

Role of Mitochondrial ROS for Calcium Alternans in Atrial Myocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Ethical Approval
2.2. Chemicals, Solutions and Experimental Conditions
2.3. Isolation of Atrial Myocytes
2.4. Cytosolic Ca ([Ca]i) Measurements
2.5. ROS Measurements
2.6. CaT Alternans
2.7. Data Presentation and Statistics
3. Results
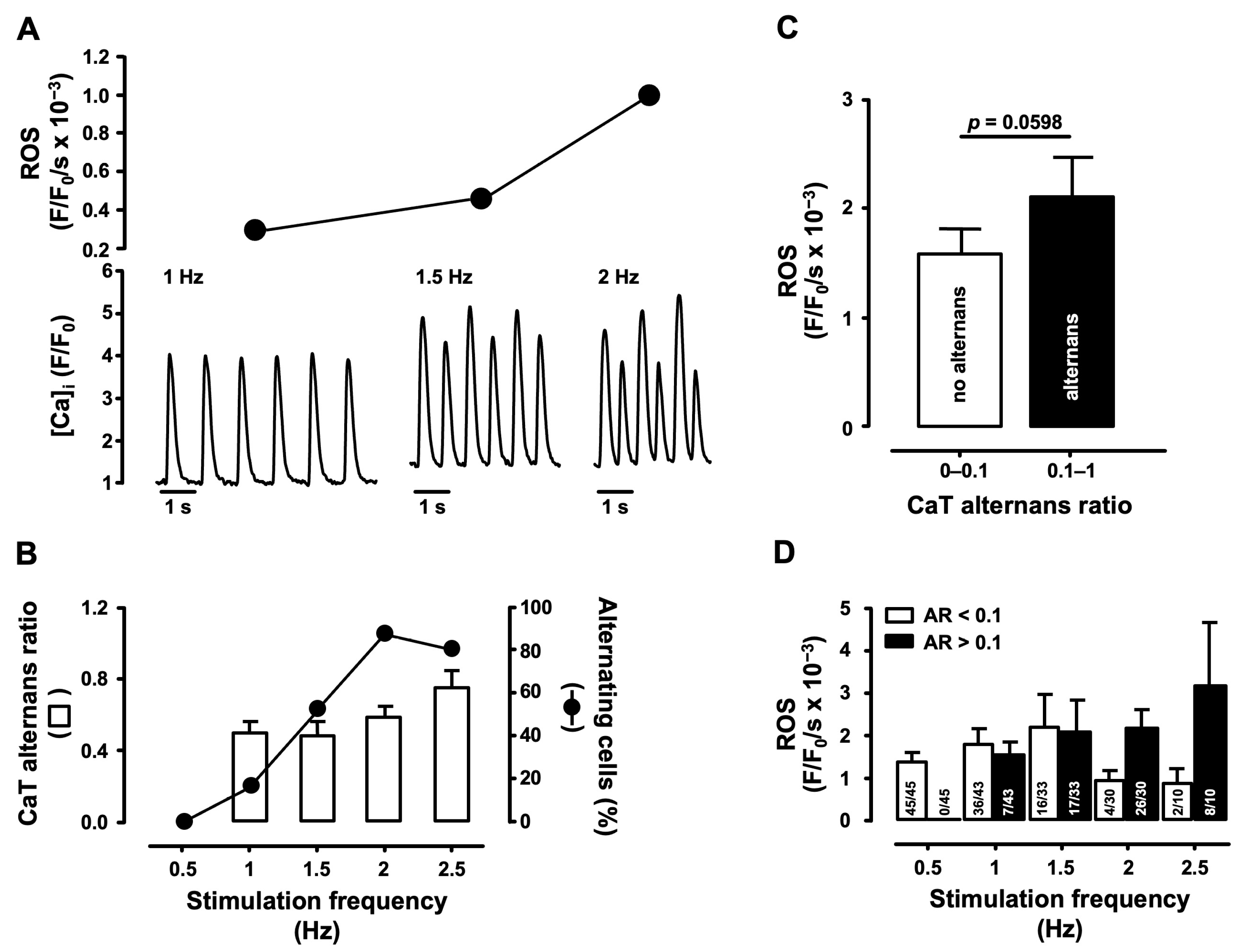
3.1. Correlation of Cellular ROS Production and CaT Alternans
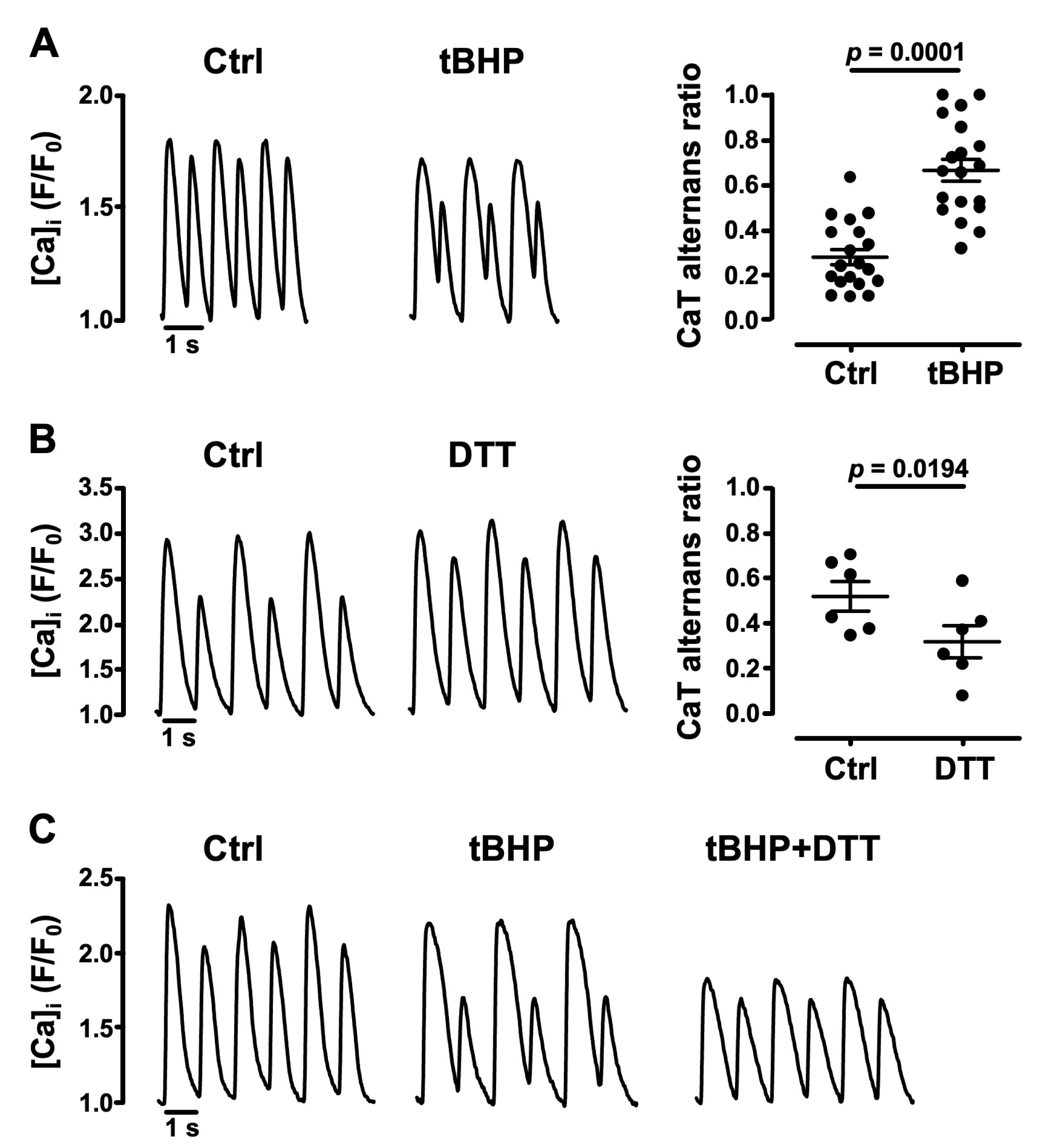
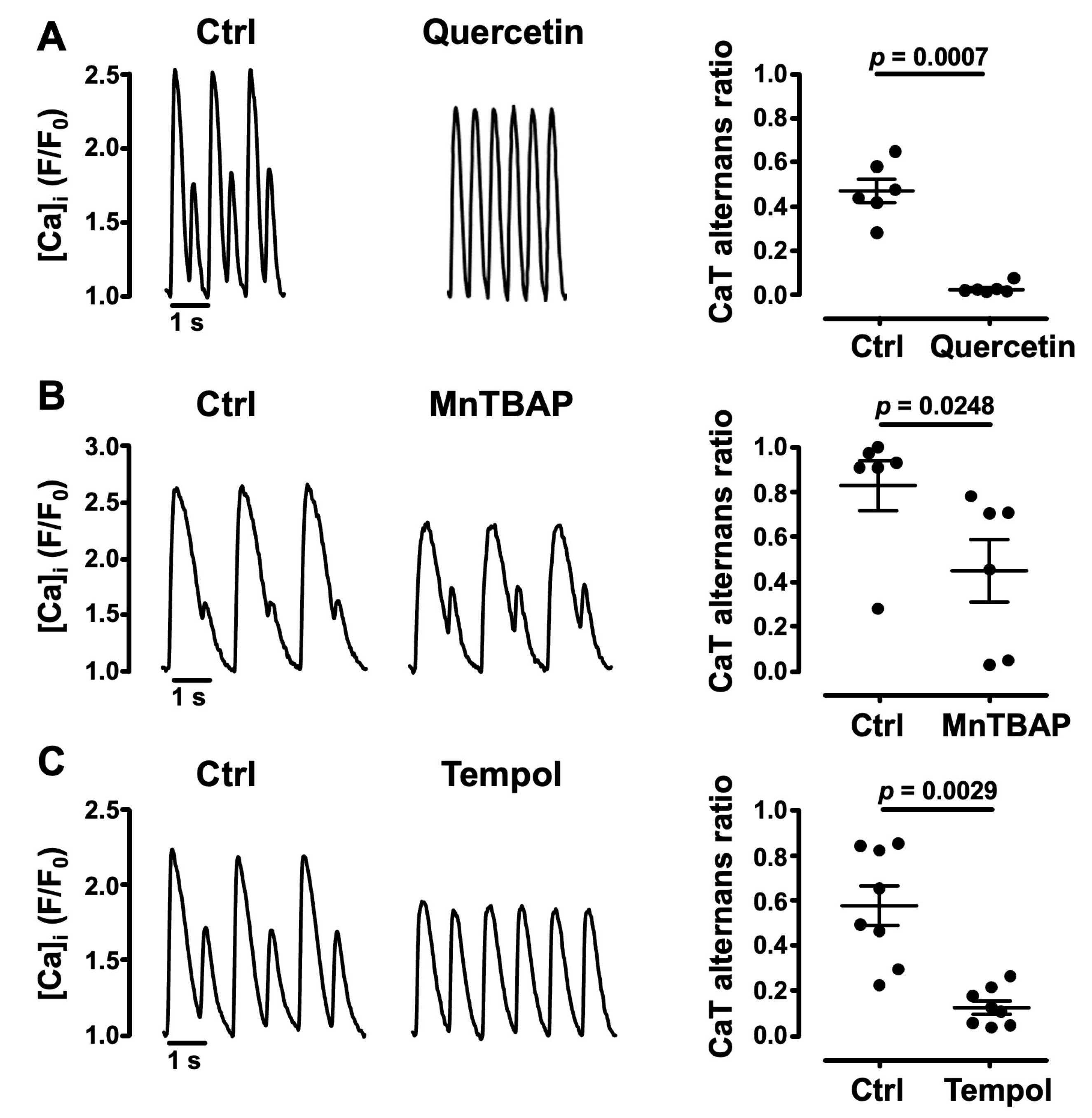
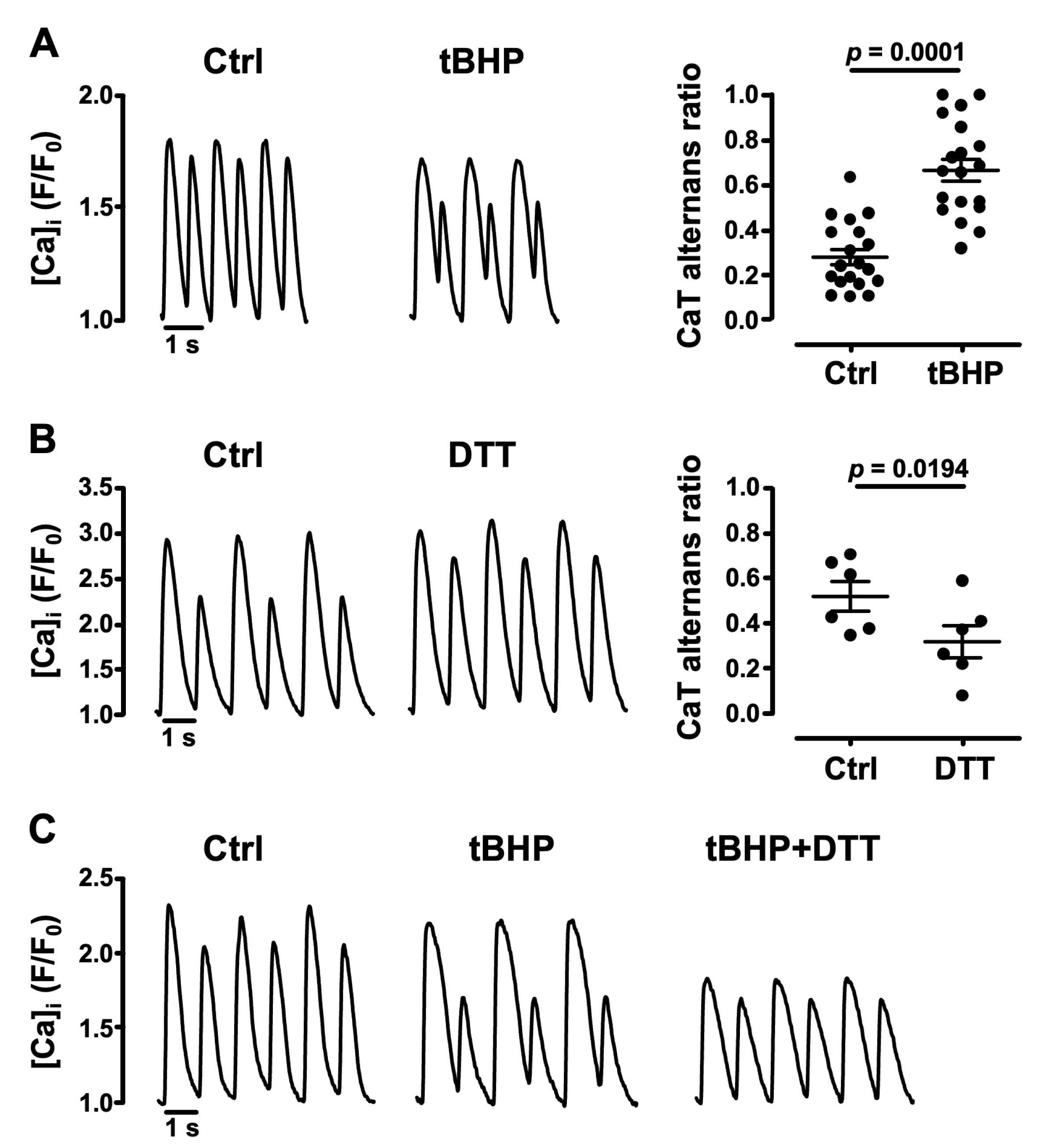
3.2. Oxidative Stress Facilitates CaT Alternans
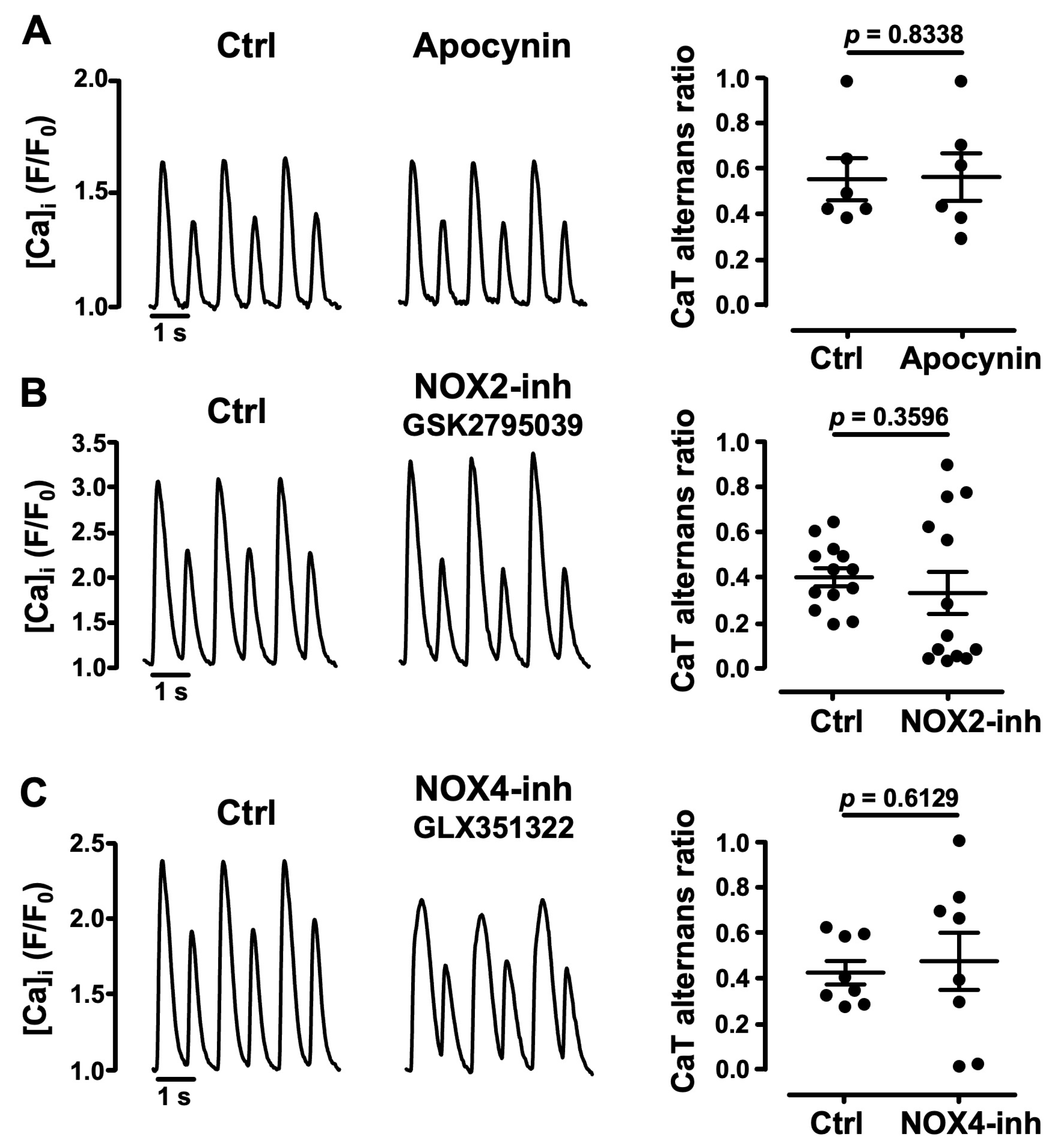
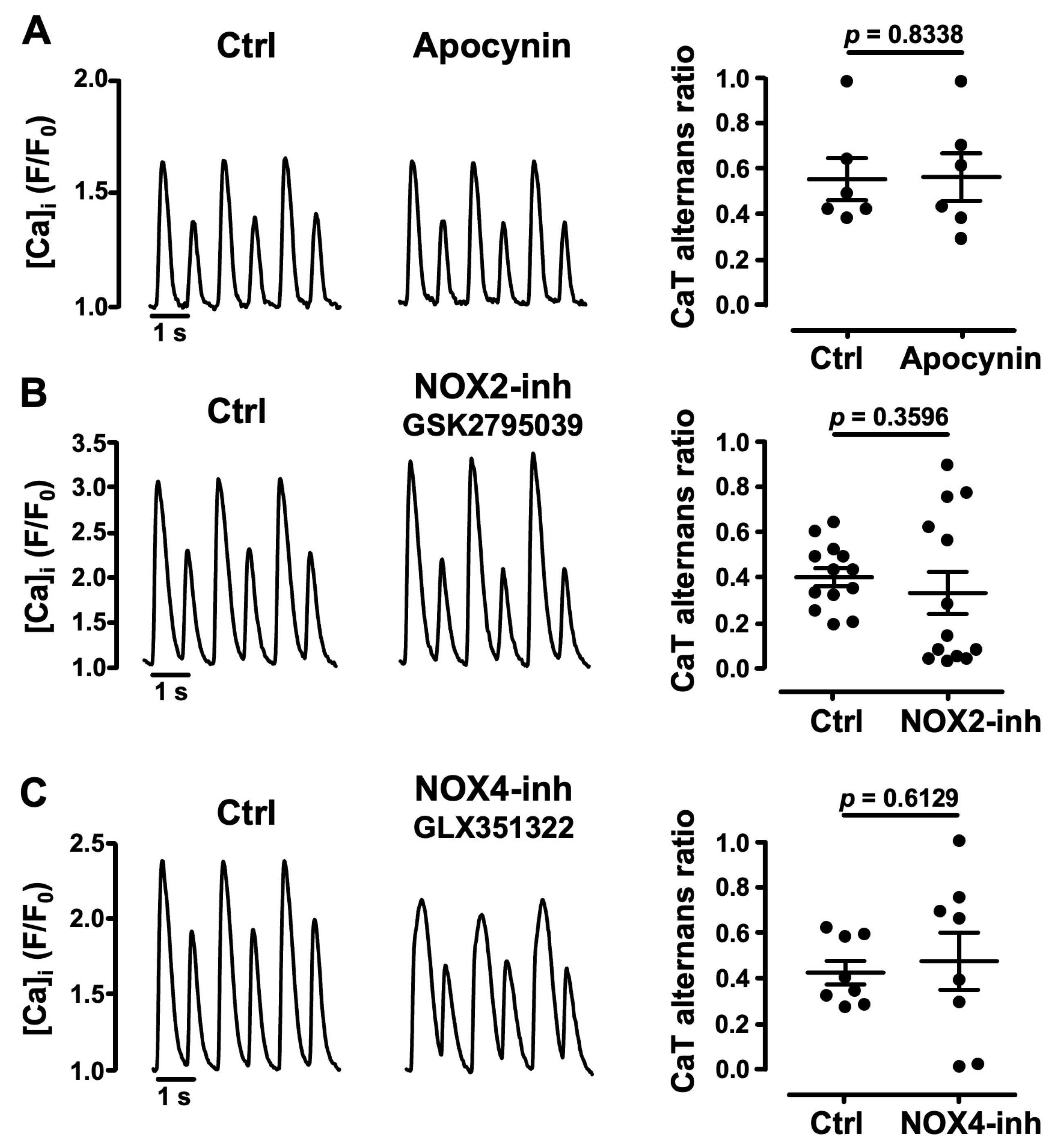
3.3. Role of NADPH Oxidases in Atrial CaT Alternans
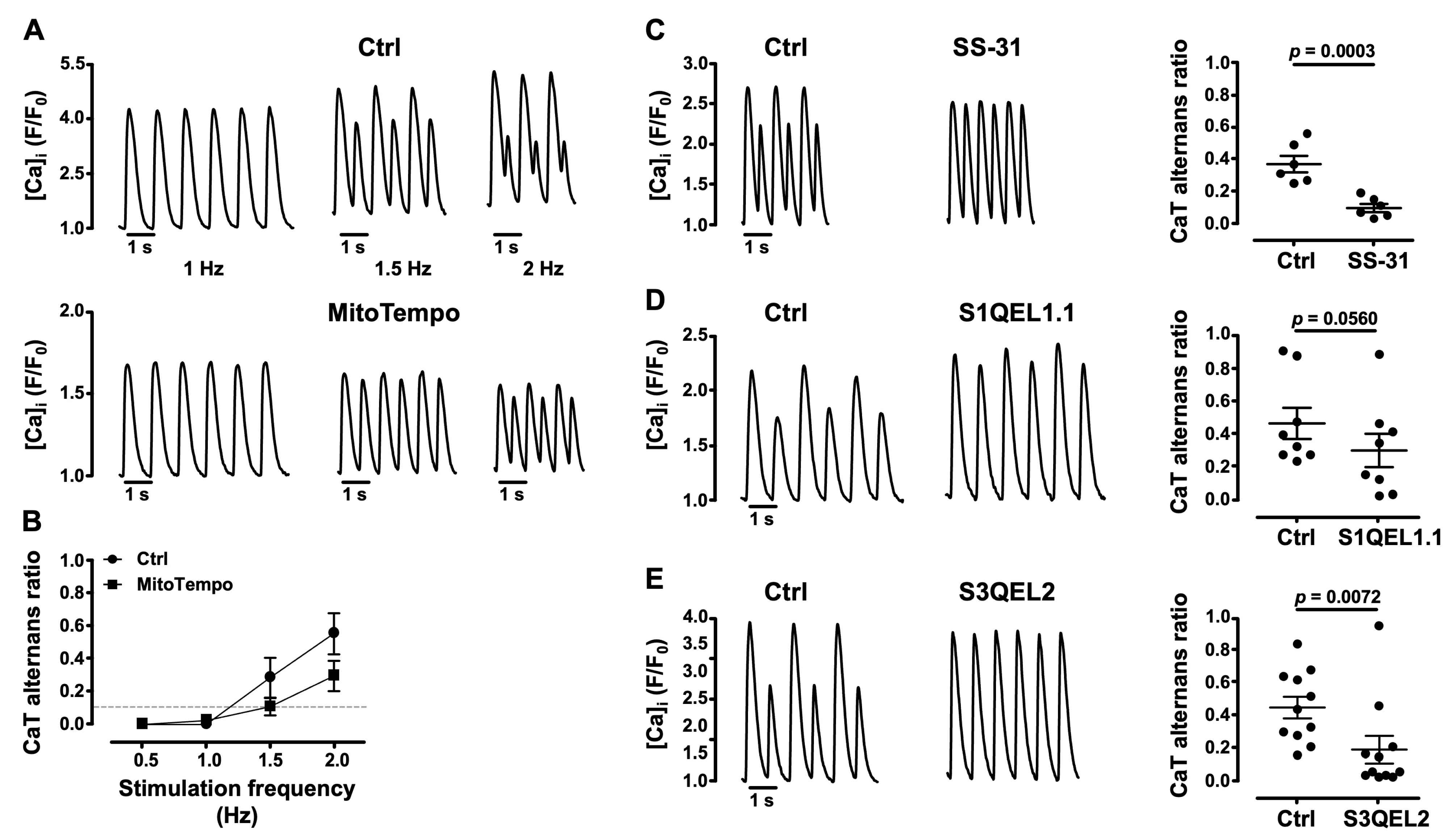
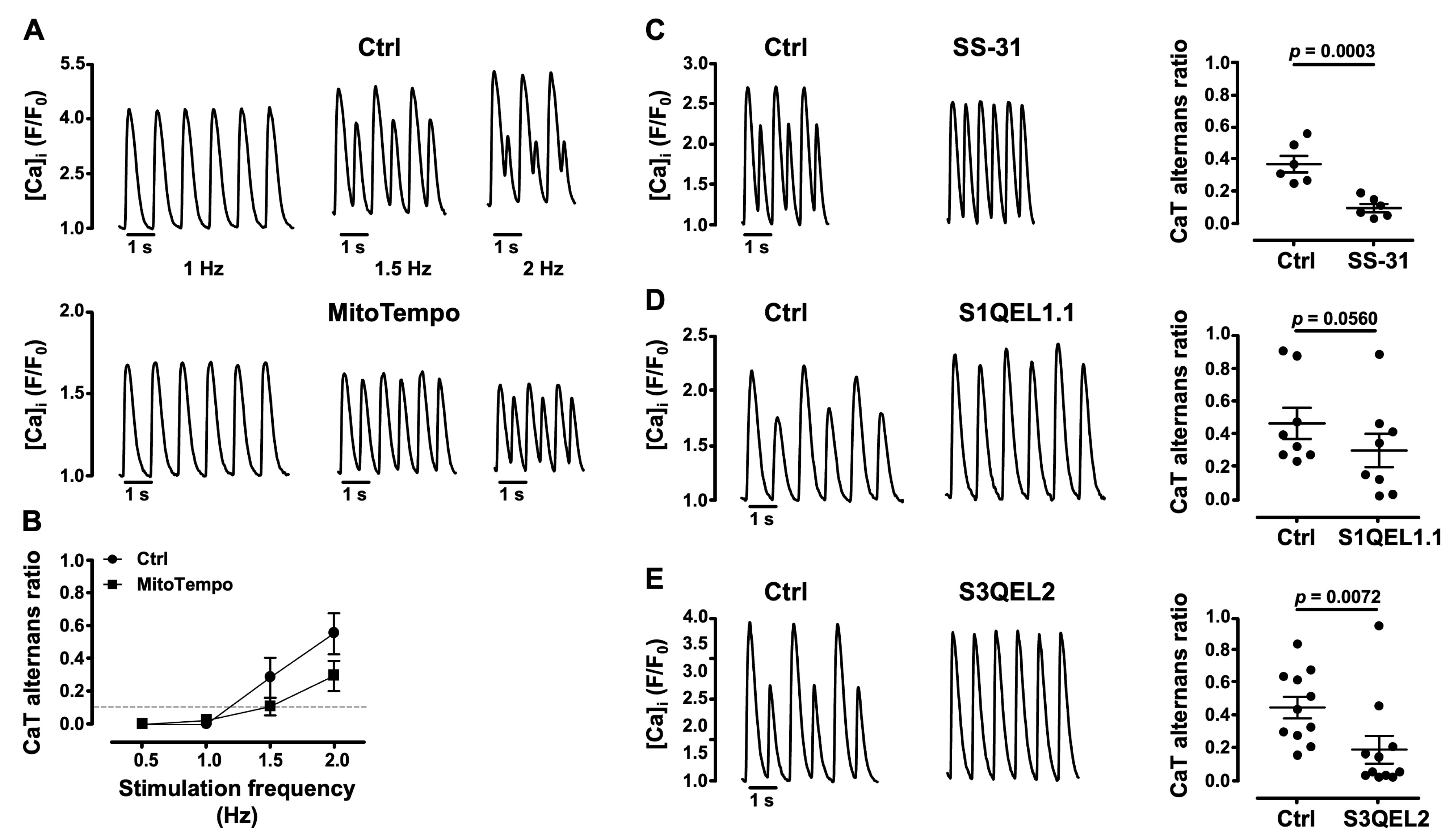
3.4. Mitochondrial ROS (ROSm) and CaT Alternans
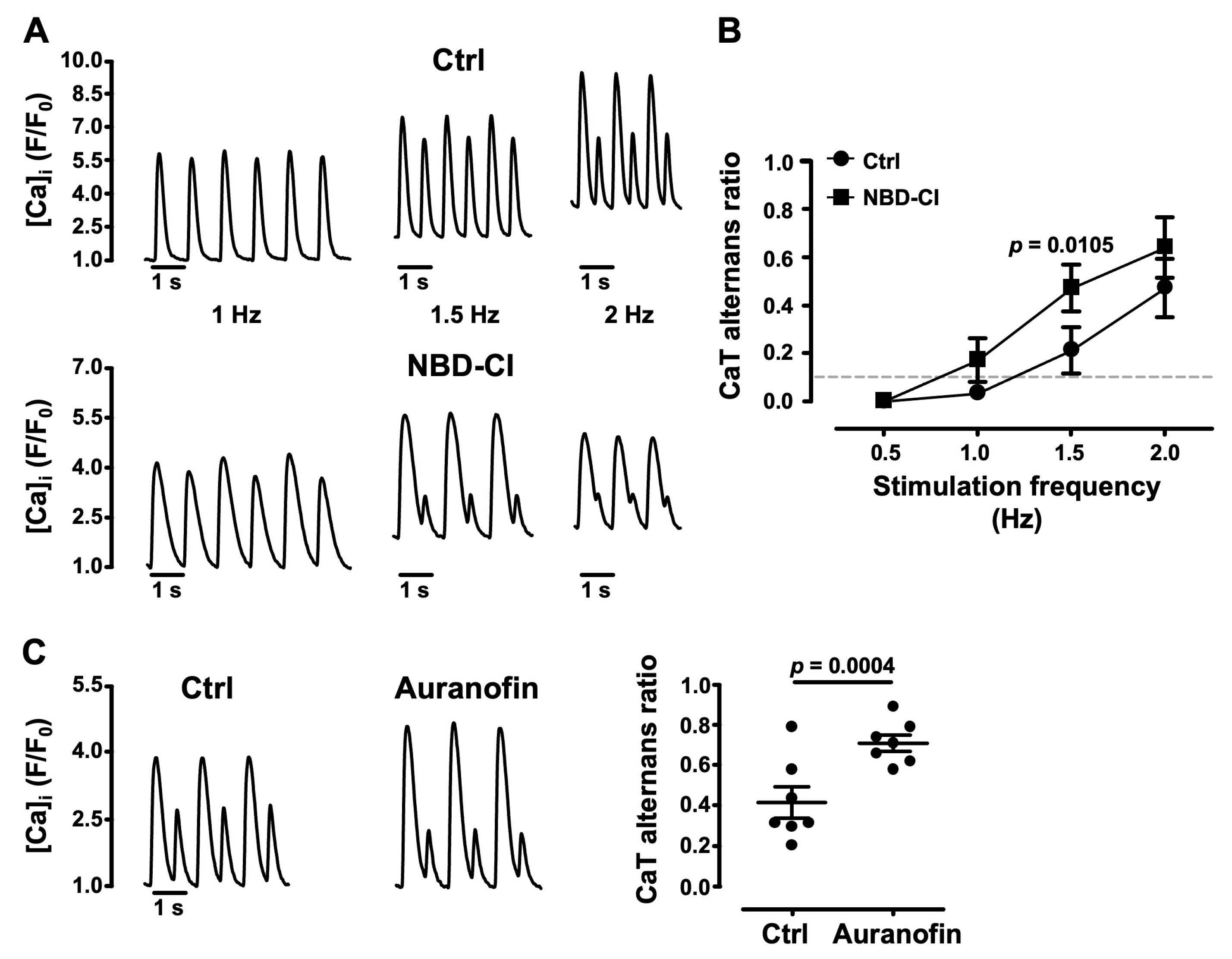
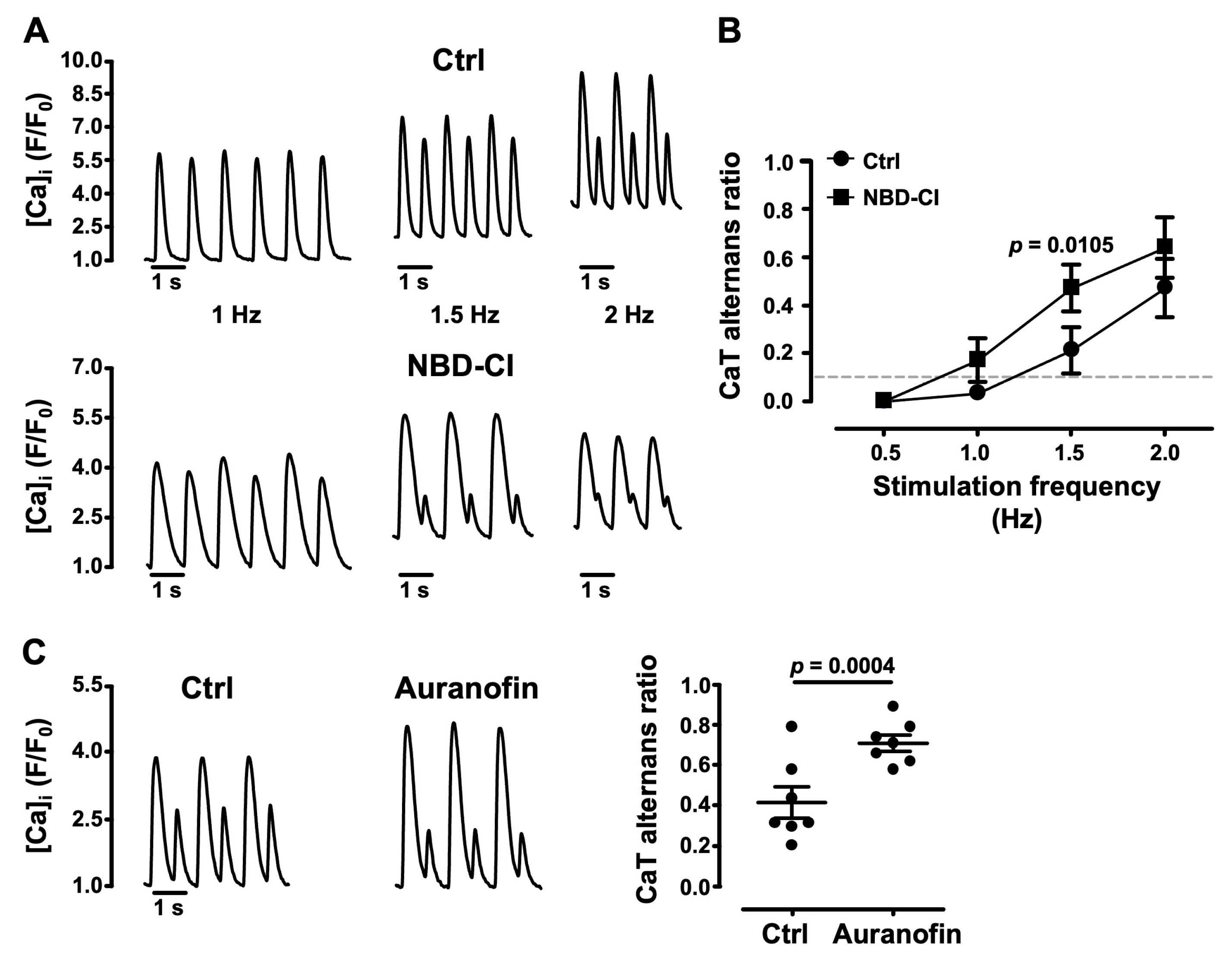
3.5. Impairment of Mitochondrial Antioxidant Defense Promotes CaT Alternans
4. Discussion
- (1)
- the cellular redox environment modulates pacing-induced CaT alternans development: exogenous ROS (applied in the form of tBHP) enhanced CaT alternans whereas ROS scavengers and reducing agents decreased AR;
- (2)
- cellular ROS production is increased during CaT alternans and the degree of alternans and rate of ROS production correlate with the pacing rate;
- (3)
- pharmacological inhibitors of NADPH oxidases, including specific inhibitors of NOX2 and NOX4, failed to affect alternans and excluded an important role of these specific ROS sources for alternans regulation;
- (4)
- several lines of evidence point to the important role of ROSm for the modulation of alternans: MitoTempo, a mitochondrial SOD mimetic, shifted the alternans pacing threshold to higher frequencies and decreased the degree of alternans; in the presence of the inhibitor of cyt c peroxidase SS-31 and during the inhibition of ROS production at ETC Complexes I and III with the small molecules S1QEL1.1 and S3QEL2, AR decreased or alternans even disappeared; the impairment of mitochondrial antioxidant defense by THD inhibition with NBD-Cl and the inhibition of TR2 with auranofin enhanced alternans.
4.1. Correlation of Pacing-Induced ROS Production and CaT Alternans Development
4.2. ROSm Production and CaT Alternans in Atrial Myocytes Are Interconnected
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kornej, J.; Börschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Blatter, L.A. Alternans in Atria: Mechanisms and Clinical Relevance. Medicina 2017, 53, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.N.; Blatter, L.A. Proceedings of the Australian Physiological Society Symposium: New Insights into Cardiac Ca2+ Handling and Excitability Cardiac Alternans and Intracellular Calcium Cycling. Clin. Exp. Pharmacol. Physiol. 2014, 41, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Karma, A.; Shiferaw, Y.; Chen, P.; Garfinkel, A.; Qu, Z. From Pulsus to Pulseless The Saga of Cardiac Alternans. Circ. Res. 2006, 98, 1244–1253. [Google Scholar] [CrossRef]
- Blatter, L.A. The Intricacies of Atrial Calcium Cycling during Excitation-Contraction Coupling. J. Physiol. 2017, 149, 857–865. [Google Scholar] [CrossRef]
- Hohendanner, F.; Maxwell, J.T.; Blatter, L.A. Cytosolic and Nuclear Calcium Signaling in Atrial Myocytes: IP3-Mediated Calcium Release and the Role of Mitochondria. Channels 2015, 9, 129–138. [Google Scholar] [CrossRef]
- Hüser, J.; Lipsius, S.L.; Blatter, L. Calcium Gradients during Excitation-Contraction Coupling in Cat Atrial Myocytes. J. Physiol. 1996, 494, 641–651. [Google Scholar] [CrossRef]
- Balaban, R.S. The Role of Ca2+ Signaling in the Coordination of Mitochondrial ATP Production with Cardiac Work. Biochim. Biophys. Acta BBA—Bioenerg. 2009, 1787, 1334–1341. [Google Scholar] [CrossRef]
- Brandes, R.; Bers, D.M. Intracellular Ca2+ Increases the Mitochondrial NADH Concentration during Elevated Work in Intact Cardiac Muscle. Circ. Res. 1997, 80, 82–87. [Google Scholar] [CrossRef]
- Murphy, M. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Brand, M. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.G.; Kohlhaas, M.; Maack, C. Mitochondrial Reactive Oxygen Species Production and Elimination. J. Mol. Cell Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabò, R.; Giorgio, M. Mitochondrial Pathways for ROS Formation and Myocardial Injury: The Relevance of P66Shc and Monoamine Oxidase. Basic Res. Cardiol. 2009, 104, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Characteristics and Function of Cardiac Mitochondrial Nitric Oxide Synthase. J. Physiol. 2009, 587, 851–872. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Aon, M.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/Thioredoxin Systems Modulate Mitochondrial H2O2 Emission: An Experimental-Computational Study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef]
- Nickel, A.G.; Von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Zhang, H.; Gomez, A.M.; Wang, X.; Yan, Y.; Zheng, M.; Cheng, H. ROS Regulation of Microdomain Ca2+ Signalling at the Dyads. Cardiovasc. Res. 2013, 98, 248–258. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M. Reactive Oxygen Species Production by Mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef]
- Jeong, E.-M.; Liu, M.; Sturdy, M.; Gao, G.; Sovari, A.A.; Dudley, S.C. Metabolic Stress, Reactive Oxygen Species, and Arrhythmia. J. Mol. Cell Cardiol. 2012, 52, 454–463. [Google Scholar] [CrossRef]
- Zima, A.V.; Blatter, L.A. Redox Regulation of Cardiac Calcium Channels and Transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Viatchenko-Karpinski, S.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; Wilson, L.D.; Cardounel, A.J.; Laurita, K.R.; Carnes, C.A.; et al. Redox Modification of Ryanodine Receptors Underlies Calcium Alternans in a Canine Model of Sudden Cardiac Death. Cardiovasc. Res. 2009, 84, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Mazurek, S.R.; Zima, A.V. The Role of RyR2 Oxidation in the Blunted Frequency-Dependent Facilitation of Ca2+ Transient Amplitude in Rabbit Failing Myocytes. Pflug. Arch. 2018, 470, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Subramanyam, P.; Radlicz, C.; Trent, C.M.; Iyer, V.; Colecraft, H.M.; Morrow, J.P. Mitochondrial Oxidative Stress during Cardiac Lipid Overload Causes Intracellular Calcium Leak and Arrhythmia. Heart Rhythm 2016, 13, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Sovari, A.A.; Xie, Y.; Fishbein, M.C.; Mandel, W.J.; Garfinkel, A.; Lin, S.F.; Chen, P.S.; Xie, L.H.; Chen, F.; et al. Increased Susceptibility of Aged Hearts to Ventricular Fibrillation during Oxidative Stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, 1594–1605. [Google Scholar] [CrossRef]
- Plummer, B.N.; Liu, H.; Wan, X.; Deschênes, I.; Laurita, K.R. Targeted Antioxidant Treatment Decreases Cardiac Alternans Associated with Chronic Myocardial Infarction. Circ. Arrhythm. Electrophysiol. 2015, 8, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Choi, E.; Park, S.H.; Lee, S.H.; Cho, H.; Ho, W.K.; Ryu, S.Y. Sustained CaMKII Activity Mediates Transient Oxidative Stress-Induced Long-Term Facilitation of L-Type Ca2+ Current in Cardiomyocytes. Free Radic. Biol. Med. 2011, 51, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative Stress Creates a Unique, CaMKII-Mediated Substrate for Atrial Fibrillation in Heart Failure. JCI Insight 2018, 3, e120728. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial Oxidative Stress Promotes Atrial Fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Eustress: On Constant Alert for Redox Homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Dey, S.; Sidor, A.; O’Rourke, B. Compartment-Specific Control of Reactive Oxygen Species Scavenging by Antioxidant Pathway Enzymes. J. Biol. Chem. 2016, 291, 11185–11197. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Kalik, Z.M.; Blatter, L.A. Action Potential Shortening Rescues Atrial Calcium Alternans. J. Physiol. 2019, 597, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Griendling, K.K.; Harrison, D.G. Measurement of Reactive Oxygen Species in Cardiovascular Studies. Hypertension 2007, 49, 717–727. [Google Scholar] [CrossRef]
- Kockskämper, J.; Blatter, L.A. Subcellular Ca2+ Alternans Represents a Novel Mechanism for the Generation of Arrhythmogenic Ca2+ Waves in Cat Atrial Myocytes. J. Physiol. 2002, 545, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Clusin, W.T. Calcium Transient Alternans in Blood-Perfused Ischemic Hearts: Observations with Fluorescent Indicator Fura Red. Am. J. Physiol. Heart Circ. Physiol. 1997, 273, 2161–2169. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. The Mechanisms of Calcium Cycling and Action Potential Dynamics in Cardiac Alternans. Circ. Res. 2015, 116, 846–856. [Google Scholar] [CrossRef]
- Murphy, E.; Liu, J. Mitochondrial Calcium and Reactive Oxygen Species in Cardiovascular Disease. Cardiovasc. Res. 2023, 119, 1105–1116. [Google Scholar] [CrossRef]
- Euler, D.E. Cardiac Alternans: Mechanisms and Pathophysiological Significance. Cardiovasc. Res. 1999, 42, 583–590. [Google Scholar] [CrossRef]
- Dey, S.; DeMazumder, D.; Sidor, A.; Brian Foster, D.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Varma, D.; Almeida, J.F.Q.; DeSantiago, J.; Blatter, L.A.; Banach, K. Inositol 1,4,5-Trisphosphate Receptor—Reactive Oxygen Signaling Domain Regulates Excitation-Contraction Coupling in Atrial Myocytes. J. Mol. Cell. Cardiol. 2022, 163, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, T.; Long, M.; Li, P. Quercetin: Its Main Pharmacological Activity and Potential Application in Clinical Medicine. Oxid. Med. Cell Longev. 2020, 2020, 8825387. [Google Scholar] [CrossRef] [PubMed]
- Day, B.J.; Fridovich, I.; Crapo, J.D. Manganic Porphyrins Possess Catalase Activity and Protect Endothelial Cells against Hydrogen Peroxide-Mediated Injury. Arch Biochem. Biophys. 1997, 347, 256–262. [Google Scholar] [CrossRef]
- Wilcox, C.S. Effects of Tempol and Redox-Cycling Nitroxides in Models of Oxidative Stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Jing, Z.; Pinfang, K.; Chao, S.; Shaohuan, Q. Quercetin Inhibits Pyroptosis in Diabetic Cardiomyopathy through the Nrf2 Pathway. Genet. Res. (Camb.) 2022, 2022, 9723632. [Google Scholar] [CrossRef]
- Fiordaliso, F.; De Angelis, N.; Bai, A.; Cuccovillo, I.; Salio, M.; Maria Serra, D.; Bianchi, R.; Razzetti, R.; Latini, R.; Masson, S. Effect of β-Adrenergic and Renin-Angiotensin System Blockade on Myocyte Apoptosis and Oxidative Stress in Diabetic Hypertensive Rats. Life Sci. 2007, 81, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH Oxidases and Oxidase Crosstalk in Cardiovascular Diseases: Novel Therapeutic Targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX Signaling in Molecular Cardiovascular Mechanisms Involved in the Blood Pressure Homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef]
- Santos, C.X.C.; Anilkumar, N.; Zhang, M.; Brewer, A.C.; Shah, A.M. Redox Signaling in Cardiac Myocytes. Free Radic. Biol. Med. 2011, 50, 777–793. [Google Scholar] [CrossRef]
- Hirano, K.; Chen, W.S.; Chueng, A.L.W.; Dunne, A.A.; Seredenina, T.; Filippova, A.; Ramachandran, S.; Bridges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by Hypertrophic Stimuli Promotes Apoptosis and Mitochondrial Dysfunction in Cardiac Myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.; Weissmann, N.; Schröder, K. Nox Family NADPH Oxidases: Molecular Mechanisms of Activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.H. NOX4 Activity Is Determined by MRNA Levels and Reveals a Unique Pattern of ROS Generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef]
- Anvari, E.; Wikström, P.; Walum, E.; Welsh, N. The Novel NADPH Oxidase 4 Inhibitor GLX351322 Counteracts Glucose Intolerance in High-Fat Diet-Treated C57BL/6 Mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.J.; Murphy, M.P. A Mitochondria-Targeted Nitroxide Is Reduced to Its Hydroxylamine by Ubiquinol in Mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Kirilyuk, I.A.; Dikalov, S.I. Antihypertensive Effect of Mitochondria-Targeted Proxyl Nitroxides. Redox Biol. 2015, 4, 355–362. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyeva, R.; Martin, B.; Perger, F.; Li, J.; Stepanov, A.; Bonilla, I.M.; Knollmann, B.C.; Radwański, P.B.; Györke, S.; et al. Increased RyR2 Activity Is Exacerbated by Calcium Leak-Induced Mitochondrial ROS. Basic Res. Cardiol. 2020, 115, 1–20. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H. Targeting Mitochondrial Cardiolipin and the Cytochrome c/Cardiolipin Complex to Promote Electron Transport and Optimize Mitochondrial ATP Synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Zhang, H.; Alder, N.N.; Wang, W.; Szeto, H.; Marcinek, D.J.; Rabinovitch, P.S. Reduction of Elevated Proton Leak Rejuvenates Mitochondria in the Aged Cardiomyocyte. eLife 2020, 9, e60827. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Brand, M.; Goncalves, R.L.S.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef]
- Orr, A.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.S.; et al. Suppressors of Superoxide Production from Mitochondrial Complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Aon, M.; Cortassa, S.; Maack, C.; O’Rourke, B. Sequential Opening of Mitochondrial Ion Channels as a Function of Glutathione Redox Thiol Status. J. Biol. Chem. 2007, 282, 21889–21900. [Google Scholar] [CrossRef]
- Stanley, B.A.; Sivakumaran, V.; Shi, S.; McDonald, I.; Lloyd, D.; Watson, W.H.; Aon, M.; Paolocci, N. Thioredoxin Reductase-2 Is Essential for Keeping Low Levels of H2O2 Emission from Isolated Heart Mitochondria. J. Biol. Chem. 2011, 286, 33669–33677. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Folda, A.; Baldoin, M.C.; Scutari, G.; Bindoli, A. Effect of Auranofin on the Mitochondrial Generation of Hydrogen Peroxide. Role of Thioredoxin Reductase. Free Radic. Res. 2005, 39, 687–695. [Google Scholar] [CrossRef]
- Heinzel, F.; Luo, Y.; Dodoni, G.; Boengler, K.; Petrat, F.; Di Lisa, F.; de Groot, H.; Schulz, R.; Heusch, G. Formation of Reactive Oxygen Species at Increased Contraction Frequency in Rat Cardiomyocytes. Cardiovasc. Res. 2006, 71, 374–382. [Google Scholar] [CrossRef]
- Oropeza-Almazán, Y.; Blatter, L. Mitochondrial Calcium Uniporter Complex Activation Protects against Calcium Alternans in Atrial Myocytes. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H873–H881. [Google Scholar] [CrossRef]
- Florea, S.M.; Blatter, L.A. The Role of Mitochondria for the Regulation of Cardiac Alternans. Front Physiol. 2010, 1, 141. [Google Scholar] [CrossRef]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and Mitochondrial Mechanisms of Atrial Fibrillation. Basic Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Monternier, P.A.; Brand, M.D. S1QELs Suppress Mitochondrial Superoxide/Hydrogen Peroxide Production from Site IQ without Inhibiting Reverse Electron Flow through Complex I. Free Radic. Biol. Med. 2019, 143, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Dröse, S. Superoxide Generation by Complex III: From Mechanistic Rationales to Functional Consequences. Biochim. Biophys. Acta Rev. Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Copello, J.A.; Blatter, L.A. Effects of Cytosolic NADH/NAD+ Levels on Sarcoplasmic Reticulum Ca2+ Release in Permeabilized Rat Ventricular Myocytes. J. Physiol. 2004, 555, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Györke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; De Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox Modification of Ryanodine Receptors Contributes to Sarcoplasmic Reticulum Ca2+ Leak in Chronic Heart Failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H. First-in-class Cardiolipin-protective Compound as a Therapeutic Agent to Restore Mitochondrial Bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; van Marion, D.M.S.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; de Groot, N.M.S.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial Dysfunction and Oxidative Stress in Heart Disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting Mitochondria for Cardiovascular Disorders: Therapeutic Potential and Obstacles. Nat. Rev. Cardiol. 2018, 16, 33–55. [Google Scholar] [CrossRef]
- Mitchell, W.; Ng, E.A.; Tamucci, J.D.; Boyd, K.J.; Sathappa, M.; Coscia, A.; Pan, M.; Han, X.; Eddy, N.A.; May, E.R.; et al. The Mitochondria-Targeted Peptide SS-31 Binds Lipid Bilayers and Modulates Surface Electrostatics as a Key Component of Its Mechanism of Action. J. Biol. Chem. 2020, 295, 7452–7469. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative Lipidomics of Apoptosis: Redox Catalytic Interactions of Cytochrome c with Cardiolipin and Phosphatidylserine. Free Radic. Biol. Med. 2004, 37, 1963–1985. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H. Cell-Permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane Inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.; Kruse, S.; Percival, J.; Goh, J.; White, C.; Hopkins, H.; Kavanagh, T.; Szeto, H.; Rabinovich, P.; Marcinek, D. Mitochondrial-Targeted Peptide Rapidly Improves Mitochondrial Energetics and Skeletal Muscle Performance in Aged Mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Ruggiero, F.M.; Di Venosa, N.; Paradies, G. Decreased Complex III Activity in Mitochondria Isolated from Rat Heart Subjected to Ischemia and Reperfusion: Role of Reactive Oxygen Species and Cardiolipin. FASEB J. 2003, 17, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Pabbidi, M.R.; Lipsius, S.L.; Blatter, L.A. Effects of Mitochondrial Uncoupling on Ca2+ Signaling during Excitation-Contraction Coupling in Atrial Myocytes. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; O’Neill, S.C. A Comparison of the Effects of ATP and Tetracaine on Spontaneous Ca2+ Release from Rat Permeabilised Cardiac Myocytes. J. Physiol. 2001, 534, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Esworthy, R.S.; Ho, Y.S.; Chu, F.F. The Gpx1 Gene Encodes Mitochondrial Glutathione Peroxidase in the Mouse Liver. Arch. Biochem. Biophys. 1997, 340, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, J.H.; Lim, D.S.; Shim, W.J.; Ro, Y.M.; Park, G.H.; Becker, K.G.; Cho-Chung, Y.S.; Kim, M.K. Gene Expression Profiling of Oxidative Stress on Atrial Fibrillation in Humans. Exp. Mol. Med. 2003, 35, 336–349. [Google Scholar] [CrossRef]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Szekeres, F.L.M.; Walum, E.; Wikström, P.; Arner, A. A Small Molecule Inhibitor of Nox2 and Nox4 Improves Contractile Function after Ischemia–Reperfusion in the Mouse Heart. Sci. Rep. 2021, 11, 11970. [Google Scholar] [CrossRef]
- Silberman, G.A.; Fan, T.H.M.; Liu, H.; Jiao, Z.; Xiao, H.D.; Lovelock, J.D.; Boulden, B.M.; Widder, J.; Fredd, S.; Bernstein, K.E.; et al. Uncoupled Cardiac Nitric Oxide Synthase Mediates Diastolic Dysfunction. Circulation 2010, 121, 519–528. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oropeza-Almazán, Y.; Blatter, L.A. Role of Mitochondrial ROS for Calcium Alternans in Atrial Myocytes. Biomolecules 2024, 14, 144. https://doi.org/10.3390/biom14020144
Oropeza-Almazán Y, Blatter LA. Role of Mitochondrial ROS for Calcium Alternans in Atrial Myocytes. Biomolecules. 2024; 14(2):144. https://doi.org/10.3390/biom14020144
Chicago/Turabian StyleOropeza-Almazán, Yuriana, and Lothar A. Blatter. 2024. "Role of Mitochondrial ROS for Calcium Alternans in Atrial Myocytes" Biomolecules 14, no. 2: 144. https://doi.org/10.3390/biom14020144
APA StyleOropeza-Almazán, Y., & Blatter, L. A. (2024). Role of Mitochondrial ROS for Calcium Alternans in Atrial Myocytes. Biomolecules, 14(2), 144. https://doi.org/10.3390/biom14020144