; Ghezraoui, H.; Roobol, S.J.; Lutz, C.; Frankum, J.; Song, F.F.; Brough, R.; et al. The CST Complex Mediates End Protection at Double-Strand Breaks and Promotes PARP Inhibitor Sensitivity in BRCA1-Deficient Cells. Cell Rep. 2018, 23, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1–RIF1–Shieldin Counteracts DSB Resection through CST- and Polα-Dependent Fill-In. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Mirman, Z.; Sasi, N.K.; King, A.; Chapman, J.R.; De Lange, T. 53BP1–Shieldin-Dependent DSB Processing in BRCA1-Deficient Cells Requires CST–Polα–Primase Fill-in Synthesis. Nat. Cell Biol. 2022, 24, 51–61. [Google Scholar] [CrossRef]
- Vouzas, A.E.; Gilbert, D.M. Mammalian DNA Replication Timing. Cold Spring Harb. Perspect. Biol. 2021, 13, a040162. [Google Scholar] [CrossRef]
- Fu, H.; Baris, A.; Aladjem, M.I. Replication Timing and Nuclear Structure. Curr. Opin. Cell Biol. 2018, 52, 43–50. [Google Scholar] [CrossRef]
- Her, J.; Bunting, S.F. How Cells Ensure Correct Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef]
- Gu, P.; Min, J.-N.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 Deletion Results in Defective Telomere Replication, Leading to Catastrophic Telomere Loss and Stem Cell Exhaustion. EMBO J. 2012, 31, 2309–2321. [Google Scholar] [CrossRef]
- Boccardi, V.; Razdan, N.; Kaplunov, J.; Mundra, J.J.; Kimura, M.; Aviv, A.; Herbig, U. Stn1 Is Critical for Telomere Maintenance and Long-term Viability of Somatic Human Cells. Aging Cell 2015, 14, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Y.; Majerská, J.; Lingner, J. Molecular Basis of Telomere Syndrome Caused by CTC1 Mutations. Genes Dev. 2013, 27, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Chang, S. Functional Characterization of Human CTC 1 Mutations Reveals Novel Mechanisms Responsible for the Pathogenesis of the Telomere Disease C Oats Plus. Aging Cell 2013, 12, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sahoo, S.S.; Honda, M.; Granger, S.L.; Goodings, C.; Sanchez, L.; Künstner, A.; Busch, H.; Beier, F.; Pruett-Miller, S.M.; et al. Gain-of-Function Mutations in RPA1 Cause a Syndrome with Short Telomeres and Somatic Genetic Rescue. Blood 2022, 139, 1039–1051. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olson, C.L.; Wuttke, D.S. Guardians of the Genome: How the Single-Stranded DNA-Binding Proteins RPA and CST Facilitate Telomere Replication. Biomolecules 2024, 14, 263. https://doi.org/10.3390/biom14030263
Olson CL, Wuttke DS. Guardians of the Genome: How the Single-Stranded DNA-Binding Proteins RPA and CST Facilitate Telomere Replication. Biomolecules. 2024; 14(3):263. https://doi.org/10.3390/biom14030263
Chicago/Turabian StyleOlson, Conner L., and Deborah S. Wuttke. 2024. "Guardians of the Genome: How the Single-Stranded DNA-Binding Proteins RPA and CST Facilitate Telomere Replication" Biomolecules 14, no. 3: 263. https://doi.org/10.3390/biom14030263
APA StyleOlson, C. L., & Wuttke, D. S. (2024). Guardians of the Genome: How the Single-Stranded DNA-Binding Proteins RPA and CST Facilitate Telomere Replication. Biomolecules, 14(3), 263. https://doi.org/10.3390/biom14030263