
Calcium-Associated Proteins in Neuroregeneration


Abstract
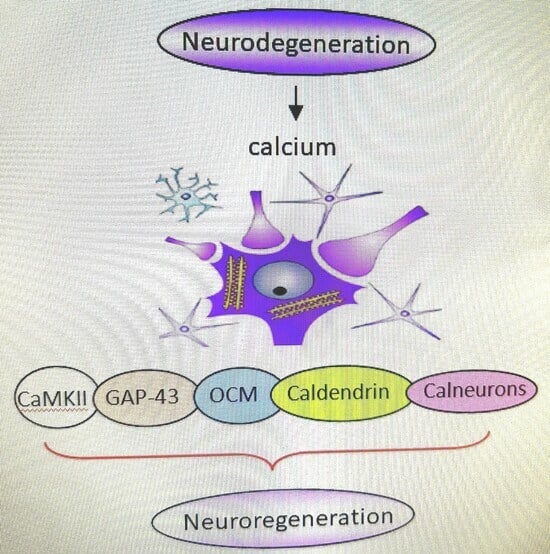
{kind=link}
{kind=link}
{kind=link}
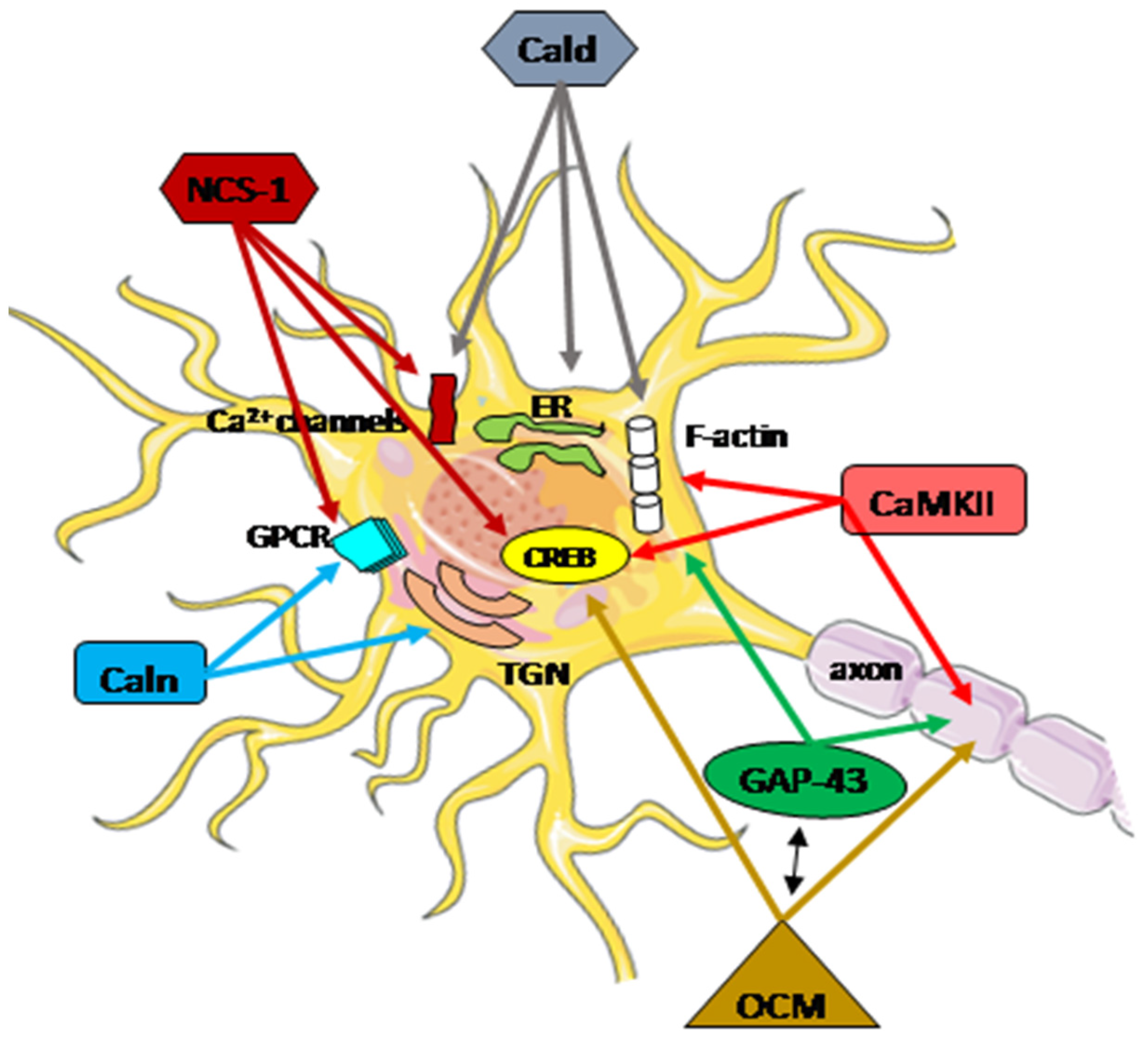{kind=link}
1. Introduction
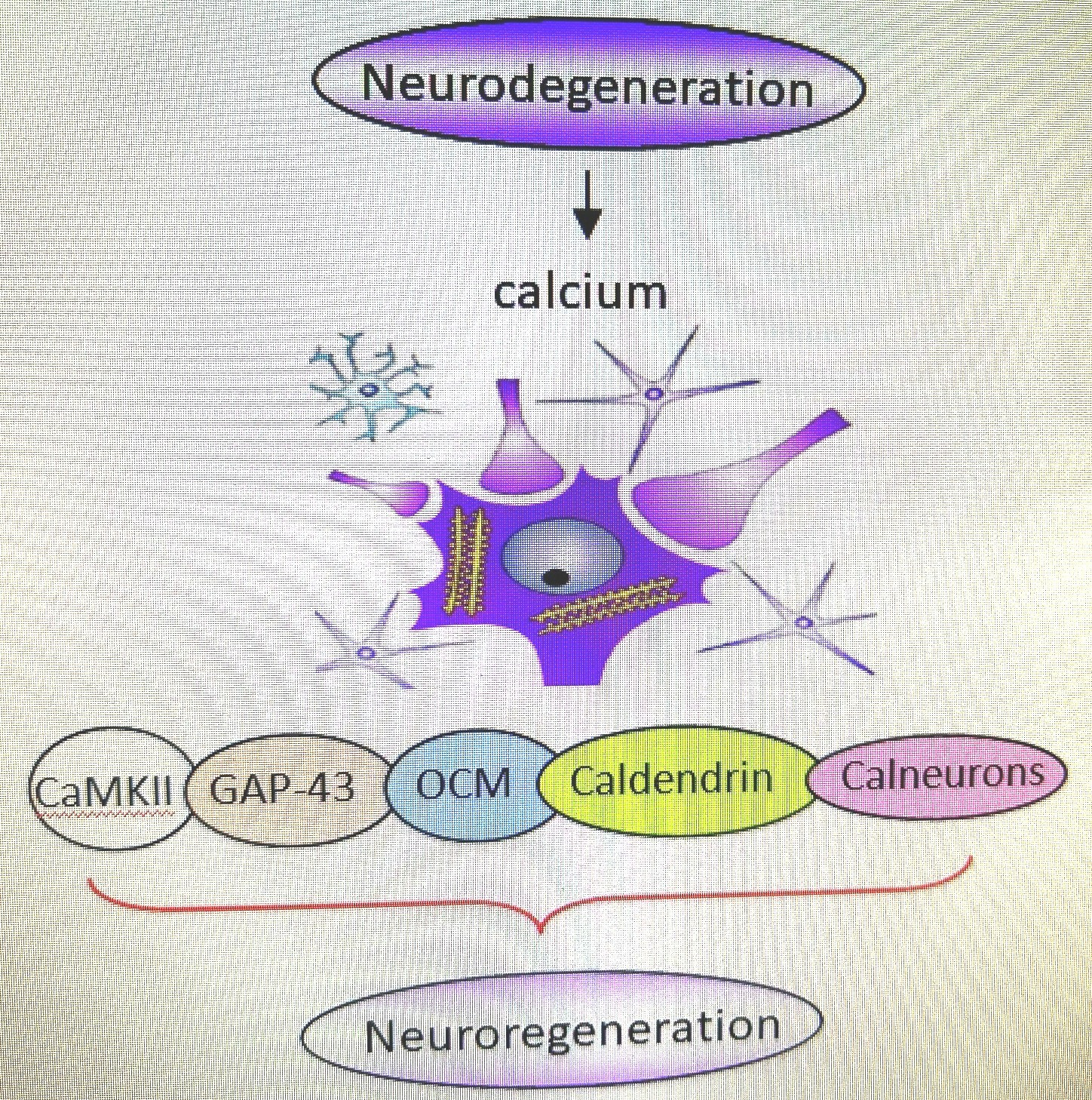
2. CaMKII
3. GAP-43
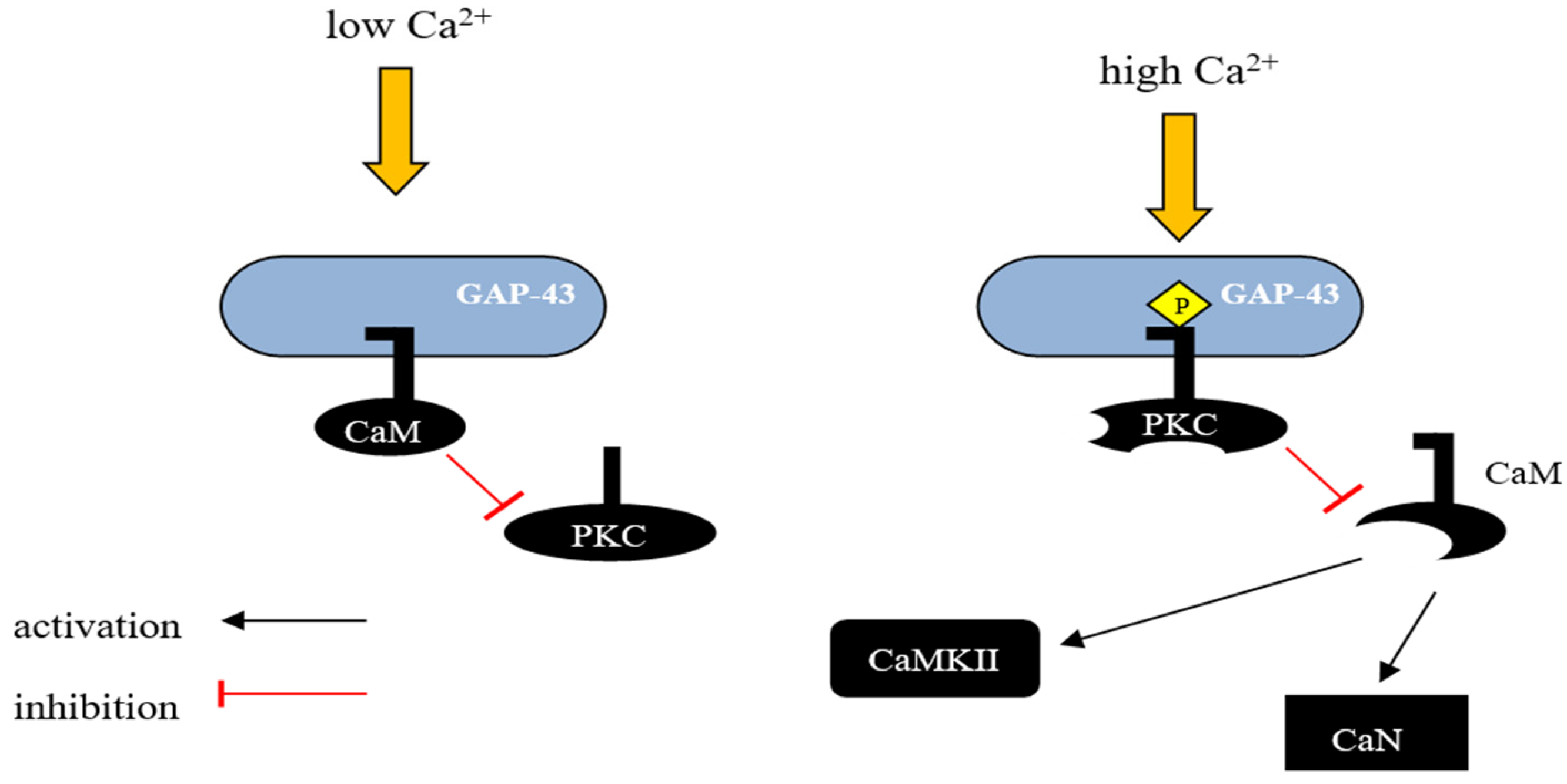
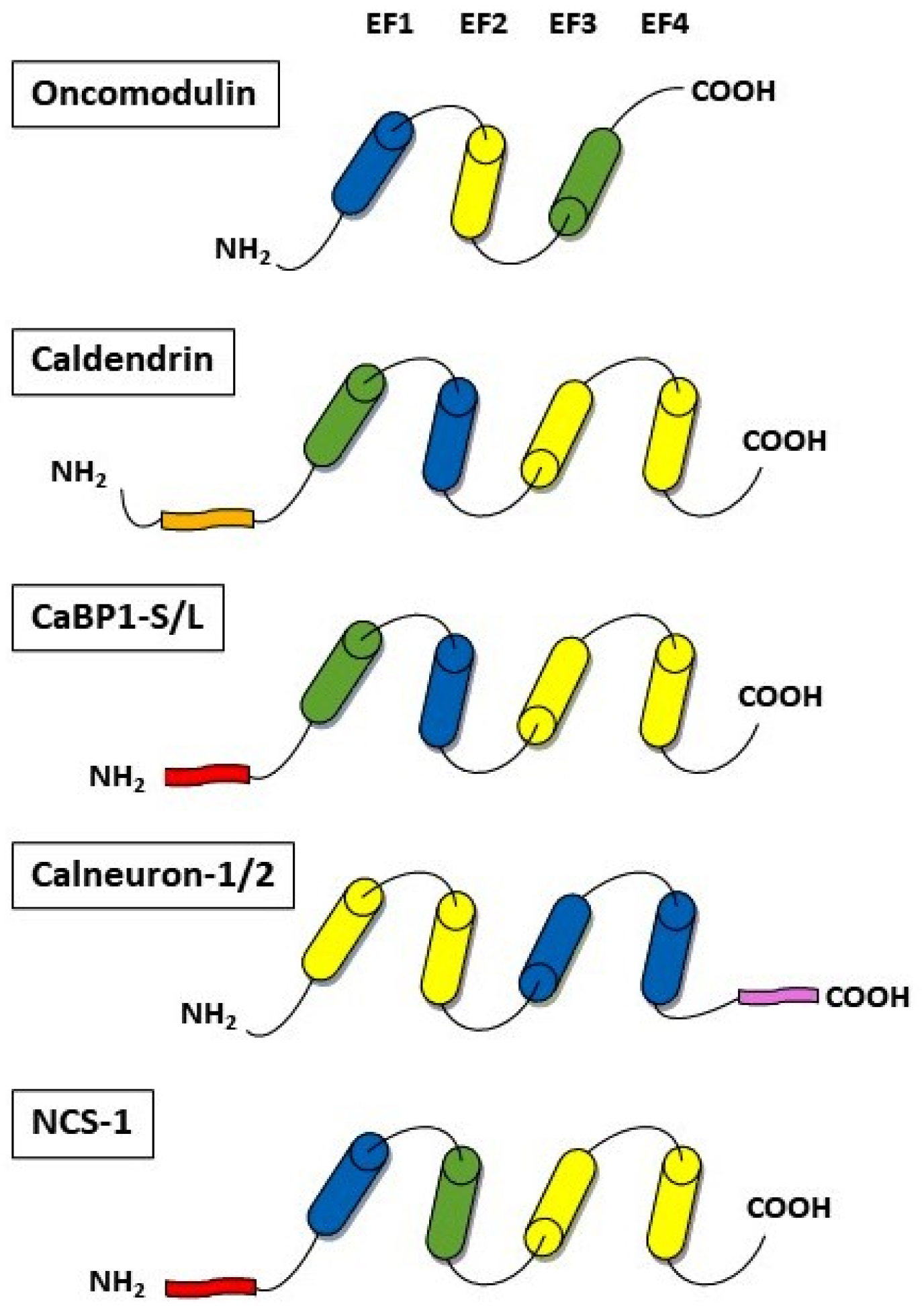
4. Calcium-Binding Proteins
4.1. Oncomodulin
4.2. Caldendrin
4.3. Calneurons
4.4. Neuronal Calcium Sensor-1
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tedeschi, A.; Bradke, F. Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regeneration. Curr. Opin. Neurobiol. 2017, 42, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Shamoun, F.; Shamoun, V.; Akhavan, A.; Tuffaha, S.H. Target Receptors of Regenerating Nerves: Neuroma Formation and Current Treatment Options. Front. Mol. Neurosci. 2022, 15, 859221. [Google Scholar] [CrossRef] [PubMed]
- Bradke, F. Mechanisms of Axon Growth and Regeneration: Moving between Development and Disease. J. Neurosci. 2022, 42, 8393–8405. [Google Scholar] [CrossRef] [PubMed]
- Dubový, P.; Klusáková, I.; Hradilová-Svíženská, I.; Joukal, M. Expression of Regeneration-Associated Proteins in Primary Sensory Neurons and Regenerating Axons After Nerve Injury—An Overview. Anat. Rec. 2018, 301, 1618–1627. [Google Scholar] [CrossRef]
- Makwana, M.; Raivich, G. Molecular mechanisms in successful peripheral regeneration. FEBS J. 2005, 272, 2628–2638. [Google Scholar] [CrossRef]
- Raivich, G.; Makwana, M. The making of successful axonal regeneration: Genes, molecules and signal transduction pathways. Brain Res. Rev. 2007, 53, 287–311. [Google Scholar] [CrossRef]
- Woodworth, M.B.; Greig, L.C.; Goldberg, J.L. Intrinsic and Induced Neuronal Regeneration in the Mammalian Retina. Antioxid. Redox Signal. 2023, 39, 1039–1052. [Google Scholar] [CrossRef]
- Di Giaimo, R.; Penna, E.; Pizzella, A.; Cirillo, R.; Perrone-Capano, C.; Crispino, M. Cross Talk at the Cytoskeleton-Plasma Membrane Interface: Impact on Neuronal Morphology and Functions. Int. J. Mol. Sci. 2020, 21, 9133. [Google Scholar] [CrossRef]
- Blanquie, O.; Bradke, F. Cytoskeleton dynamics in axon regeneration. Curr. Opin. Neurobiol. 2018, 51, 60–69. [Google Scholar] [CrossRef]
- An, J.; Chen, B.; Tian, D.; Guo, Y.; Yan, Y.; Yang, H. Regulation of Neurogenesis and Neuronal Differentiation by Natural Compounds. Curr. Stem Cell Res. Ther. 2022, 17, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Patodia, S.; Raivich, G. Role of transcription factors in peripheral nerve regeneration. Front. Mol. Neurosci. 2012, 5, 8. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Q.; Chen, Q.; Xu, L.; Yi, S. Transcriptional Control of Peripheral Nerve Regeneration. Mol. Neurobiol. 2023, 60, 329–341. [Google Scholar] [CrossRef]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Nedergaard, M.; Verkhratsky, A. Calcium dyshomeostasis and pathological calcium signalling in neurological diseases. Cell Calcium 2010, 47, 101–102. [Google Scholar] [CrossRef]
- Uryash, A.; Flores, V.; Adams, J.A.; Allen, P.D.; Lopez, J.R. Memory and Learning Deficits Are Associated With Ca. Front. Aging Neurosci. 2020, 12, 224. [Google Scholar] [CrossRef]
- Glaser, T.; Arnaud Sampaio, V.F.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2019, 95, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Conte, I.; Carter, T.; Bayer, K.U.; Molloy, J.E. Multiple CaMKII Binding Modes to the Actin Cytoskeleton Revealed by Single-Molecule Imaging. Biophys. J. 2016, 111, 395–408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, K.; Lakhanpal, G.; Lu, H.E.; Khan, M.; Suzuki, A.; Hayashi, M.K.; Narayanan, R.; Luyben, T.T.; Matsuda, T.; Nagai, T.; et al. A Temporary Gating of Actin Remodeling during Synaptic Plasticity Consists of the Interplay between the Kinase and Structural Functions of CaMKII. Neuron 2015, 87, 813–826. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, M.; Schafer, N.P.; Bueno, C.; Song, S.S.; Hudmon, A.; Wolynes, P.G.; Waxham, M.N.; Cheung, M.S. Assemblies of calcium/calmodulin-dependent kinase II with actin and their dynamic regulation by calmodulin in dendritic spines. Proc. Natl. Acad. Sci. USA 2019, 116, 18937–18942. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef]
- Eisner, D.; Neher, E.; Taschenberger, H.; Smith, G. Physiology of intracellular calcium buffering. Physiol. Rev. 2023, 103, 2767–2845. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chu, X.; Lu, H.P.; Wang, J. Molecular mechanism of multispecific recognition of Calmodulin through conformational changes. Proc. Natl. Acad. Sci. USA 2017, 114, E3927–E3934. [Google Scholar] [CrossRef] [PubMed]
- Kursula, P. The many structural faces of calmodulin: A multitasking molecular jackknife. Amino Acids 2014, 46, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Peck, A.; Pande, V.S. Conformational heterogeneity of the calmodulin binding interface. Nat. Commun. 2016, 7, 10910. [Google Scholar] [CrossRef] [PubMed]
- Westerlund, A.M.; Delemotte, L. Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput. Biol. 2018, 14, e1006072. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193. [Google Scholar] [CrossRef]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Mantilla, G.; Peréz-Gordones, M.C.; Cisneros-Montufar, S.; Benaim, G.; Navarro, J.C.; Mendoza, M.; Ramírez-Iglesias, J.R. Structural Analysis and Diversity of Calmodulin-Binding Domains in Membrane and Intracellular Ca. J. Membr. Biol. 2023, 256, 159–174. [Google Scholar] [CrossRef]
- Denny, J.B. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr. Neuropharmacol. 2006, 4, 293–304. [Google Scholar] [CrossRef]
- Chung, D.; Shum, A.; Caraveo, G. GAP-43 and BASP1 in Axon Regeneration: Implications for the Treatment of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 567537. [Google Scholar] [CrossRef]
- Zhang, X.; Connelly, J.; Levitan, E.S.; Sun, D.; Wang, J.Q. Calcium/Calmodulin-Dependent Protein Kinase II in Cerebrovascular Diseases. Transl. Stroke Res. 2021, 12, 513–529. [Google Scholar] [CrossRef]
- Anderson, M.E. Calmodulin kinase signaling in heart: An intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol. Ther. 2005, 106, 39–55. [Google Scholar] [CrossRef]
- Lin, M.Y.; Zal, T.; Ch’en, I.L.; Gascoigne, N.R.; Hedrick, S.M. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: From activation to unresponsiveness. J. Immunol. 2005, 174, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- McGargill, M.A.; Sharp, L.L.; Bui, J.D.; Hedrick, S.M.; Calbo, S. Active Ca2+/calmodulin-dependent protein kinase II gamma B impairs positive selection of T cells by modulating TCR signaling. J. Immunol. 2005, 175, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Xi, F.; Xu, R.J.; Xu, J.H.; Ma, J.J.; Wang, W.H.; Wang, F.; Ma, Y.X.; Qi, S.B.; Zhang, H.C.; Zhang, H.N.; et al. Calcium/calmodulin-dependent protein kinase II regulates mammalian axon growth by affecting F-actin length in growth cone. J. Cell. Physiol. 2019, 234, 23053–23065. [Google Scholar] [CrossRef] [PubMed]
- Nicole, O.; Pacary, E. CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases. Int. J. Mol. Sci. 2020, 21, 7272. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.A.P.; Skelding, K.A. Calcium/Calmodulin-Stimulated Protein Kinase II (CaMKII): Different Functional Outcomes from Activation, Depending on the Cellular Microenvironment. Cells 2023, 12, 401. [Google Scholar] [CrossRef] [PubMed]
- Mar, F.M.; Bonni, A.; Sousa, M.M. Cell intrinsic control of axon regeneration. EMBO Rep. 2014, 15, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Roy, A.; Wu, Z.; Goncharov, A.; Jin, Y.; Chisholm, A.D. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. 2010, 30, 3175–3183. [Google Scholar] [CrossRef]
- Goshima, Y.; Ohsako, S.; Yamauchi, T. Overexpression of Ca2+/calmodulin-dependent protein kinase II in Neuro2a and NG108-15 neuroblastoma cell lines promotes neurite outgrowth and growth cone motility. J. Neurosci. 1993, 13, 559–567. [Google Scholar] [CrossRef]
- Sogawa, Y.; Yoshimura, Y.; Otaka, A.; Yamauchi, T. Ca2+-independent activity of Ca2+/calmodulin-dependent protein kinase II involved in stimulation of neurite outgrowth in neuroblastoma cells. Brain Res. 2000, 881, 165–175. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Jia, W.; Zhao, D.; Boczek, T.; Gao, Q.; Wang, Q.; Fu, Y.; He, M.; Shi, R.; et al. Calcium-/Calmodulin-Dependent Protein Kinase II (CaMKII) Inhibition Induces Learning and Memory Impairment and Apoptosis. Oxid. Med. Cell. Longev. 2021, 2021, 4635054. [Google Scholar] [CrossRef]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.M.; Yang, I.H.; Kim, D.H.; Byun, J.; Saijilafu; Xu, W.L.; Nicovich, P.R.; Cheong, R.; Levchenko, A.; Thakor, N.; et al. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 1998, 21, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Easley, C.A.; Faison, M.O.; Kirsch, T.L.; Lee, J.A.; Seward, M.E.; Tombes, R.M. Laminin activates CaMK-II to stabilize nascent embryonic axons. Brain Res. 2006, 1092, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Fink, C.C.; Bayer, K.U.; Myers, J.W.; Ferrell, J.E.; Schulman, H.; Meyer, T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 2003, 39, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, R.; Hayashi, Y.; Hell, J.W. CaMKII: A central molecular organizer of synaptic plasticity, learning and memory. Nat. Rev. Neurosci. 2022, 23, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.W.; Gao, Z.L.; Ji, Q.Y.; Wang, H.; Zhang, H.Y.; Yang, Y.D.; Xing, F.J.; Meng, L.J.; Wang, Y. Regulation of cofilin activity by CaMKII and calcineurin. Am. J. Med. Sci. 2012, 344, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Donai, H.; Sugiura, H.; Ara, D.; Yoshimura, Y.; Yamagata, K.; Yamauchi, T. Interaction of Arc with CaM kinase II and stimulation of neurite extension by Arc in neuroblastoma cells expressing CaM kinase II. Neurosci. Res. 2003, 47, 399–408. [Google Scholar] [CrossRef]
- Goebel, D.J. Selective blockade of CaMKII-alpha inhibits NMDA-induced caspase-3-dependent cell death but does not arrest PARP-1 activation or loss of plasma membrane selectivity in rat retinal neurons. Brain Res. 2009, 1256, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Laabich, A.; Li, G.; Cooper, N.G. Calcium/calmodulin-dependent protein kinase II containing a nuclear localizing signal is altered in retinal neurons exposed to N-methyl-D-aspartate. Brain Res. Mol. Brain Res. 2000, 76, 253–265. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Song, W.; Brustovetsky, T.; Engleman, E.A.; Brustovetsky, N.; Cummins, T.R.; Hudmon, A. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J. Biol. Chem. 2012, 287, 8495–8506. [Google Scholar] [CrossRef]
- Musada, G.R.; Carmy-Bennun, T.; Hackam, A.S. Identification of a Novel Axon Regeneration Role for Noncanonical Wnt Signaling in the Adult Retina after Injury. eNeuro 2022, 9, ENEURO.0182-22.2022. [Google Scholar] [CrossRef]
- Griem-Krey, N.; Clarkson, A.N.; Wellendorph, P. CaMKIIα as a Promising Drug Target for Ischemic Grey Matter. Brain Sci. 2022, 12, 1639. [Google Scholar] [CrossRef] [PubMed]
- Rumian, N.L.; Chalmers, N.E.; Tullis, J.E.; Herson, P.S.; Bayer, K.U. CaMKIIα knockout protects from ischemic neuronal cell death after resuscitation from cardiac arrest. Brain Res. 2021, 1773, 147699. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Vest, R.S.; Ashpole, N.M.; Hudmon, A.; Bayer, K.U. CaMKII in cerebral ischemia. Acta Pharmacol. Sin. 2011, 32, 861–872. [Google Scholar] [CrossRef]
- Ye, J.; Das, S.; Roy, A.; Wei, W.; Huang, H.; Lorenz-Guertin, J.M.; Xu, Q.; Jacob, T.C.; Wang, B.; Sun, D.; et al. Ischemic Injury-Induced CaMKIIδ and CaMKIIγ Confer Neuroprotection Through the NF-κB Signaling Pathway. Mol. Neurobiol. 2019, 56, 2123–2136. [Google Scholar] [CrossRef]
- Frey, D.; Laux, T.; Xu, L.; Schneider, C.; Caroni, P. Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J. Cell Biol. 2000, 149, 1443–1454. [Google Scholar] [CrossRef]
- McGuire, C.B.; Snipes, G.J.; Norden, J.J. Light-microscopic immunolocalization of the growth- and plasticity-associated protein GAP-43 in the developing rat brain. Brain Res. 1988, 469, 277–291. [Google Scholar] [CrossRef]
- Benowitz, L.I.; Apostolides, P.J.; Perrone-Bizzozero, N.; Finklestein, S.P.; Zwiers, H. Anatomical distribution of the growth-associated protein GAP-43/B-50 in the adult rat brain. J. Neurosci. 1988, 8, 339–352. [Google Scholar] [CrossRef]
- Widmer, F.; Caroni, P. Identification, localization, and primary structure of CAP-23, a particle-bound cytosolic protein of early development. J. Cell Biol. 1990, 111, 3035–3047. [Google Scholar] [CrossRef]
- Meiri, K.F.; Pfenninger, K.H.; Willard, M.B. Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc. Natl. Acad. Sci. USA 1986, 83, 3537–3541. [Google Scholar] [CrossRef]
- Shen, Y.; Mani, S.; Donovan, S.L.; Schwob, J.E.; Meiri, K.F. Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J. Neurosci. 2002, 22, 239–247. [Google Scholar] [CrossRef]
- Metz, G.A.; Schwab, M.E. Behavioral characterization in a comprehensive mouse test battery reveals motor and sensory impairments in growth-associated protein-43 null mutant mice. Neuroscience 2004, 129, 563–574. [Google Scholar] [CrossRef]
- Mishra, R.; Gupta, S.K.; Meiri, K.F.; Fong, M.; Thostrup, P.; Juncker, D.; Mani, S. GAP-43 is key to mitotic spindle control and centrosome-based polarization in neurons. Cell Cycle 2008, 7, 348–357. [Google Scholar] [CrossRef][Green Version]
- Mosevitsky, M.I. Nerve ending “signal” proteins GAP-43, MARCKS, and BASP1. Int. Rev. Cytol. 2005, 245, 245–325. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Dent, E.W.; Meiri, K.F. Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J. Neurosci. 1997, 17, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Korshunova, I.; Novitskaya, V.; Kiryushko, D.; Pedersen, N.; Kolkova, K.; Kropotova, E.; Mosevitsky, M.; Rayko, M.; Morrow, J.S.; Ginzburg, I.; et al. GAP-43 regulates NCAM-180-mediated neurite outgrowth. J. Neurochem. 2007, 100, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Haruta, T.; Takami, N.; Ohmura, M.; Misumi, Y.; Ikehara, Y. Ca2+-dependent interaction of the growth-associated protein GAP-43 with the synaptic core complex. Biochem. J. 1997, 325, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.W.; Meiri, K.F. Distribution of phosphorylated GAP-43 (neuromodulin) in growth cones directly reflects growth cone behavior. J. Neurobiol. 1998, 35, 287–299. [Google Scholar] [CrossRef]
- Dekker, L.V.; De Graan, P.N.; Versteeg, D.H.; Oestreicher, A.B.; Gispen, W.H. Phosphorylation of B-50 (GAP43) is correlated with neurotransmitter release in rat hippocampal slices. J. Neurochem. 1989, 52, 24–30. [Google Scholar] [CrossRef]
- Hulo, S.; Alberi, S.; Laux, T.; Muller, D.; Caroni, P. A point mutant of GAP-43 induces enhanced short-term and long-term hippocampal plasticity. Eur. J. Neurosci. 2002, 15, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kawagoe, Y.; Takasugi, T.; Nozumi, M.; Ito, Y.; Fukusumi, H.; Kanemura, Y.; Fujii, Y.; Igarashi, M. JNK1-Dependent Phosphorylation of GAP-43 Serine 142 is a Novel Molecular Marker for Axonal Growth. Neurochem. Res. 2022, 47, 2668–2682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lu, C.; Severin, C.; Sretavan, D.W. GAP-43 mediates retinal axon interaction with lateral diencephalon cells during optic tract formation. Development 2000, 127, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, S.M.; Fankhauser, C.; Huang, P.L.; Mashimo, H.; Fishman, M.C. Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell 1995, 80, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.L.; Mamounas, L.A.; Andrews, A.M.; Blue, M.E.; McCasland, J.S. GAP-43 is critical for normal development of the serotonergic innervation in forebrain. J. Neurosci. 2002, 22, 3543–3552. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Kim, K.Y.; Lee, E.J.; Park, S.J.; Kwon, S.O.; Jung, C.S.; Lee, M.Y.; Chun, M.H. Inhibition of nitric oxide synthase induces increased production of growth-associated protein 43 in the developing retina of the postnatal rat. Brain Res. Dev. Brain Res. 2002, 136, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Cen, L.P.; Liu, L.; Leaver, S.G.; Harvey, A.R.; Cui, Q.; Pang, C.P.; Zhang, M. Adeno-associated virus-mediated expression of growth-associated protein-43 aggravates retinal ganglion cell death in experimental chronic glaucomatous injury. Mol. Vis. 2013, 19, 1422–1432. [Google Scholar]
- Skene, J.H.; Willard, M. Changes in axonally transported proteins during axon regeneration in toad retinal ganglion cells. J. Cell Biol. 1981, 89, 86–95. [Google Scholar] [CrossRef]
- Carmichael, S.T.; Archibeque, I.; Luke, L.; Nolan, T.; Momiy, J.; Li, S. Growth-associated gene expression after stroke: Evidence for a growth-promoting region in peri-infarct cortex. Exp. Neurol. 2005, 193, 291–311. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Wang, E.H.; Woodson, W.J.; Wang, S.; Sun, G.; Lee, A.G.; Arac, A.; Fenno, L.E.; Deisseroth, K.; Steinberg, G.K. Optogenetic neuronal stimulation promotes functional recovery after stroke. Proc. Natl. Acad. Sci. USA 2014, 111, 12913–12918. [Google Scholar] [CrossRef]
- Kawamata, T.; Ren, J.; Cha, J.H.; Finklestein, S.P. Intracisternal antisense oligonucleotide to growth associated protein-43 blocks the recovery-promoting effects of basic fibroblast growth factor after focal stroke. Exp. Neurol. 1999, 158, 89–96. [Google Scholar] [CrossRef]
- Allegra Mascaro, A.L.; Cesare, P.; Sacconi, L.; Grasselli, G.; Mandolesi, G.; Maco, B.; Knott, G.W.; Huang, L.; De Paola, V.; Strata, P.; et al. In vivo single branch axotomy induces GAP-43-dependent sprouting and synaptic remodeling in cerebellar cortex. Proc. Natl. Acad. Sci. USA 2013, 110, 10824–10829. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Mandolesi, G.; Strata, P.; Cesare, P. Impaired sprouting and axonal atrophy in cerebellar climbing fibres following in vivo silencing of the growth-associated protein GAP-43. PLoS ONE 2011, 6, e20791. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Strata, P. Structural plasticity of climbing fibers and the growth-associated protein GAP-43. Front. Neural Circuits 2013, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Aigner, L.; Caroni, P. Absence of persistent spreading, branching, and adhesion in GAP-43-depleted growth cones. J. Cell Biol. 1995, 128, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Buffo, A.; Holtmaat, A.J.; Savio, T.; Verbeek, J.S.; Oberdick, J.; Oestreicher, A.B.; Gispen, W.H.; Verhaagen, J.; Rossi, F.; Strata, P. Targeted overexpression of the neurite growth-associated protein B-50/GAP-43 in cerebellar Purkinje cells induces sprouting after axotomy but not axon regeneration into growth-permissive transplants. J. Neurosci. 1997, 17, 8778–8791. [Google Scholar] [CrossRef] [PubMed]
- Gudasheva, T.A.; Povarnina, P.Y.; Volkova, A.A.; Kruglov, S.V.; Antipova, T.A.; Seredenin, S.B. A Nerve Growth Factor Dipeptide Mimetic Stimulates Neurogenesis and Synaptogenesis in the Hippocampus and Striatum of Adult Rats with Focal Cerebral Ischemia. Acta Naturae 2019, 11, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Ouyang, C.; Wang, Y.; Zhang, S.; Ma, X.; Song, Y.; Yu, H.; Tang, J.; Fu, W.; Sheng, L.; et al. HO-1 attenuates hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt signaling pathway in stroke. Brain Res. 2014, 1577, 69–76. [Google Scholar] [CrossRef]
- Wu, H.; Lu, D.; Jiang, H.; Xiong, Y.; Qu, C.; Li, B.; Mahmood, A.; Zhou, D.; Chopp, M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J. Neurotrauma 2008, 25, 130–139. [Google Scholar] [CrossRef]
- Gupta, S.K.; Mishra, R.; Kusum, S.; Spedding, M.; Meiri, K.F.; Gressens, P.; Mani, S. GAP-43 is essential for the neurotrophic effects of BDNF and positive AMPA receptor modulator S18986. Cell Death Differ. 2009, 16, 624–637. [Google Scholar] [CrossRef]
- Henikoff, S. ENCODE and our very busy genome. Nat. Genet. 2007, 39, 817–818. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca. Cold Spring Harb. Perspect. Biol. 2020, 12, a035543. [Google Scholar] [CrossRef]
- Climer, L.K.; Cox, A.M.; Reynolds, T.J.; Simmons, D.D. Oncomodulin: The Enigmatic Parvalbumin Protein. Front. Mol. Neurosci. 2019, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Thalmann, I.; Thalmann, R.; Simmons, D.D. Expression of alpha and beta parvalbumin is differentially regulated in the rat organ of corti during development. J. Neurobiol. 2004, 58, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Cui, Q.; Gilbert, H.Y.; Yang, Y.; Yang, Z.; Berlinicke, C.; Li, Z.; Zaverucha-do-Valle, C.; He, H.; Petkova, V.; et al. Oncomodulin links inflammation to optic nerve regeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 19587–19592. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, T.; Yin, Y.; Habboub, G.; Gilbert, H.Y.; Li, Y.; Nakao, S.; Hafezi-Moghadam, A.; Benowitz, L.I. Neutrophils express oncomodulin and promote optic nerve regeneration. J. Neurosci. 2013, 33, 14816–14824. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Hornak, A.J.; Maison, S.F.; Ohlemiller, K.K.; Liberman, M.C.; Simmons, D.D. Oncomodulin, an EF-Hand Ca2+ Buffer, Is Critical for Maintaining Cochlear Function in Mice. J. Neurosci. 2016, 36, 1631–1635. [Google Scholar] [CrossRef]
- Hackney, C.M.; Mahendrasingam, S.; Penn, A.; Fettiplace, R. The concentrations of calcium buffering proteins in mammalian cochlear hair cells. J. Neurosci. 2005, 25, 7867–7875. [Google Scholar] [CrossRef]
- Yang, Y.; Murtha, K.; Climer, L.K.; Ceriani, F.; Thompson, P.; Hornak, A.J.; Marcotti, W.; Simmons, D.D. Oncomodulin regulates spontaneous calcium signalling and maturation of afferent innervation in cochlear outer hair cells. J. Physiol. 2023, 601, 4291–4308. [Google Scholar] [CrossRef] [PubMed]
- Murtha, K.E.; Yang, Y.; Ceriani, F.; Jeng, J.Y.; Climer, L.K.; Jones, F.; Charles, J.; Devana, S.K.; Hornak, A.J.; Marcotti, W.; et al. Oncomodulin (OCM) uniquely regulates calcium signaling in neonatal cochlear outer hair cells. Cell Calcium 2022, 105, 102613. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.; Bradke, F. Axon Regeneration in the Central Nervous System: Facing the Challenges from the Inside. Annu. Rev. Cell Dev. Biol. 2018, 34, 495–521. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Verhaagen, J. Intrinsic Determinants of Axon Regeneration. Dev. Neurobiol. 2018, 78, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, L.I.; Xie, L.; Yin, Y. Inflammatory Mediators of Axon Regeneration in the Central and Peripheral Nervous Systems. Int. J. Mol. Sci. 2023, 24, 15359. [Google Scholar] [CrossRef]
- Yang, X.; Liu, R.; Xu, Y.; Ma, X.; Zhou, B. The Mechanisms of Peripheral Nerve Preconditioning Injury on Promoting Axonal Regeneration. Neural Plast. 2021, 2021, 6648004. [Google Scholar] [CrossRef]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediat. Inflamm. 2013, 2013, 342931. [Google Scholar] [CrossRef]
- Xanthos, D.N.; Sandkühler, J. Neurogenic neuroinflammation: Inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 2014, 15, 43–53. [Google Scholar] [CrossRef]
- Moore, D.L.; Goldberg, J.L. Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 2011, 71, 1186–1211. [Google Scholar] [CrossRef]
- Fagoe, N.D.; van Heest, J.; Verhaagen, J. Spinal cord injury and the neuron-intrinsic regeneration-associated gene program. Neuromol. Med. 2014, 16, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Niemi, J.P.; DeFrancesco-Oranburg, T.; Cox, A.; Lindborg, J.A.; Echevarria, F.D.; McCluskey, J.; Simmons, D.D.; Zigmond, R.E. The Conditioning Lesion Response in Dorsal Root Ganglion Neurons Is Inhibited in Oncomodulin Knock-Out Mice. eNeuro 2022, 9, ENEURO.0477-21.2022. [Google Scholar] [CrossRef] [PubMed]
- Niemi, J.P.; DeFrancesco-Lisowitz, A.; Roldán-Hernández, L.; Lindborg, J.A.; Mandell, D.; Zigmond, R.E. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J. Neurosci. 2013, 33, 16236–16248. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.P.; Horn, K.P.; Tom, V.J.; Miller, J.H.; Busch, S.A.; Nair, D.; Silver, D.J.; Silver, J. Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J. Neurosci. 2005, 25, 8066–8076. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, L.I.; Popovich, P.G. Inflammation and axon regeneration. Curr. Opin. Neurol. 2011, 24, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Kim, J.; Shin, H.; Jeong, S.R.; Kang, Y.M.; Choi, J.Y.; Hwang, D.H.; Kim, B.G. Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J. Neurosci. 2013, 33, 15095–15108. [Google Scholar] [CrossRef] [PubMed]
- Harel, R.; Iannotti, C.A.; Hoh, D.; Clark, M.; Silver, J.; Steinmetz, M.P. Oncomodulin affords limited regeneration to injured sensory axons in vitro and in vivo. Exp. Neurol. 2012, 233, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Yoon, H.J.; Kim, B.G. Regeneration-associated macrophages: A novel approach to boost intrinsic regenerative capacity for axon regeneration. Neural Regen. Res. 2016, 11, 1368–1371. [Google Scholar] [CrossRef]
- Kwon, M.J.; Seo, Y.; Cho, H.; Kim, H.S.; Oh, Y.J.; Genişcan, S.; Kim, M.; Park, H.H.; Joe, E.H.; Kwon, M.H.; et al. Nanogel-mediated delivery of oncomodulin secreted from regeneration-associated macrophages promotes sensory axon regeneration in the spinal cord. Theranostics 2022, 12, 5856–5876. [Google Scholar] [CrossRef]
- Yin, Y.; Henzl, M.T.; Lorber, B.; Nakazawa, T.; Thomas, T.T.; Jiang, F.; Langer, R.; Benowitz, L.I. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat. Neurosci. 2006, 9, 843–852. [Google Scholar] [CrossRef]
- Fine, N.; Tasevski, N.; McCulloch, C.A.; Tenenbaum, H.C.; Glogauer, M. The Neutrophil: Constant Defender and First Responder. Front. Immunol. 2020, 11, 571085. [Google Scholar] [CrossRef]
- Kurimoto, T.; Yin, Y.; Omura, K.; Gilbert, H.Y.; Kim, D.; Cen, L.P.; Moko, L.; Kügler, S.; Benowitz, L.I. Long-distance axon regeneration in the mature optic nerve: Contributions of oncomodulin, cAMP, and pten gene deletion. J. Neurosci. 2010, 30, 15654–15663. [Google Scholar] [CrossRef]
- Xu, K.; Yu, L.; Wang, Z.; Lin, P.; Zhang, N.; Xing, Y.; Yang, N. Use of gene therapy for optic nerve protection: Current concepts. Front. Neurosci. 2023, 17, 1158030. [Google Scholar] [CrossRef]
- Xie, L.; Yin, Y.; Jayakar, S.; Kawaguchi, R.; Wang, Q.; Peterson, S.; Shi, C.; Turnes, B.L.; Zhang, Z.; Oses-Prieto, J.; et al. The oncomodulin receptor ArmC10 enables axon regeneration in mice after nerve injury and neurite outgrowth in human iPSC-derived sensory neurons. Sci. Transl. Med. 2023, 15, eadg6241. [Google Scholar] [CrossRef]
- Takemura, M.; Mishima, T.; Wang, Y.; Kasahara, J.; Fukunaga, K.; Ohashi, K.; Mizuno, K. Ca2+/calmodulin-dependent protein kinase IV-mediated LIM kinase activation is critical for calcium signal-induced neurite outgrowth. J. Biol. Chem. 2009, 284, 28554–28562. [Google Scholar] [CrossRef]
- Alberini, C.M. Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 2009, 89, 121–145. [Google Scholar] [CrossRef]
- Landeira, B.S.; Santana, T.T.D.S.; Araújo, J.A.M.; Tabet, E.I.; Tannous, B.A.; Schroeder, T.; Costa, M.R. Activity-Independent Effects of CREB on Neuronal Survival and Differentiation during Mouse Cerebral Cortex Development. Cereb. Cortex 2018, 28, 538–548. [Google Scholar] [CrossRef]
- Kitagawa, H.; Sugo, N.; Morimatsu, M.; Arai, Y.; Yanagida, T.; Yamamoto, N. Activity-Dependent Dynamics of the Transcription Factor of cAMP-Response Element Binding Protein in Cortical Neurons Revealed by Single-Molecule Imaging. J. Neurosci. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Willis, D.E. What makes a RAG regeneration associated? Front. Mol. Neurosci. 2015, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Deng, K.; Hou, J.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.A.; Benowitz, L.I. Retinal Ganglion Cell Survival and Axon Regeneration after Optic Nerve Injury: Role of Inflammation and Other Factors. Int. J. Mol. Sci. 2022, 23, 10179. [Google Scholar] [CrossRef]
- Seidenbecher, C.I.; Langnaese, K.; Sanmartí-Vila, L.; Boeckers, T.M.; Smalla, K.H.; Sabel, B.A.; Garner, C.C.; Gundelfinger, E.D.; Kreutz, M.R. Caldendrin, a novel neuronal calcium-binding protein confined to the somato-dendritic compartment. J. Biol. Chem. 1998, 273, 21324–21331. [Google Scholar] [CrossRef]
- Laube, G.; Seidenbecher, C.I.; Richter, K.; Dieterich, D.C.; Hoffmann, B.; Landwehr, M.; Smalla, K.H.; Winter, C.; Böckers, T.M.; Wolf, G.; et al. The neuron-specific Ca2+-binding protein caldendrin: Gene structure, splice isoforms, and expression in the rat central nervous system. Mol. Cell Neurosci. 2002, 19, 459–475. [Google Scholar] [CrossRef]
- Seidenbecher, C.I.; Reissner, C.; Kreutz, M.R. Caldendrins in the inner retina. Adv. Exp. Med. Biol. 2002, 514, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Sokal, I.; Verlinde, C.L.; Erdjument-Bromage, H.; Tempst, P.; Pronin, A.N.; Benovic, J.L.; Fariss, R.N.; Palczewski, K. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 2000, 275, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Seidenbecher, C.I.; Smalla, K.H.; Gundelfinger, E.D.; Bogerts, B.; Kreutz, M.R. Distribution and cellular localization of caldendrin immunoreactivity in adult human forebrain. J. Histochem. Cytochem. 2003, 51, 1109–1112. [Google Scholar] [CrossRef]
- Kim, K.Y.; Scholl, E.S.; Liu, X.; Shepherd, A.; Haeseleer, F.; Lee, A. Localization and expression of CaBP1/caldendrin in the mouse brain. Neuroscience 2014, 268, 33–47. [Google Scholar] [CrossRef]
- Mundhenk, J.; Fusi, C.; Kreutz, M.R. Caldendrin and Calneurons-EF-Hand CaM-Like Calcium Sensors With Unique Features and Specialized Neuronal Functions. Front. Mol. Neurosci. 2019, 12, 16. [Google Scholar] [CrossRef]
- Kiran, U.; Regur, P.; Kreutz, M.R.; Sharma, Y.; Chakraborty, A. Intermotif Communication Induces Hierarchical Ca. Biochemistry 2017, 56, 2467–2476. [Google Scholar] [CrossRef]
- Haynes, L.P.; McCue, H.V.; Burgoyne, R.D. Evolution and functional diversity of the Calcium Binding Proteins (CaBPs). Front. Mol. Neurosci. 2012, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- McCue, H.V.; Haynes, L.P.; Burgoyne, R.D. Bioinformatic analysis of CaBP/calneuron proteins reveals a family of highly conserved vertebrate Ca2+-binding proteins. BMC Res. Notes 2010, 3, 118. [Google Scholar] [CrossRef]
- Mikhaylova, M.; Bär, J.; van Bommel, B.; Schätzle, P.; YuanXiang, P.; Raman, R.; Hradsky, J.; Konietzny, A.; Loktionov, E.Y.; Reddy, P.P.; et al. Caldendrin Directly Couples Postsynaptic Calcium Signals to Actin Remodeling in Dendritic Spines. Neuron 2018, 97, 1110–1125.e14. [Google Scholar] [CrossRef]
- Hall, D.D.; Dai, S.; Tseng, P.Y.; Malik, Z.; Nguyen, M.; Matt, L.; Schnizler, K.; Shephard, A.; Mohapatra, D.P.; Tsuruta, F.; et al. Competition between α-actinin and Ca2+-calmodulin controls surface retention of the L-type Ca2+ channel Ca(V)1.2. Neuron 2013, 78, 483–497. [Google Scholar] [CrossRef]
- Oliveria, S.F.; Dittmer, P.J.; Youn, D.H.; Dell’Acqua, M.L.; Sather, W.A. Localized calcineurin confers Ca2+-dependent inactivation on neuronal L-type Ca2+ channels. J. Neurosci. 2012, 32, 15328–15337. [Google Scholar] [CrossRef] [PubMed]
- Christel, C.; Lee, A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 2012, 1820, 1243–1252. [Google Scholar] [CrossRef]
- Zhou, H.; Yu, K.; McCoy, K.L.; Lee, A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J. Biol. Chem. 2005, 280, 29612–29619. [Google Scholar] [CrossRef]
- Tippens, A.L.; Lee, A. Caldendrin, a neuron-specific modulator of Cav/1.2 (L-type) Ca2+ channels. J. Biol. Chem. 2007, 282, 8464–8473. [Google Scholar] [CrossRef]
- Oz, S.; Benmocha, A.; Sasson, Y.; Sachyani, D.; Almagor, L.; Lee, A.; Hirsch, J.A.; Dascal, N. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+ channels by calmodulin and Ca2+-binding protein 1. J. Biol. Chem. 2013, 288, 12680–12691. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.B. L-Type Ca. Biomolecules 2021, 11, 1811. [Google Scholar] [CrossRef]
- Findeisen, F.; Minor, D.L. Structural basis for the differential effects of CaBP1 and calmodulin on Ca(V)1.2 calcium-dependent inactivation. Structure 2010, 18, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Westenbroek, R.E.; Haeseleer, F.; Palczewski, K.; Scheuer, T.; Catterall, W.A. Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat. Neurosci. 2002, 5, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita-Kawada, M.; Tang, J.; Xiao, R.; Kaneko, S.; Foskett, J.K.; Zhu, M.X. Inhibition of TRPC5 channels by Ca2+-binding protein 1 in Xenopus oocytes. Pflug. Arch. 2005, 450, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Enomoto, M.; Rossi, A.M.; Seo, M.D.; Rahman, T.; Stathopulos, P.B.; Taylor, C.W.; Ikura, M.; Ames, J.B. CaBP1, a neuronal Ca2+ sensor protein, inhibits inositol trisphosphate receptors by clamping intersubunit interactions. Proc. Natl. Acad. Sci. USA 2013, 110, 8507–8512. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; McBride, S.; Mak, D.O.; Vardi, N.; Palczewski, K.; Haeseleer, F.; Foskett, J.K. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. USA 2002, 99, 7711–7716. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.P.; Tepikin, A.V.; Burgoyne, R.D. Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J. Biol. Chem. 2004, 279, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Kapitein, L.C.; van Bergeijk, P.; Lipka, J.; Keijzer, N.; Wulf, P.S.; Katrukha, E.A.; Akhmanova, A.; Hoogenraad, C.C. Myosin-V opposes microtubule-based cargo transport and drives directional motility on cortical actin. Curr. Biol. 2013, 23, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Konietzny, A.; Grendel, J.; Kadek, A.; Bucher, M.; Han, Y.; Hertrich, N.; Dekkers, D.H.W.; Demmers, J.A.A.; Grünewald, K.; Uetrecht, C.; et al. Caldendrin and myosin V regulate synaptic spine apparatus localization via ER stabilization in dendritic spines. EMBO J. 2022, 41, e106523. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Choi, J.E.; Soh, D.; Tobin, K.; Joiner, M.L.; Hansen, M.; Lee, A. CaBP1 regulates Ca. Mol. Cell. Neurosci. 2018, 88, 342–352. [Google Scholar] [CrossRef]
- Lopez, J.A.; Yamamoto, A.; Vecchi, J.T.; Hagen, J.; Lee, K.; Sonka, M.; Hansen, M.R.; Lee, A. Caldendrin represses neurite regeneration and growth in dorsal root ganglion neurons. Sci. Rep. 2023, 13, 2608. [Google Scholar] [CrossRef]
- Wu, J.; Xie, X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006, 7, R85. [Google Scholar] [CrossRef]
- Wild, A.R.; Dell’Acqua, M.L. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther. 2018, 185, 99–121. [Google Scholar] [CrossRef]
- Zhang, J.; Shapiro, M.S. Mechanisms and dynamics of AKAP79/150-orchestrated multi-protein signalling complexes in brain and peripheral nerve. J. Physiol. 2016, 594, 31–37. [Google Scholar] [CrossRef]
- Seeger, C.; Gorny, X.; Reddy, P.P.; Seidenbecher, C.; Danielson, U.H. Kinetic and mechanistic differences in the interactions between caldendrin and calmodulin with AKAP79 suggest different roles in synaptic function. J. Mol. Recognit. 2012, 25, 495–503. [Google Scholar] [CrossRef]
- Gorny, X.; Mikhaylova, M.; Seeger, C.; Reddy, P.P.; Reissner, C.; Schott, B.H.; Helena Danielson, U.; Kreutz, M.R.; Seidenbecher, C. AKAP79/150 interacts with the neuronal calcium-binding protein caldendrin. J. Neurochem. 2012, 122, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Karpova, A.; Mikhaylova, M.; Bera, S.; Bär, J.; Reddy, P.P.; Behnisch, T.; Rankovic, V.; Spilker, C.; Bethge, P.; Sahin, J.; et al. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 2013, 152, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylova, M.; Karpova, A.; Bär, J.; Bethge, P.; YuanXiang, P.; Chen, Y.; Zuschratter, W.; Behnisch, T.; Kreutz, M.R. Cellular distribution of the NMDA-receptor activated synapto-nuclear messenger Jacob in the rat brain. Brain Struct. Funct. 2014, 219, 843–860. [Google Scholar] [CrossRef] [PubMed]
- Grochowska, K.M.; Bär, J.; Gomes, G.M.; Kreutz, M.R.; Karpova, A. Jacob, a Synapto-Nuclear Protein Messenger Linking N-methyl-D-aspartate Receptor Activation to Nuclear Gene Expression. Front. Synaptic Neurosci. 2021, 13, 787494. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Dieterich, D.C.; Karpova, A.; Mikhaylova, M.; Zdobnova, I.; König, I.; Landwehr, M.; Kreutz, M.; Smalla, K.H.; Richter, K.; Landgraf, P.; et al. Caldendrin-Jacob: A protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 2008, 6, e34. [Google Scholar] [CrossRef] [PubMed]
- Landwehr, M.; Redecker, P.; Dieterich, D.C.; Richter, K.; Böckers, T.M.; Gundelfinger, E.D.; Kreutz, M.R. Association of Caldendrin splice isoforms with secretory vesicles in neurohypophyseal axons and the pituitary. FEBS Lett. 2003, 547, 189–192. [Google Scholar] [CrossRef]
- Mikhaylova, M.; Sharma, Y.; Reissner, C.; Nagel, F.; Aravind, P.; Rajini, B.; Smalla, K.H.; Gundelfinger, E.D.; Kreutz, M.R. Neuronal Ca2+ signaling via caldendrin and calneurons. Biochim. Biophys. Acta 2006, 1763, 1229–1237. [Google Scholar] [CrossRef]
- Hradsky, J.; Bernstein, H.G.; Marunde, M.; Mikhaylova, M.; Kreutz, M.R. Alternative splicing, expression and cellular localization of Calneuron-1 in the rat and human brain. J. Histochem. Cytochem. 2015, 63, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylova, M.; Reddy, P.P.; Munsch, T.; Landgraf, P.; Suman, S.K.; Smalla, K.H.; Gundelfinger, E.D.; Sharma, Y.; Kreutz, M.R. Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc. Natl. Acad. Sci. USA 2009, 106, 9093–9098. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Q.; Lin, X.; Liu, C.M.; Jamrich, M.; Shaffer, L.G. Identification of a human brain-specific gene, calneuron 1, a new member of the calmodulin superfamily. Mol. Genet. Metab. 2001, 72, 343–350. [Google Scholar] [CrossRef]
- McCue, H.V.; Burgoyne, R.D.; Haynes, L.P. Membrane targeting of the EF-hand containing calcium-sensing proteins CaBP7 and CaBP8. Biochem. Biophys. Res. Commun. 2009, 380, 825–831. [Google Scholar] [CrossRef][Green Version]
- McCue, H.V.; Burgoyne, R.D.; Haynes, L.P. Determination of the membrane topology of the small EF-hand Ca2+-sensing proteins CaBP7 and CaBP8. PLoS ONE 2011, 6, e17853. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Raghuram, V.; Mikhaylova, M.; Kreutz, M.R. A plasmid-based expression system to study protein-protein interactions at the Golgi in vivo. Anal. Biochem. 2016, 502, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Rajamanoharan, D.; McCue, H.V.; Burgoyne, R.D.; Haynes, L.P. Modulation of phosphatidylinositol 4-phosphate levels by CaBP7 controls cytokinesis in mammalian cells. Mol. Biol. Cell 2015, 26, 1428–1439. [Google Scholar] [CrossRef]
- de Barry, J.; Janoshazi, A.; Dupont, J.L.; Procksch, O.; Chasserot-Golaz, S.; Jeromin, A.; Vitale, N. Functional implication of neuronal calcium sensor-1 and phosphoinositol 4-kinase-beta interaction in regulated exocytosis of PC12 cells. J. Biol. Chem. 2006, 281, 18098–18111. [Google Scholar] [CrossRef]
- Yip, P.K.; Wong, L.F.; Sears, T.A.; Yáñez-Muñoz, R.J.; McMahon, S.B. Cortical overexpression of neuronal calcium sensor-1 induces functional plasticity in spinal cord following unilateral pyramidal tract injury in rat. PLoS Biol. 2010, 8, e1000399. [Google Scholar] [CrossRef]
- Neumann, B.; Walter, T.; Hériché, J.K.; Bulkescher, J.; Erfle, H.; Conrad, C.; Rogers, P.; Poser, I.; Held, M.; Liebel, U.; et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010, 464, 721–727. [Google Scholar] [CrossRef]
- Navarro, G.; Aguinaga, D.; Moreno, E.; Hradsky, J.; Reddy, P.P.; Cortés, A.; Mallol, J.; Casadó, V.; Mikhaylova, M.; Kreutz, M.R.; et al. Intracellular calcium levels determine differential modulation of allosteric interactions within G protein-coupled receptor heteromers. Chem. Biol. 2014, 21, 1546–1556. [Google Scholar] [CrossRef]
- Angelats, E.; Requesens, M.; Aguinaga, D.; Kreutz, M.R.; Franco, R.; Navarro, G. Neuronal Calcium and cAMP Cross-Talk Mediated by Cannabinoid CB. Front. Cell Dev. Biol. 2018, 6, 67. [Google Scholar] [CrossRef]
- Li, Z.; Xiang, Y.; Chen, J.; Li, Q.; Shen, J.; Liu, Y.; Li, W.; Xing, Q.; Wang, Q.; Wang, L.; et al. Loci with genome-wide associations with schizophrenia in the Han Chinese population. Br. J. Psychiatry 2015, 207, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhou, X.; Wang, T.; Zhang, Q.; Li, Q.; Liu, Y.; Xing, Q.; Wang, L.; He, L.; Zhao, X. Experimental validation of candidate schizophrenia gene CALN1 as a target for microRNA-137. Neurosci. Lett. 2015, 602, 110–114. [Google Scholar] [CrossRef]
- Roussos, P.; Giakoumaki, S.G.; Zouraraki, C.; Fullard, J.F.; Karagiorga, V.E.; Tsapakis, E.M.; Petraki, Z.; Siever, L.J.; Lencz, T.; Malhotra, A.; et al. The Relationship of Common Risk Variants and Polygenic Risk for Schizophrenia to Sensorimotor Gating. Biol. Psychiatry 2016, 79, 988–996. [Google Scholar] [CrossRef]
- Bourne, Y.; Dannenberg, J.; Pollmann, V.; Marchot, P.; Pongs, O. Immunocytochemical localization and crystal structure of human frequenin (neuronal calcium sensor 1). J. Biol. Chem. 2001, 276, 11949–11955. [Google Scholar] [CrossRef]
- McFerran, B.W.; Weiss, J.L.; Burgoyne, R.D. Neuronal Ca2+)sensor 1. Characterization of the myristoylated protein, its cellular effects in permeabilized adrenal chromaffin cells, Ca2+-independent membrane association, and interaction with binding proteins, suggesting a role in rapid Ca2+ signal transduction. J. Biol. Chem. 1999, 274, 30258–30265. [Google Scholar] [CrossRef] [PubMed]
- Aravind, P.; Chandra, K.; Reddy, P.P.; Jeromin, A.; Chary, K.V.; Sharma, Y. Regulatory and structural EF-hand motifs of neuronal calcium sensor-1: Mg 2+ modulates Ca2+ binding, Ca2+-induced conformational changes, and equilibrium unfolding transitions. J. Mol. Biol. 2008, 376, 1100–1115. [Google Scholar] [CrossRef]
- Chen, C.; Yu, L.; Zhang, P.; Jiang, J.; Zhang, Y.; Chen, X.; Wu, Q.; Zhao, S. Human neuronal calcium sensor-1 shows the highest expression level in cerebral cortex. Neurosci. Lett. 2002, 319, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Saab, B.J.; Georgiou, J.; Nath, A.; Lee, F.J.; Wang, M.; Michalon, A.; Liu, F.; Mansuy, I.M.; Roder, J.C. NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 2009, 63, 643–656. [Google Scholar] [CrossRef]
- Reynolds, A.J.; Bartlett, S.E.; Morgans, C. The distribution of neuronal calcium sensor-1 protein in the developing and adult rat retina. Neuroreport 2001, 12, 725–728. [Google Scholar] [CrossRef]
- Kawasaki, T.; Nishio, T.; Kurosawa, H.; Roder, J.; Jeromin, A. Spatiotemporal distribution of neuronal calcium sensor-1 in the developing rat spinal cord. J. Comp. Neurol. 2003, 460, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Treloar, H.B.; Uboha, U.; Jeromin, A.; Greer, C.A. Expression of the neuronal calcium sensor protein NCS-1 in the developing mouse olfactory pathway. J. Comp. Neurol. 2005, 482, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Dason, J.S.; Romero-Pozuelo, J.; Atwood, H.L.; Ferrús, A. Multiple roles for frequenin/NCS-1 in synaptic function and development. Mol. Neurobiol. 2012, 45, 388–402. [Google Scholar] [CrossRef]
- O’Callaghan, D.W.; Ivings, L.; Weiss, J.L.; Ashby, M.C.; Tepikin, A.V.; Burgoyne, R.D. Differential use of myristoyl groups on neuronal calcium sensor proteins as a determinant of spatio-temporal aspects of Ca2+ signal transduction. J. Biol. Chem. 2002, 277, 14227–14237. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Helassa, N.; McCue, H.V.; Haynes, L.P. Calcium Sensors in Neuronal Function and Dysfunction. Cold Spring Harb. Perspect. Biol. 2019, 11, a035154. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.L.; Hui, H.; Burgoyne, R.D. Neuronal calcium sensor-1 regulation of calcium channels, secretion, and neuronal outgrowth. Cell. Mol. Neurobiol. 2010, 30, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.Y.; Pandalaneni, S.R.; Todd, P.A.; Martin, V.M.; Burgoyne, R.D.; Haynes, L.P. Demonstration of binding of neuronal calcium sensor-1 to the cav2.1 p/q-type calcium channel. Biochemistry 2014, 53, 6052–6062. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Jeromin, A.; Saitoh, N.; Roder, J.C.; Takahashi, T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science 2002, 295, 2276–2279. [Google Scholar] [CrossRef]
- Iketani, M.; Imaizumi, C.; Nakamura, F.; Jeromin, A.; Mikoshiba, K.; Goshima, Y.; Takei, K. Regulation of neurite outgrowth mediated by neuronal calcium sensor-1 and inositol 1,4,5-trisphosphate receptor in nerve growth cones. Neuroscience 2009, 161, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, C.; Boehmerle, W.; Jeromin, A.; DeGray, B.; Varshney, A.; Sharma, Y.; Szigeti-Buck, K.; Ehrlich, B.E. Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J. Clin. Investig. 2006, 116, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.D.; Petri, E.T.; Huynh, L.K.; Ehrlich, B.E. Characterization of NCS1-InsP3R1 interaction and its functional significance. J. Biol. Chem. 2019, 294, 18923–18933. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.P.; Thomas, G.M.; Burgoyne, R.D. Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J. Biol. Chem. 2005, 280, 6047–6054. [Google Scholar] [CrossRef] [PubMed]
- Kabbani, N.; Negyessy, L.; Lin, R.; Goldman-Rakic, P.; Levenson, R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J. Neurosci. 2002, 22, 8476–8486. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.P.; Sherwood, M.W.; Dolman, N.J.; Burgoyne, R.D. Specificity, promiscuity and localization of ARF protein interactions with NCS-1 and phosphatidylinositol-4 kinase-III beta. Traffic 2007, 8, 1080–1092. [Google Scholar] [CrossRef]
- Hui, H.; McHugh, D.; Hannan, M.; Zeng, F.; Xu, S.Z.; Khan, S.U.; Levenson, R.; Beech, D.J.; Weiss, J.L. Calcium-sensing mechanism in TRPC5 channels contributing to retardation of neurite outgrowth. J. Physiol. 2006, 572, 165–172. [Google Scholar] [CrossRef]
- Hui, K.; Feng, Z.P. NCS-1 differentially regulates growth cone and somata calcium channels in Lymnaea neurons. Eur. J. Neurosci. 2008, 27, 631–643. [Google Scholar] [CrossRef]
- Hasbi, A.; Fan, T.; Alijaniaram, M.; Nguyen, T.; Perreault, M.L.; O’Dowd, B.F.; George, S.R. Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. USA 2009, 106, 21377–21382. [Google Scholar] [CrossRef]
- Nakamura, T.Y.; Nakao, S.; Nakajo, Y.; Takahashi, J.C.; Wakabayashi, S.; Yanamoto, H. Possible Signaling Pathways Mediating Neuronal Calcium Sensor-1-Dependent Spatial Learning and Memory in Mice. PLoS ONE 2017, 12, e0170829. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.Y.; Jeromin, A.; Smith, G.; Kurushima, H.; Koga, H.; Nakabeppu, Y.; Wakabayashi, S.; Nabekura, J. Novel role of neuronal Ca2+ sensor-1 as a survival factor up-regulated in injured neurons. J. Cell Biol. 2006, 172, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Plotnikov, E.Y.; Turovsky, E.A. Neuronal Calcium Sensor-1 Protects Cortical Neurons from Hyperexcitation and Ca. Int. J. Mol. Sci. 2022, 23, 15675. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisek, M.; Tomczak, J.; Boczek, T.; Zylinska, L. Calcium-Associated Proteins in Neuroregeneration. Biomolecules 2024, 14, 183. https://doi.org/10.3390/biom14020183
Lisek M, Tomczak J, Boczek T, Zylinska L. Calcium-Associated Proteins in Neuroregeneration. Biomolecules. 2024; 14(2):183. https://doi.org/10.3390/biom14020183
Chicago/Turabian StyleLisek, Malwina, Julia Tomczak, Tomasz Boczek, and Ludmila Zylinska. 2024. "Calcium-Associated Proteins in Neuroregeneration" Biomolecules 14, no. 2: 183. https://doi.org/10.3390/biom14020183
APA StyleLisek, M., Tomczak, J., Boczek, T., & Zylinska, L. (2024). Calcium-Associated Proteins in Neuroregeneration. Biomolecules, 14(2), 183. https://doi.org/10.3390/biom14020183