
A Structural Proteomics Exploration of Synphilin-1 and Alpha-Synuclein Interaction in Pathogenesis of Parkinson’s Disease

 , , ,
, , ,
Abstract

1. Introduction
2. Materials and Methods
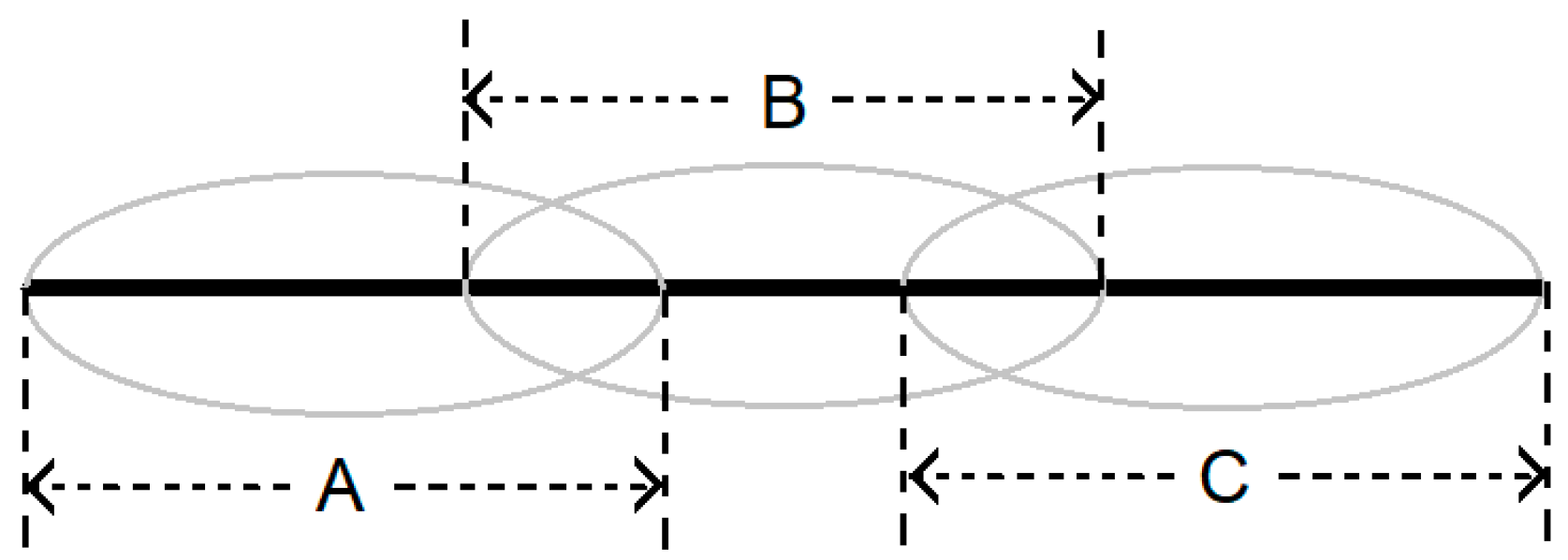
2.1. Protein Mediated Interaction Analysis (PMIA) Formalism
2.2. Building of Semi-Automatic Pipeline for Designing the Meta-Predictor AlphaLarge
2.2.1. Collection of Data for Sample Proteins and Ligands
2.2.2. Working Principle of AlphaLarge
3. Results
3.1. Results of AlphaLarge Application on the Training Samples
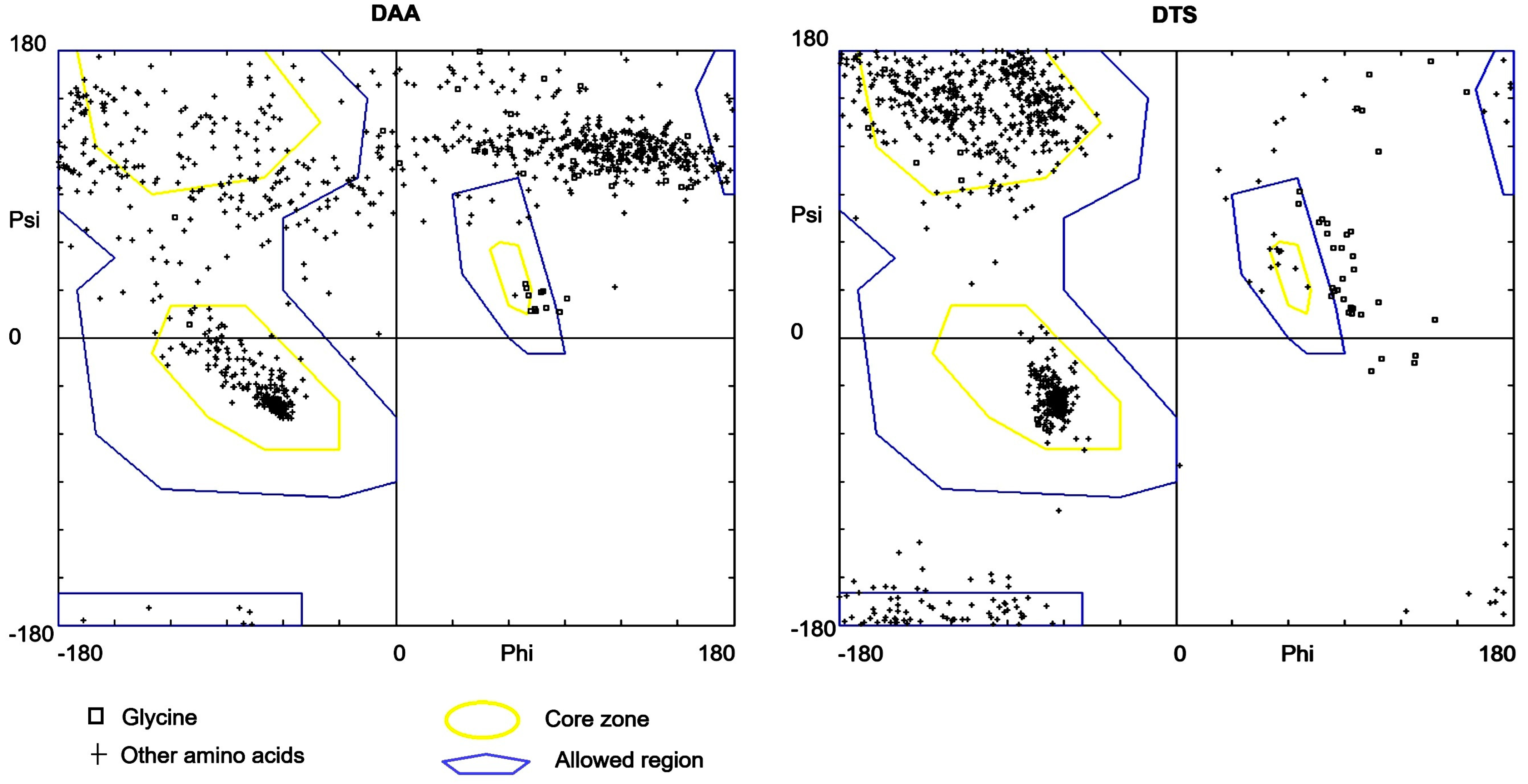
3.2. Standard Validation of Syn-1 Model Structures
3.2.1. Result of PMIA Formalism Based Re-Validation of Syn-1 Structure Using Binding Energy
3.2.2. Result of PMIA Formalism Based Re-Validation of Syn-1 Structure Using Residue Level Interaction
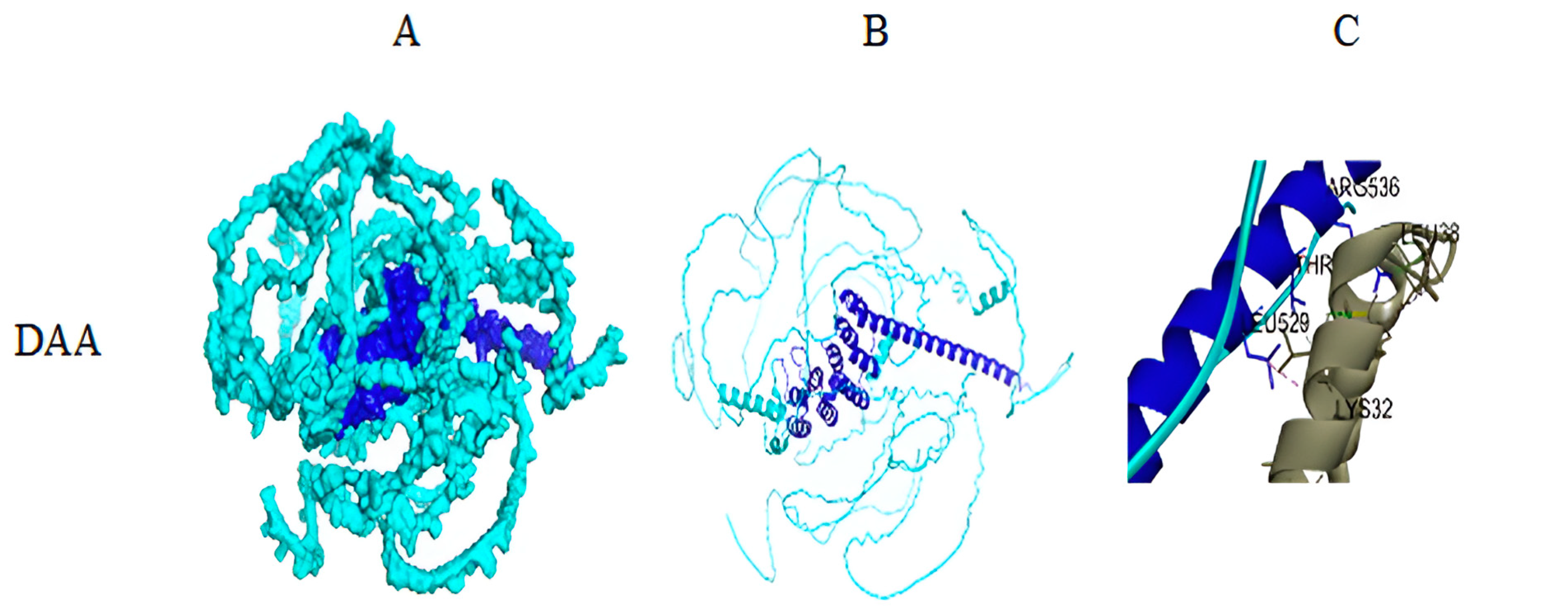
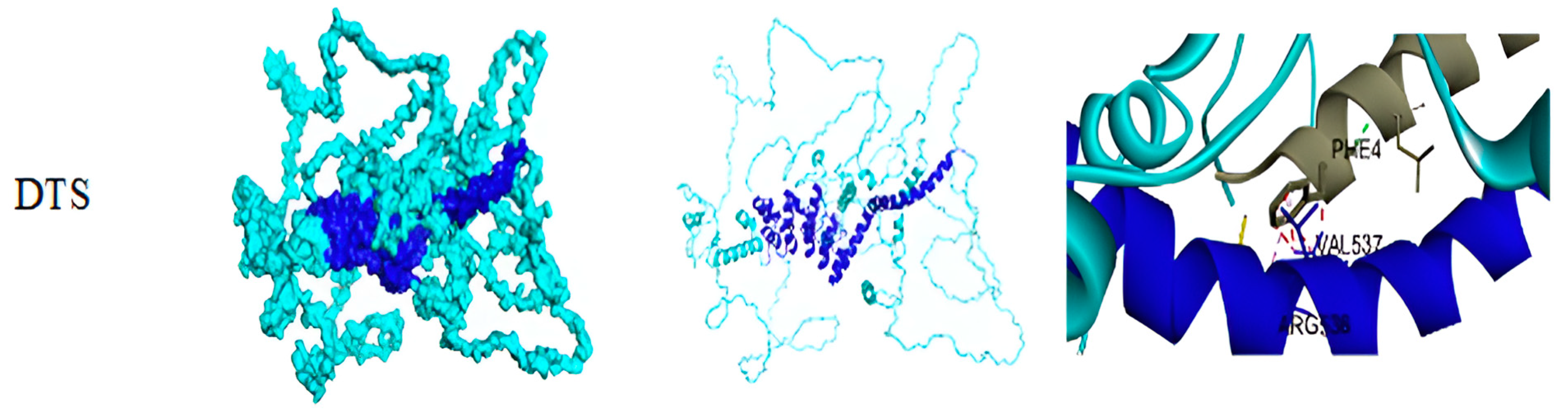
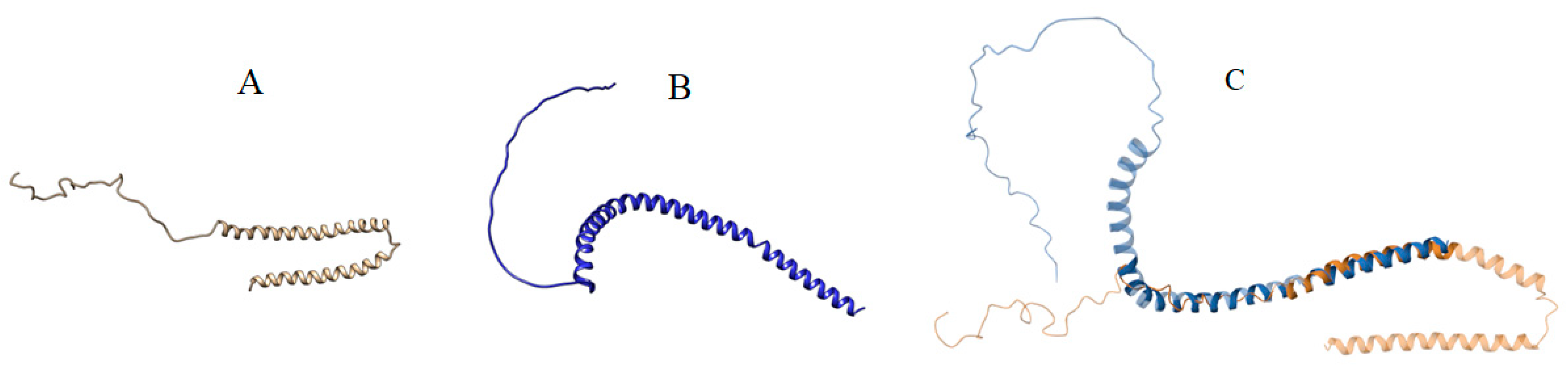
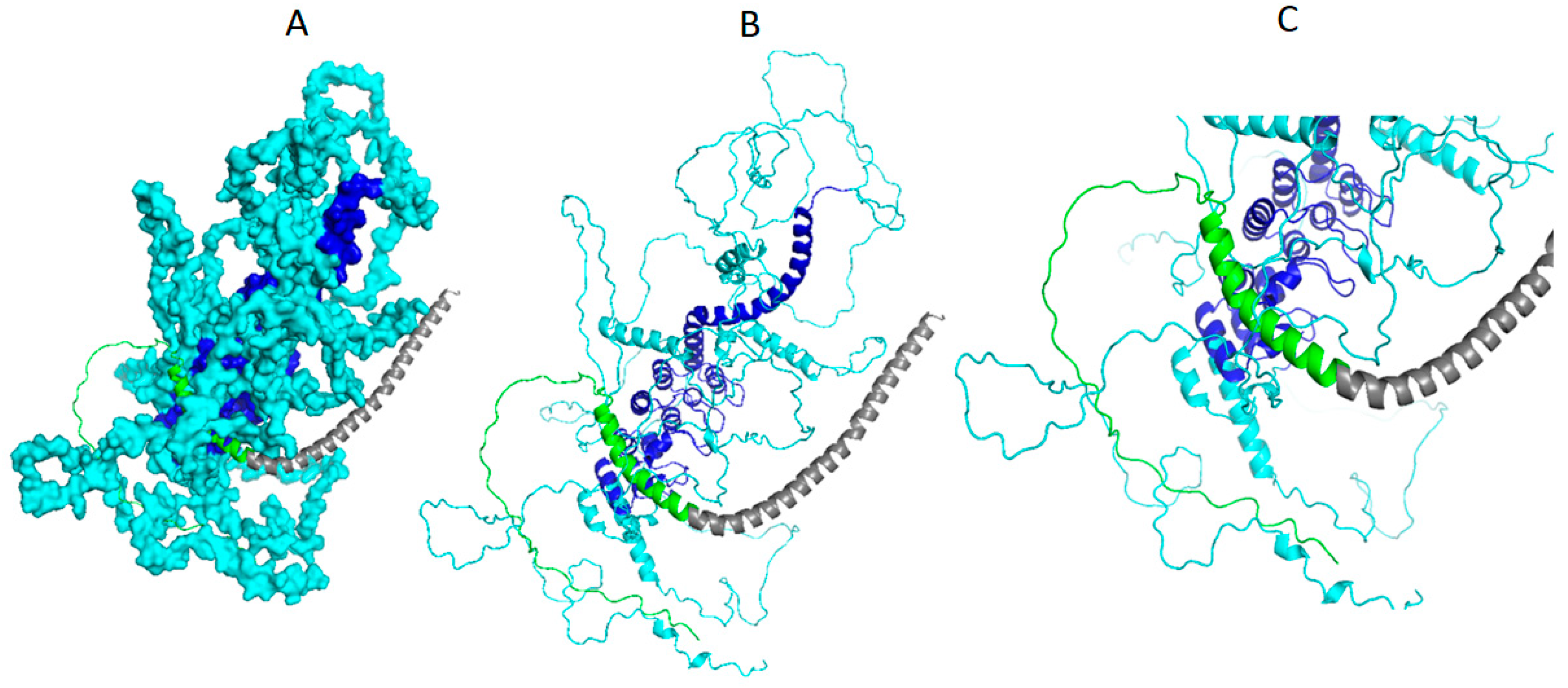
3.3. Result of Structural Details of Interaction Between Best Syn-1 and a-Syn (Both Mutated and Wild Type)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Ed.; Codon Publications: Singapore, 2018. [Google Scholar]
- Xu, L.; Pu, J. Alpha-synuclein in Parkinson’s disease: From pathogenetic dysfunction to potential clinical application. Parkinsons Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Benito, M.; Granado, N.; Garcia-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s disease with the alpha-synuclein protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Tagliafierro, L.; Chiba-Falek, O. Up-regulation of SNCA gene expression: Implications to synucleinopathies. Neurogenetics 2016, 17, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.S.; Carneiro, K.; Ross, C.A.; Menezes, J.R.L.; Engelender, S. Synphilin-1 is developmentally localized to synaptic terminals, and its association with synaptic vesicles is modulated by alpha-synuclein. J. Biol. Chem. 2002, 277, 23927–23933. [Google Scholar] [CrossRef] [PubMed]
- Richfield, E.K.; Thiruchelvam, M.J.; Cory-Slechta, D.A.; Wuertzer, C.; Gainetdinov, R.R.; Caron, M.G.; Di Monte, D.A.; Federoff, H.J. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 2002, 175, 35–48. [Google Scholar] [CrossRef]
- Engelender, S.; Wanner, T.; Kleiderlein, J.J.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Ashworth, R.; Margolis, R.L.; Ross, C.A. Organization of the human Synphilin-1 gene, a candidate for Parkinson’s disease. Mamm. Genome 2000, 11, 763–766. [Google Scholar] [CrossRef]
- Xie, Y.Y.; Zhou, C.J.; Zhou, Z.R.; Hong, J.; Che, M.X.; Fu, Q.S.; Song, A.X.; Lin, D.H.; Hu, H.Y. Interaction with synphilin-1 promotes inclusion formation of α-synuclein: Mechanistic insights and pathological implication. FASEB J. 2010, 24, 196–205. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Bisong, E. Building Machine Learning and Deep. Learning Models on Google Cloud Platform; Apress: Berkeley, CA, USA, 2019; pp. 59–64. [Google Scholar]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Abbas, U.L.; Chen, J.; Shao, Q. Assessing fairness of AlphaFold2 prediction of protein 3D structures. In Proceedings of the 14th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Houston, TX, USA, 3–6 September 2023; ACM: New York, NY, USA, 2023. [Google Scholar]
- MarcuŞtefan-Bogdan, T.; Sabin, T. An Overview of Alphafold’s Breakthrough. Front. Artif. Intell. 2022, 5, 875587. [Google Scholar]
- Meng, Q.; Guo, F.; Tang, J. Improved structure-related prediction for insufficient homologous proteins using MSA enhancement and pre-trained language model. Brief. Bioinform. 2023, 24, bbad217. [Google Scholar] [CrossRef]
- McDonald, E.F.; Jones, T.; Plate, L.; Meiler, J.; Gulsevin, A. Benchmarking AlphaFold2 on peptide structure prediction. Structure 2023, 31, 111–119.e2. [Google Scholar] [CrossRef]
- Lee, C.; Su, B.H.; Tseng, Y.J. Comparative Studies of AlphaFold, RoseTTAFold and Modeller: A Case Study Involving the Use of G-Protein-Coupled Receptors. Brief. Bioinform. 2022, 23, bbac308. [Google Scholar] [CrossRef]
- Krieger, E.; Nabuurs, S.D.; Vriend, G. Structural Bioinformatics; Bourne, P.E., Weissig, H., Eds.; Wiley-Liss: Hoboken, NJ, USA, 2012; pp. 507–520. [Google Scholar]
- Thompson, T.B.; Chaggar, P.; Kuhl, E.; Goriely, A. Alain Goriely, and Alzheimer’s Disease Neuroimaging Initiative. Protein-protein interactions in neurodegenerative diseases: A conspiracy theory. PLoS Comput. Biol. 2020, 16, e1008267. [Google Scholar] [CrossRef]
- Poluri, K.M.; Gulati, K.; Tripathi, D.K.; Nagar, N. Protein–protein interactions in neurodegenerative diseases. In Protein-Protein Interactions; Springer Nature: Singapore, 2023; pp. 101–169. [Google Scholar]
- Balint, D.; Brito, I.L. Human–gut bacterial protein–protein interactions: Understudied but impactful to human health. Trends Microbiol. 2024, 32, 325–332. [Google Scholar] [CrossRef]
- Mondal, R.; Mandal, M.; Lahiri, T. A divide and conquer approach (DACA) to predict high fidelity structure of large multidomain protein BRWD1. bioRxiv 2023. [Google Scholar] [CrossRef]
- Charan, J.; Biswas, T. How to calculate sample size for different study designs in medical research? Indian J. Psychol. Med. 2013, 35, 121–126. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 4459709, D-hexose 6-phosphate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/4459709 (accessed on 26 September 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 135509118. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/135509118 (accessed on 26 September 2024).
- Resch, M.; Dobbek, H.; Meyer, O. Structural and functional reconstruction in situ of the [CuSMoO2] active site of carbon monoxide dehydrogenase from the carbon monoxide oxidizing eubacterium Oligotrophacarboxidovorans. JBIC J. Biol. Inorg. Chem. 2005, 10, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, C.; Nielsen, D.J.; Evans, D.J.; White, J.M.; Young, C.G. Young, Models for aerobic carbon monoxide dehydrogenase: Synthesis, characterization and reactivity of paramagnetic MoVO(μ-S)CuI complexes. Chem. Sci. 2018, 9, 876–888. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5288690, 5’-O-(L-Leucylsulfamoyl)adenosine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5_-O-_L-Leucylsulfamoyl_adenosine (accessed on 26 September 2024).
- Kim, S.; Yoon, I.; Son, J.; Park, J.; Kim, K.; Lee, J.H.; Park, S.Y.; Kang, B.S.; Han, J.M.; Hwang, K.Y.; et al. Leucine-sensing mechanism of leucyl-tRNA synthetase 1 for mTORC1 activation. Cell Rep. 2021, 35, 109031. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell. 2000, 6, 909–919. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 312145, Wortmannin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Wortmannin (accessed on 26 September 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 24139, N-ACETYL-beta-D-GLUCOSAMINE. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/N-ACETYL-beta-D-GLUCOSAMINE (accessed on 26 September 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 164513459. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/164513459 (accessed on 26 September 2024).
- Winkelmann, D.A.; Forgacs, E.; Miller, M.T.; Stock, A.M. Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat. Commun. 2015, 6, 7974. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 11689883, Omecamtivmecarbil. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/11689883 (accessed on 26 September 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 65482, Sinefungin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Sinefungin (accessed on 26 September 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 86208110, 3-Amino-N-{(1r)-2-(Hydroxyamino)-2-Oxo-1-[4-(1h-Pyrazol-1-Yl)phenyl]ethyl}benzamide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/86208110 (accessed on 26 September 2024).
- Mistry, S.N.; Drinkwater, N.; Ruggeri, C.; Sivaraman, K.K.; Loganathan, S.; Fletcher, S.; Drag, M.; Paiardini, A.; Avery, V.M.; Scammells, P.J.; et al. Two-pronged attack: Dual inhibition of Plasmodium falciparum M1 and M17 metalloaminopeptidases by a novel series of hydroxamic acid-based inhibitors. J. Med. Chem. 2014, 57, 9168–9183. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 91532, Phosphomethylphosphonic Acid Adenylate Ester. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Phosphomethylphosphonic-acid-adenylate-ester (accessed on 26 September 2024).
- Inoue, M.; Sakuta, N.; Watanabe, S.; Zhang, Y.; Yoshikaie, K.; Tanaka, Y.; Ushioda, R.; Kato, Y.; Takagi, J.; Tsukazaki, T.; et al. Structural Basis of Sarco/Endoplasmic Reticulum Ca2+-ATPase 2b Regulation via Transmembrane Helix Interplay. Cell Rep. 2019, 27, 1221–1230. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 8117, Diethylene Glycol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Diethylene-glycol (accessed on 26 September 2024).
- Jiang, K.; Zhang, Y.; Chen, Z.; Wu, D.; Cai, J.; Gao, X. Structural and Functional Insights into the C-terminal Fragment of Insecticidal Vip3A Toxin of Bacillus thuringiensis. Toxins 2020, 12, 438. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 155804422. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/155804422 (accessed on 26 September 2024).
- Gao, Y.; Hu, Y.; Liu, Q.; Li, X.; Li, X.; Kim, C.Y.; James, T.D.; Li, J.; Chen, X.; Guo, Y. Two-Dimensional Design Strategy to Construct Smart Fluorescent Probes for the Precise Tracking of Senescence. Angew. Chem. Int. Ed. Engl. 2021, 60, 10756–10765. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6022, Adenosine-5′-diphosphate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Adenosine-5_-diphosphate (accessed on 26 September 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 644100, 2-(4-((2,5-Difluorobenzyl)oxy)phenoxy)-5-ethoxyaniline. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/sea0400 (accessed on 26 September 2024).
- Xue, Z.; Xu, D.; Wang, Y.; Zhang, Y. ThreaDom: Extracting protein domain boundary information from multiple threading alignments. Bioinformatics 2013, 29, 247–256. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, D.; Xu, D. DeepDom: Predicting protein domain boundary from sequence alone using stacked bidirectional LSTM. Pac. Symp. Biocomput. 2019, 24, 66–75. [Google Scholar] [PubMed]
- Rost, B.; Schneider, R.; Sander, C. Protein fold recognition by prediction-based threading. J. Mol. Biol. 1997, 270, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Pal, M.K.; Lahiri, T.; Tanwar, G.; Kumar, R. An improved protein structure evaluation using a semi-empirically derived structure property. BMC Struct. Biol. 2018, 18, 16. [Google Scholar] [CrossRef]
- Tripathi, A.; Mondal, R.; Lahiri, T.; Chaurasiya, D.; Pal, M.K. TemPred: A novel protein template search engine to improve protein structure prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 2112–2121. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017.0: Maestro; Schrödinger LLC: New York, NY, USA, 2017.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes, Discovery Studio, 4.5; Dassault Systèmes: San Diego, CA, USA, 2021.
- Kawamata, H.; McLean, P.J.; Sharma, N.; Hyman, B.T. Interaction of alpha-synuclein and synphilin-1: Effect of Parkinson’s disease-associated mutations. J. Neurochem. 2001, 77, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Camargos, S.T.; Dornas, L.O.; Momeni, P.; Lees, A.; Hardy, J.; Singleton, A.; Cardoso, F. Familial Parkinsonism and early onset Parkinson’s disease in a Brazilian movement disorders clinic: Phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov. Disord. 2009, 24, 662–666. [Google Scholar] [CrossRef]
- Hattori, N.; Kitada, T.; Matsumine, H.; Asakawa, S.; Yamamura, Y.; Yoshino, H.; Kobayashi, T.; Yokochi, M.; Wang, M.; Yoritaka, A.; et al. Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: Evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann. Neurol. 1998, 44, 935–941. [Google Scholar] [CrossRef]
- Bras, J.; Guerreiro, R.; Ribeiro, M.; Morgadinho, A.; Januario, C.; Dias, M.; Calado, A.; Semedo, C.; Oliveira, C.; Hardy, J.; et al. Analysis of Parkinson disease patients from Portugal for mutations in SNCA, PRKN, PINK1 and LRRK2. BMC Neurol. 2008, 8, 1. [Google Scholar] [CrossRef]
- Golbe, L.I.; Di Iorio, G.; Bonavita, V.; Miller, D.C.; Duvoisin, R.C. A large kindred with autosomal dominant Parkinson’s disease. Ann. Neurol. 1990, 27, 276–282. [Google Scholar] [CrossRef]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef]
- Oczkowska, A.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Mutations in PRKN and SNCA Genes Important for the Progress of Parkinson’s Disease. Curr. Genom. 2014, 14, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, U.; Chakrabarti, S.S.; Kaur, U.; Mukherjee, A.; Chakrabarti, S. Alpha-synuclein, proteotoxicity and Parkinson’s disease: Search for neuroprotective therapy. Curr. Neuropharmacol. 2018, 16, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mondal, R.; Lahiri, T.; Pal, M.K. Application of sequence semantic and integrated cellular geography approach to study alternative biogenesis of exonic circular RNA. BMC Bioinform. 2023, 24, 148. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef]
- Qin, K.; Dong, C.; Wu, G.; Lambert, N.A. Inactive-state preassembly of G(q)-coupled receptors and G(q) heterotrimers. Nat. Chem. Biol. 2011, 7, 740–747. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins (PDB ID) | Ligands (Chemical ID) |
|---|---|
| Mutant monomer of recombinant human hexokinase Type I complexed with Glucose, Glucose-6-Phosphate, and ADP (1CZA) | alpha-D-glucose 6-phosphate(G6P) [26] |
| Crystal structure of the aldehyde oxidoreductase from desulfovibriodesulfuricansatcc 27774(1DGJ) | Pterin cytosine dinucleotide(MCN) [27,28,29] |
| Crystal structure of human leucyl-tRNA synthetase, Leu-AMS-bound form(6KIE) | 5′-O-(L-leucylsulfamoyl)adenosine(LSS) [30,31] |
| Structure determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin and staurosporine(1E7U) | Wortmannin (KWT) [32,33] |
| X-ray crystal structure of human ceruloplasmin at 3.0 angstroms(1KCW) | 2-acetamido-2-deoxy-beta-D-glucopyranose(NAG) [34] |
| Crystal structure of endoplasmic reticulum aminopeptidase 2 (erap2) complex with a highly selective and potent small molecule(7SH0) | (2S)-N-hydroxy-3-(4-methoxyphenyl)-2-[4-({[5-(pyridin-2-yl)thiophene-2-sulfonyl]amino}methyl)-1H-1,2,3-triazol-1-yl]propenamide (GIY) [35] |
| OmecamtivMercarbil binding site on the Human Beta-Cardiac Myosin Motor Domain(4PA0) | methyl 4-(2-fluoro-3-{[(6-methylpyridin-3-yl)carbamoyl]amino}benzyl)piperazine-1-carboxylate(2OW) [36,37] |
| Structure of human DNMT1 (601-1600) in complex with Sinefungin(3SWR) | SINEFUNGIN(SFG) [38] |
| Structure of the m1 alanylaminopeptidase from malaria complexed with a hydroxamic acid-based inhibitor(4R5X) | 3-amino-N-{(1R)-2-(hydroxyamino)-2-oxo-1-[4-(1H-pyrazol-1-yl)phenyl]ethyl}benzamide(R5X)[39,40] |
| Structure of Ca2+ ATPase(5ZTF) | Phosphomethylphosphonic acid adenylate ester(ACP) [41,42] |
| Structure of C-terminal fragment of Vip3A toxin(6VLS) | DI(HYDROXYETHYL)ETHER(PEG) [43,44] |
| E.coli beta-galactosidase (E537Q) in complex with fluorescent probe KSA02(7BRS) | 8-[2-[(E)-2-[4-[(2S,3R,4S,5R,6R)-6-(hydroxymethyl)-3,4,5-tris(oxidanyl)oxan-2-yl]oxyphenyl]ethenyl]-3,3-dimethyl-indol-1-ium-1-yl]octanoic acid(F4X) [45,46] |
| Crystal structure of human MTR4(6IEG) | ADENOSINE-5′-DIPHOSPHATE(ADP) [47] |
| Structure of human sodium-calcium exchanger NCX1(8JP0) | 2-{4-[(2,5-difluorophenyl)methoxy]phenoxy}-5-ethoxyaniline(EKY) [48] |
| Standard Structure Validation Metrics | Tie | ||
|---|---|---|---|
| Ramachandran Score | 100 | 0 | 0 |
| G-factor | 93 | 7 | 0 |
| GDT–TS | 7 | 36 | 57 |
| PAL_RMSD | 43 | 28 | 29 |
| QMEAN | 50 | 43 | 7 |
| SRI | 43 | 43 | 14 |
| ERRAT | 7 | 93 | 0 |
| TM-Score | 7 | 43 | 50 |
| Proteins | Ligands | Interactions Details | ||||
|---|---|---|---|---|---|---|
| Proximity of Binding Site of Experimental Structure in Å with That of | Binding Energy in kJ/mol | |||||
| DAA | DTS | BIExperimental | BIDAA | BIDTS | ||
| 1CZA | G6P | 33.3 | 27.8 | −25.9408 | −28.0328 | −27.6144 |
| 1DGJ | MCN | 29.2 | 28.6 | −59.8312 | −10.8784 | −24.2672 |
| 6KIE | LSS | 13.0 | 9.0 | −19.2464 | −23.4304 | −12.9704 |
| 1E7U | KWT | 28.9 | 24.3 | −23.4304 | −15.4808 | −15.4808 |
| 1KCW | NAG | 11.6 | 16.5 | −17.9912 | −22.1752 | −22.5936 |
| 7SH0 | GIY | 11.8 | 6.2 | −33.0536 | −27.196 | −26.7776 |
| 4PA0 | 2OW | 11.9 | 15.9 | −30.1248 | −23.4304 | −25.9408 |
| 3SWR | SFG | 11.2 | 18.5 | −40.1664 | −22.1752 | −18.4096 |
| 4R5X | R5X | 22.9 | 36.5 | −22.5936 | −17.1544 | −16.3176 |
| 5ZTF | ACP | 11.6 | 7.7 | −30.5432 | −17.5728 | −26.7776 |
| 6VLS | PEG | 31.3 | 10.1 | −1.6736 | −0.4184 | −6.6944 |
| 7BRS | F4X | 21.4 | 4.7 | −22.5936 | −25.5224 | −20.0832 |
| 6IEG | ADP | 13.9 | 22.6 | −21.3384 | −23.8488 | −15.0624 |
| 8JP0 | EKY | 16.8 | 10.5 | −32.6352 | −22.5936 | −10.46 |
| Models | Ramachandran Score in % for Different Regions | G-Factor | QMEAN Score | % Coil | ERRAT | |||
|---|---|---|---|---|---|---|---|---|
| Core | Allowed | Generously Allowed | Disallowed | |||||
| DTS | 82.1 | 14.9 | 2.1 | 0.9 | −0.15 | −5.97 | 65.6 | 72.48 |
| DAA | 32.2 | 17 | 18.3 | 32.5 | −2.55 | −21.82 | 72.7 | 60.27 |
| Structure Model of a-Syn | Structure Models of Syn-1 | BI in kJ/mol |
|---|---|---|
| Wild type (PDB ID: 1XQ8) | DTS | −58.576 |
| DAA | −56.0656 |
| Structure Model of Syn-1 | Structure Models of a-Syn | BI in kJ/mol | PDB–RMSD (R) in Å with 11% Identity |
|---|---|---|---|
| DTS | Wild type (PDB ID: 1XQ8) | −58.576 | 3.35 |
| Mutated a-Syn | −110.0392 |
| Models | Ramachandran Score in % for Different Regions | G-Factor | QMEAN Score | % Coil | ERRAT | |||
|---|---|---|---|---|---|---|---|---|
| Core | Allowed | Generously Allowed | Disallowed | |||||
| Wild Type (PDB ID: 1XQ8) | 77.4 | 13 | 7.8 | 1.7 | −0.05 | −5.59 | 25 | 73.01 |
| Mutated a-Syn | 67.5 | 14.9 | 11.4 | 6.1 | −1.95 | −11.25 | 34.2 | 0 |
| Interaction of Syn-1 with a-Syn (Wild Type) | Interaction of Syn-1 with a-syn (Mutated) |
|---|---|
| A: LEU547—B: LEU8 A: LYS551—B: MET1 A: LYS626—B: VAL3 A: ILE619—B: VAL3 A: LEU623—B: VAL3 A: VAL537—B: LYS12 A: VAL816—B: ALA18 A: ARG536—B: PHE4 A: GLU627—B: MET1 A: GLU630—B: MET1 | A:PRO699—C: ALA11 A: ARG700—C: ALA11 A: ALA696—C: ALA18 A: ARG899—C: LYS58 A: PRO907—C: GLY73 A: GLY52—C: VAL118 A: ARG700—C: GLY7 A: ARG899—C: THR54 A: THR901—C: VAL55 A: SER19—C: TYR133 A: ARG34—C: TYR125 A: ARG34—C: GLU126 A: CYS51—C: ASN122 A: TYR158—C: THR81 A: LYS160—C: LYS80 A: SER200—C:ASP98 A: ARG700—C: GLY7 A: ARG899—C: THR54 A: THR901—C: VAL55 A: SER21—C: GLU130 A: ARG33—C: TYR125 A: TRP49—C: ALA124 A: GLN307—C: TYR133 A: SER912—C: THR75 A: GLN164—C: THR92 A: LEU165—C: THR92 A: GLN164—C: GLY93 A: ASP36—C: SER129 A: SER21—C: GLN134 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, A.; Mondal, R.; Mandal, M.; Lahiri, T.; Pal, M.K. A Structural Proteomics Exploration of Synphilin-1 and Alpha-Synuclein Interaction in Pathogenesis of Parkinson’s Disease. Biomolecules 2024, 14, 1588. https://doi.org/10.3390/biom14121588
Tripathi A, Mondal R, Mandal M, Lahiri T, Pal MK. A Structural Proteomics Exploration of Synphilin-1 and Alpha-Synuclein Interaction in Pathogenesis of Parkinson’s Disease. Biomolecules. 2024; 14(12):1588. https://doi.org/10.3390/biom14121588
Chicago/Turabian StyleTripathi, Asmita, Rajkrishna Mondal, Malay Mandal, Tapobrata Lahiri, and Manoj Kumar Pal. 2024. "A Structural Proteomics Exploration of Synphilin-1 and Alpha-Synuclein Interaction in Pathogenesis of Parkinson’s Disease" Biomolecules 14, no. 12: 1588. https://doi.org/10.3390/biom14121588
APA StyleTripathi, A., Mondal, R., Mandal, M., Lahiri, T., & Pal, M. K. (2024). A Structural Proteomics Exploration of Synphilin-1 and Alpha-Synuclein Interaction in Pathogenesis of Parkinson’s Disease. Biomolecules, 14(12), 1588. https://doi.org/10.3390/biom14121588