
RIPK4 Downregulation Reduces ABCG2 Expression, Increasing BRAF-Mutated Melanoma Cell Susceptibility to Cisplatin- and Doxorubicin-Induced Apoptosis
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Transfection with Small Interfering RNA (siRNA)
2.3. Overexpression of RIPK4
2.4. MTT
2.5. Apoptosis Assays
2.6. Quantitative Real-Time PCR
2.7. Western Blot
2.8. Statistical Analysis
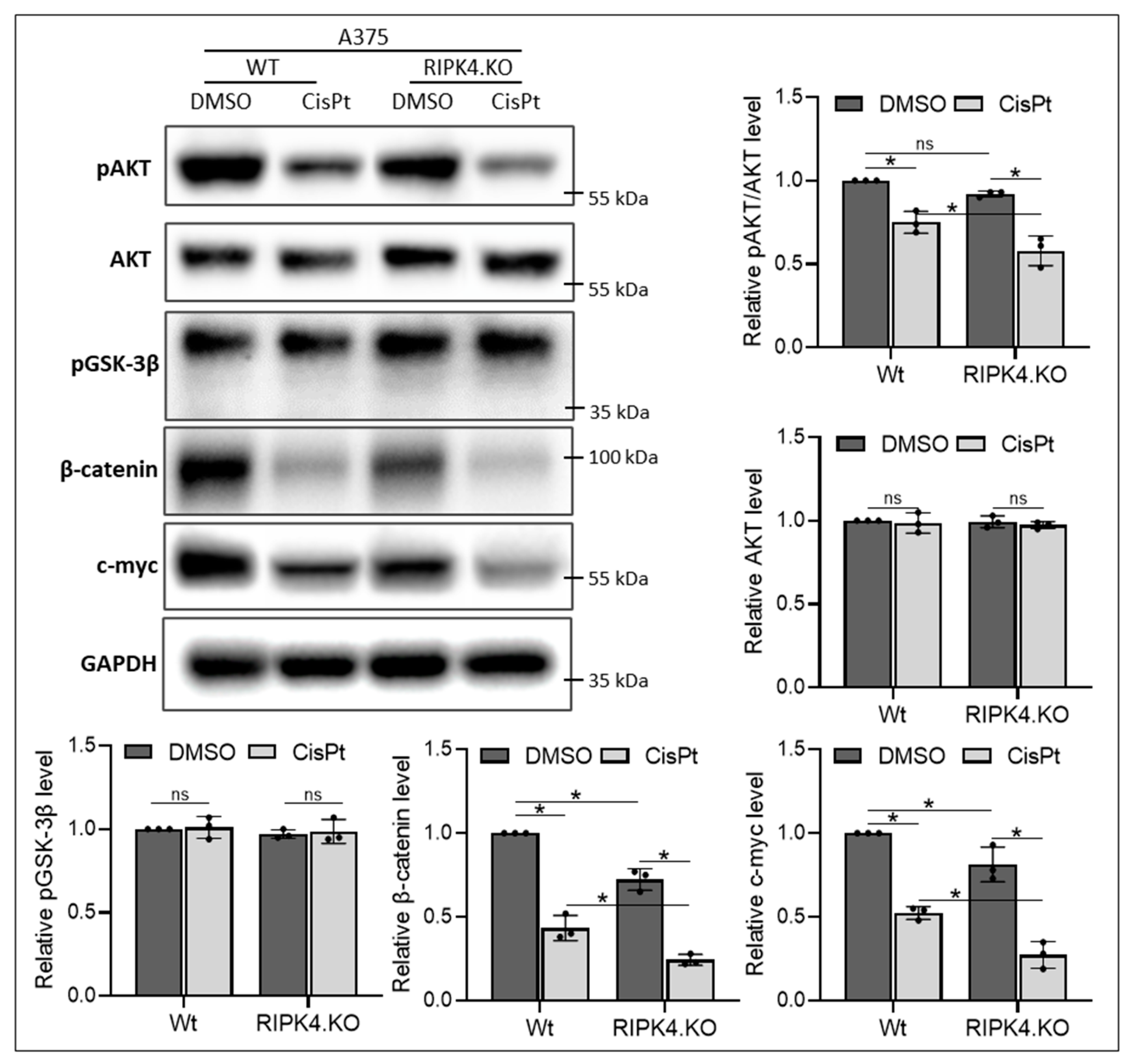
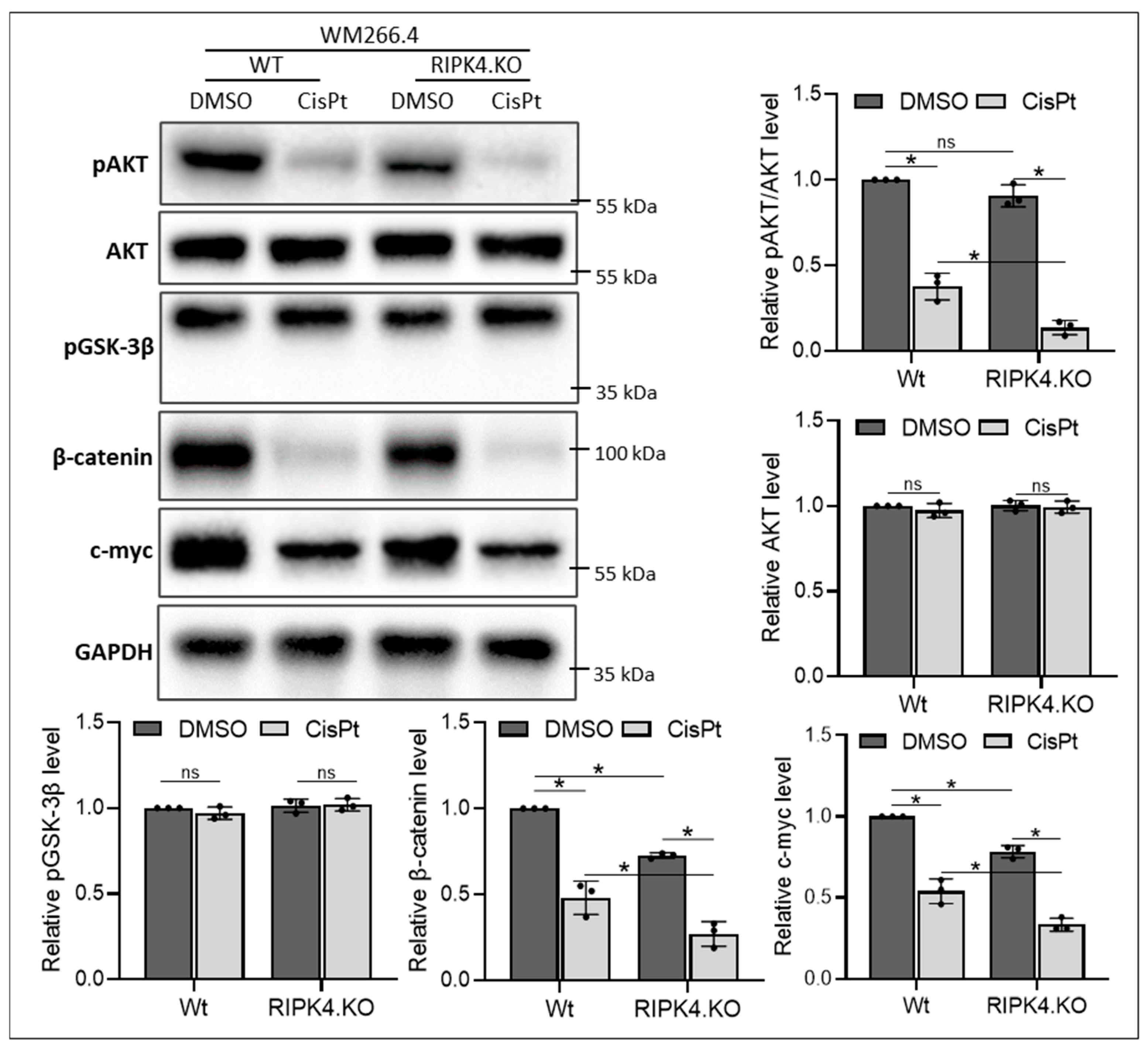
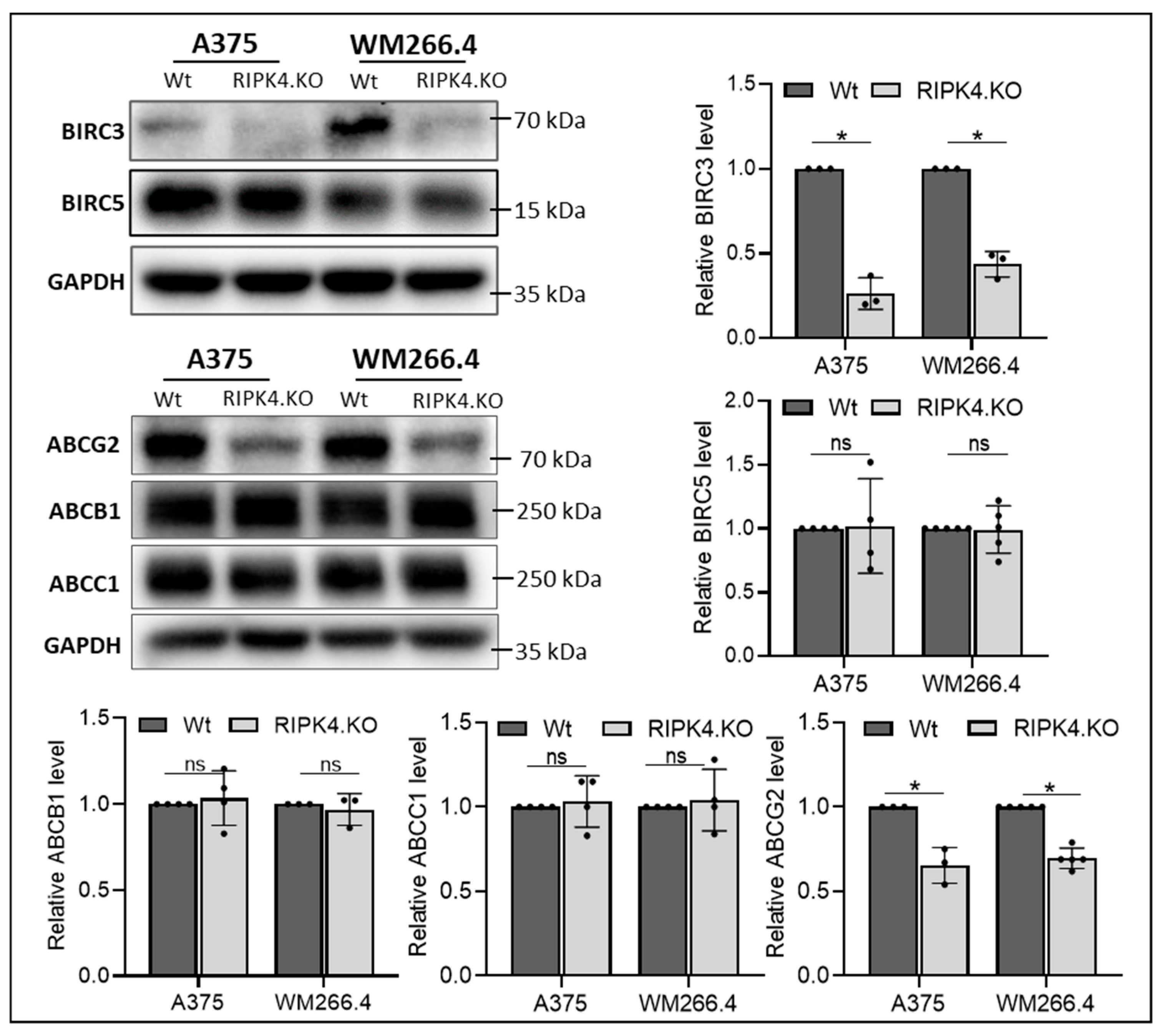
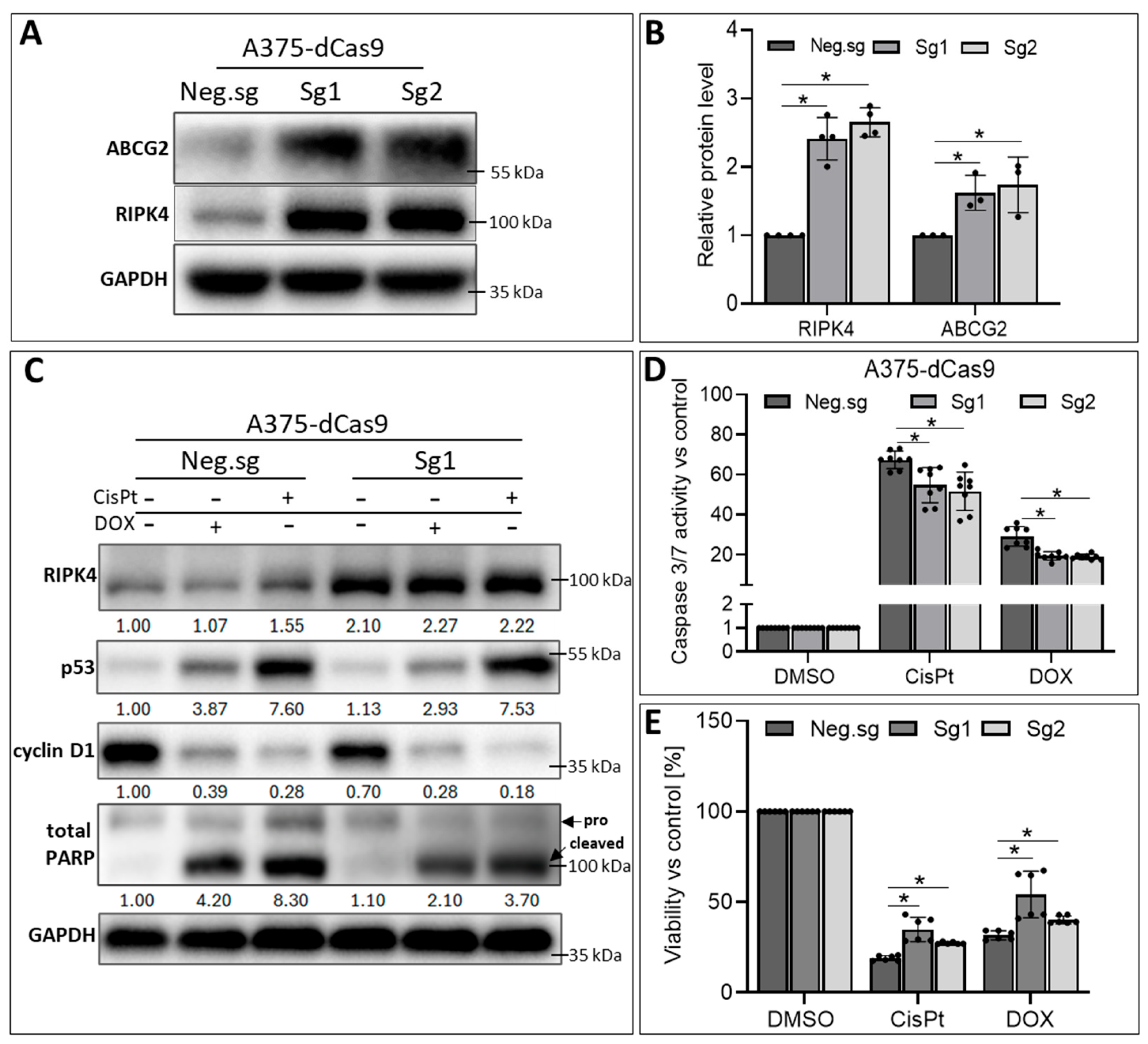
3. Results
3.1. RIPK4 Downregulation Enhances Sensitivity of Melanoma Cells Towards CisPt Treatment
3.2. RIPK4 Knockout Regulates the Levels of Apoptosis-Related Genes and the ABCG2 Protein in Melanoma Cells
3.3. Downregulation of RIPK4 Enhanced the Sensitivity of Melanoma Cells to CisPt-Induced Apoptosis, Similar to Cyclosporin A
3.4. RIPK4 Downregulation as Well as Cyclosporin A Treatment Enhanced Sensitivity of Melanoma Cells to Doxorubicin
3.5. RIPK4 Overexpression in A375-dCas9 Cells Reduces the Action of CisPt and DOX
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AnxV | Annexin V |
| ABCB1 | ATP-binding cassette super-family B member 1 (P-glycoprotein) |
| ABCC1 | ATP-binding cassette super-family C member 1 (MRP1) |
| ABCG2 | ATP-binding cassette super-family G member 2 (BCRP) |
| BIRC3 | Baculoviral IAP repeat containing 3 (c-IAP2) |
| BIRC5 | Baculoviral IAP repeat containing 5 |
| CisPt | Cisplatin |
| CsA | Cyclosporine A |
| DOX | Doxorubicin |
| IAP | Inhibitors of apoptosis proteins |
| MCL1 | Myeloid cell leukemia-1 |
| MPR | Multidrug resistance proteins |
| MT1X | Metallothionein 1X |
| 7AAD | 7-amino-actinomycin D |
| PARP | Poly (ADP-ribose) polymerase |
| RIPK4 | Receptor-interacting protein kinase |
References
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef] [PubMed]
- Hambley, T.W. Platinum binding to DNA: Structural controls and consequences. J. Chem. Soc. Dalton Trans. 2001, 2711–2718. [Google Scholar] [CrossRef]
- Siafaca, K. Oncology trends products markets–Part 1. Future Oncol. 1999, 1045–1071. [Google Scholar]
- Galanski, M.S.; Jakupec, M.A.; Keppler, B.K. Update of the Preclinical Situation of Anticancer Platinum Complexes: Novel Design Strategies and Innovative Analytical Approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef]
- Ho, G.Y.; Woodward, N.; Coward, J.I. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Adebali, O.; Wu, G.; Selby, C.P.; Chiou, Y.-Y.; Rashid, N.; Hu, J.; Hogenesch, J.B.; Sancar, A. Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc. Natl. Acad. Sci. USA 2018, 115, E4777–E4785. [Google Scholar] [CrossRef]
- Achkar, I.W.; Abdulrahman, N.; Al-Sulaiti, H.; Joseph, J.M.; Uddin, S.; Mraiche, F. Cisplatin based therapy: The role of the mitogen activated protein kinase signaling pathway. J. Transl. Med. 2018, 16, 96. [Google Scholar] [CrossRef]
- Bai, Y.; Aodeng, G.; Ga, L.; Hai, W.; Ai, J. Research Progress of Metal Anticancer Drugs. Pharmaceutics 2023, 15, 2750. [Google Scholar] [CrossRef]
- Casini, A.; Sun, R.W.Y.; Ott, I. Cisplatin and Oxaliplatin: Our Current Understanding of Their Actions. Met. Ions. Life Sci. 2018, 18, 199–217. [Google Scholar] [CrossRef]
- Kelland, L.R. Preclinical Perspectives on Platinum Resistance. Drugs 2000, 59 (Suppl. S4), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Skowron, M.A.; Melnikova, M.; Van Roermund, J.G.H.; Romano, A.; Albers, P.; Thomale, J.; Schulz, W.A.; Niegisch, G.; Hoffmann, M.J. Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines. Int. J. Mol. Sci. 2018, 19, 590. [Google Scholar] [CrossRef]
- Pérez, J.E.; Fritzell, S.; Kopecky, J.; Visse, E.; Darabi, A.; Siesjö, P. The effect of locally delivered cisplatin is dependent on an intact immune function in an experimental glioma model. Sci. Rep. 2019, 9, 5632. [Google Scholar] [CrossRef]
- Galsky, M.D.; Guan, X.; Rishipathak, D.; Rapaport, A.S.; Shehata, H.M.; Banchereau, R.; Yuen, K.; Varfolomeev, E.; Hu, R.; Han, C.-J.; et al. Immunomodulatory effects and improved outcomes with cisplatin- versus carboplatin-based chemotherapy plus atezolizumab in urothelial cancer. Cell Rep. Med. 2024, 5, 101393. [Google Scholar] [CrossRef] [PubMed]
- Boudewijns, S.; Bloemendal, M.; de Haas, N.; Westdorp, H.; Bol, K.F.; Schreibelt, G.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Gorris, M.A.J.; Croockewit, A.; et al. Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: A prospective, randomized phase 2 trial. Cancer Immunol. Immunother. 2020, 69, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, J.; Kong, Y.; Bai, Q.; Qi, P.; Zhang, L.; Wang, Q.; Zhou, X.; Chen, Y.; Zhu, X. BRAF, C-KIT, and NRAS mutations correlated with different clinicopathological features: An analysis of 691 melanoma patients from a single center. Ann. Transl. Med. 2022, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, L.D.; Gamboa, M.; Nova, J.A.; Pulido, L.; Tovar-Parra, J.D. Mutations in the BRAF, NRAS, and C-KIT Genes of Patients Diagnosed with Melanoma in Colombia Population. BioMed Res. Int. 2020, 2020, 2046947. [Google Scholar] [CrossRef]
- Geiger, C.E.; Mrabet-Dahbi, S.; Berger, I. The BRAF and NRAS status among distinct metastases of malignant melanoma differ significantly independent of tissue origin and temporal occurrence. Possible effect on clinical relevance? Melanoma Res. 2024, 34, 85–87. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, X.; Wang, Z.; Chen, X.; Wang, L.; Lu, Y.; Xiong, D.; Liu, Q.; Tian, Y.; Lin, H.; et al. The primordial differentiation of tumor-specific memory CD8+ T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 2022, 185, 4049–4066.e25. [Google Scholar] [CrossRef] [PubMed]
- Cuny, T.; Buchfelder, M.; Dufour, H.; Grossman, A.; Gatta-Cherifi, B.; Jouanneau, E.; Raverot, G.; Vasiljevic, A.; Castinetti, F. The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal? Cancers 2022, 14, 3831. [Google Scholar] [CrossRef]
- Madej, E.; Ryszawy, D.; Brożyna, A.A.; Czyz, M.; Czyz, J.; Wolnicka-glubisz, A. Deciphering the functional role of ripk4 in mel-anoma. Int. J. Mol. Sci. 2021, 22, 11504. [Google Scholar] [CrossRef]
- Wronski, N.; Madej, E.; Grabacka, M.; Brożyna, A.A.; Wolnicka-Glubisz, A. RIPK4 downregulation impairs Wnt3A-stimulated invasiveness via Wnt/β-catenin signaling in melanoma cells and tumor growth in vivo. Cell. Signal. 2024, 113, 110938. [Google Scholar] [CrossRef]
- Urwyler-Rösselet, C.; Tanghe, G.; Devos, M.; Hulpiau, P.; Saeys, Y.; Declercq, W. Functions of the RIP kinase family members in the skin. Cell. Mol. Life Sci. 2023, 80, 285. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.-H.; Xu, H.-X.; Zhang, S.-R.; Xu, J.-Z.; Li, S.; Gao, H.-L.; Jin, W.; Wang, W.-Q.; Wu, C.-T.; Ni, Q.-X.; et al. RIPK4/PEBP1 axis promotes pancreatic cancer cell migration and invasion by activating RAF1/MEK/ERK signaling. Int. J. Oncol. 2018, 52, 1105–1116. [Google Scholar] [CrossRef]
- Yi, Z.; Pu, Y.; Gou, R.; Chen, Y.; Ren, X.; Liu, W.; Dong, P. Silencing of RIPK4 inhibits epithelial-mesenchymal transition by inactivating the Wnt/β-catenin signaling pathway in osteosarcoma. Mol. Med. Rep. 2020, 21, 1154–1162. [Google Scholar] [CrossRef]
- Heim, D.; Cornils, K.; Schulze, K.; Fehse, B.; Lohse, A.W.; Brümmendorf, T.H.; Wege, H. Retroviral insertional mutagenesis in telomerase-immortalized hepatocytes identifies RIPK4 as novel tumor suppressor in human hepatocarcinogenesis. Oncogene 2015, 34, 385–393. [Google Scholar] [CrossRef]
- Madej, E.; Brożyna, A.A.; Adamczyk, A.; Wronski, N.; Harazin-Lechowska, A.; Muzyk, A.; Makuch, K.; Markiewicz, M.; Rys, J.; Wolnicka-Glubisz, A. Vemurafenib and Dabrafenib Downregulates RIPK4 Level. Cancers 2023, 15, 918. [Google Scholar] [CrossRef]
- Madej, E.; Lisek, A.; Brożyna, A.A.; Cierniak, A.; Wronski, N.; Deptula, M.; Wardowska, A.; Wolnicka-Glubisz, A. The involvement of RIPK4 in TNF-α-stimulated IL-6 and IL-8 production by melanoma cells. J. Cancer Res. Clin. Oncol. 2024, 150, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, W.; Zhou, Y.; Xu, W.; Wang, H. RIPK4 is downregulated in poorly differentiated tongue cancer and is associated with migration/invasion and cisplatin-induced apoptosis. Int. J. Biol. Markers 2014, 29, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhao, S.; Zhang, M.; Xing, J.; Zhou, J.; Gao, W.; Chen, L.; Zhang, Y.; Lin, L.; Lu, M.; et al. m6A-modified RIPK4 facilitates proliferation and cisplatin resistance in epithelial ovarian cancer. Gynecol. Oncol. 2024, 180, 99–110. [Google Scholar] [CrossRef]
- Szafraniec, M.J.; Szczygieł, M.; Urbanska, K.; Fiedor, L. Determinants of the activity and substrate recognition of breast cancer resistance protein (ABCG2). Drug Metab. Rev. 2014, 46, 459–474. [Google Scholar] [CrossRef]
- Skalniak, L.; Smejda, M.; Cierniak, A.; Adamczyk, A.; Konieczny, P.; Madej, E.; Wolnicka-Glubisz, A. p38 but not p53 is responsible for UVA-induced MCPIP1 expression. Mech. Ageing Dev. 2018, 172, 96–106. [Google Scholar] [CrossRef]
- Nomura, T.; Yamasaki, M.; Nomura, Y.; Mimata, H. Expression of the inhibitors of apoptosis proteins in cisplatin-resistant prostate cancer cells. Oncol. Rep. 2005, 14, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Petznek, H.; Kleiter, M.; Tichy, A.; Fuchs-Baumgartinger, A.; Hohenadl, C. Murine xenograft model demonstrates significant radio-sensitising effect of liposomal doxorubicin in a combination therapy for Feline Injection Site Sarcoma. Res. Vet. Sci. 2014, 97, 386–390. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Straughn, J.M.; Leath, C.A.; Kilgore, L.C.; Huh, W.K.; Barnes, M.N.; Partridge, E.E.; Alvarez, R.D. Pegylated liposomal doxorubicin consolidation therapy after platinum/paclitaxel-based chemotherapy for suboptimally debulked, advanced-stage epithelial ovarian cancer patients. Oncologist 2006, 11, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Szwed, M.; Laroche-Clary, A.; Robert, J.; Jozwiak, Z. Induction of apoptosis by doxorubicin–transferrin conjugate compared to free doxorubicin in the human leukemia cell lines. Chem. Interactions 2014, 220, 140–148. [Google Scholar] [CrossRef]
- Tiek, D.; Cheng, S.-Y. DNA damage and metabolic mechanisms of cancer drug resistance. Cancer Drug Resist. 2022, 5, 368–379. [Google Scholar] [CrossRef]
- Tasu, J.-P.; Tougeron, D.; Rols, M.-P. Irreversible electroporation and electrochemotherapy in oncology: State of the art. Diagn. Interv. Imaging 2022, 103, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Mentucci, F.M.; Nuñez, E.A.R.; Ercole, A.; Silvetti, V.; Col, J.D.; Lamberti, M.J. Impact of Genomic Mutation on Melanoma Immune Microenvironment and IFN-1 Pathway-Driven Therapeutic Responses. Cancers 2024, 16, 2568. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; España, S.; Richarz, N.; Solé-Blanch, C.; Boada, A.; Martinez-Cardús, A.; Chu, A.; Liu, Z.; Manzano, J.L. Targeted therapy or immunotherapy in BRAF-mutated metastatic melanoma: A Spanish center’s decade of experience. Front. Oncol. 2024, 14, 1322116. [Google Scholar] [CrossRef]
- Peng, B.; Gu, Y.; Xiong, Y.; Zheng, G.; He, Z. Microarray-Assisted Pathway Analysis Identifies MT1X & NFκB as Mediators of TCRP1-Associated Resistance to Cisplatin in Oral Squamous Cell Carcinoma. PLoS ONE 2012, 7, e51413. [Google Scholar] [CrossRef]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Weinlich, G. Metallothionein-overexpression as a prognostic marker in melanoma. G Ital. Dermatol. Venereol. 2009, 144, 27–38. [Google Scholar] [PubMed]
- Kaufmann, S.H.; Gores, G.J. Apoptosis in cancer: Cause and cure. Bioessays 2000, 22, 1007–1017. [Google Scholar]
- Darding, M.; Meier, P. IAPs: Guardians of RIPK1. Cell Death Differ. 2012, 19, 58–66. [Google Scholar] [CrossRef]
- Bertrand, M.J.M.; Lippens, S.; Staes, A.; Gilbert, B.; Roelandt, R.; De Medts, J.; Gevaert, K.; Declercq, W.; Vandenabeele, P. cIAP1/2 Are Direct E3 Ligases Conjugating Diverse Types of Ubiquitin Chains to Receptor Interacting Proteins Kinases 1 to 4 (RIP1–4). PLoS ONE 2011, 6, e22356. [Google Scholar] [CrossRef]
- Shen, S.; Callaghan, D.; Juzwik, C.; Xiong, H.; Huang, P.; Zhang, W. ABCG2 reduces ROS-mediated toxicity and inflammation: A potential role in Alzheimer’s disease. J. Neurochem. 2010, 114, 1590–1604. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, D.; Kuai, J.; Cheng, M.; Fang, X.; Li, G. Upregulation of miR-199a/b contributes to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling pathway in ALDHA1+colorectal cancer stem cells. Tumor Biol. 2017, 39, 1010428317715155. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Zhang, J.T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC3325772/.
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef]
- Calcagno, A.M.; Fostel, J.M.; To, K.K.W.; Salcido, C.D.; E Martin, S.; Chewning, K.J.; Wu, C.-P.; Varticovski, L.; E Bates, S.; Caplen, N.J.; et al. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br. J. Cancer 2008, 98, 1515–1524. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Rui, M.; Shen, H.; Xin, Y.; Zhang, J.; Li, J.; Yue, L.; Lai, W.; Xu, X. Tumor-specific delivery of doxorubicin through conjugation of pH-responsive peptide for overcoming drug resistance in cancer. Int. J. Pharm. 2017, 528, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, S.; Kumar, A.; Bhatkar, D.; Sharma, N.K. Molecular avenues in targeted doxorubicin cancer therapy. Futur. Oncol. 2020, 16, 687–700. [Google Scholar] [CrossRef]
- Takemura, G.; Fujiwara, H. Doxorubicin-Induced Cardiomyopathy: From the Cardiotoxic Mechanisms to Management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Ashley, N.; Poulton, J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun. 2009, 378, 450–455. [Google Scholar] [CrossRef]
- Diestra, J.E.; Scheffer, G.L.; Català, I.; Maliepaard, M.; Schellens, J.H.M.; Scheper, R.J.; Germà-Lluch, J.R.; Izquierdo, M.A. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J. Pathol. 2002, 198, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Szczygieł, M.; Markiewicz, M.; Szafraniec, M.J.; Hojda, A.; Fiedor, L.; Urbanska, K. Systemic Mobilization of Breast Cancer Re-sistance Protein in Response to Oncogenic Stress. Cancers 2022, 14, 313. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Ishii, G.; Yokose, T.; Minegishi, Y.; Tsuta, K.; Goto, K.; Nishiwaki, Y.; Kodama, T.; Suga, M.; Ochiai, A. Breast Cancer Resistance Protein Impacts Clinical Outcome in Platinum-Based Chemotherapy for Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2004, 10, 1691–1697. [Google Scholar] [CrossRef]
- Qadir, M.; O’Loughlin, K.L.; Fricke, S.M.; Williamson, N.A.; Greco, W.R.; Minderman, H.; Baer, M.R. Cyclosporin A Is a Broad-Spectrum Multidrug Resistance Modulator. Clin. Cancer Res. 2005, 11, 2320–2326. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dai, Y.; Vethanayagam, R.R.; Hebert, M.F.; Thummel, K.E.; Unadkat, J.D.; Ross, D.D.; Mao, Q. Cyclosporin A, tacrolimus and sirolimus are potent inhibitors of the human breast cancer resistance protein (ABCG2) and reverse resistance to mitoxan-trone and topotecan. Cancer Chemother Pharmacol 2006, 58, 374–383. [Google Scholar] [CrossRef]
- Yang, Y.; Li, P.; Zhang, Z.; Wang, Z.; Liu, L.; Liu, X. Prediction of Cyclosporin-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Model Characterizing Interplay of Drug Transporters and Enzymes. Int. J. Mol. Sci. 2020, 21, 7023. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olajossy, B.; Wronski, N.; Madej, E.; Komperda, J.; Szczygieł, M.; Wolnicka-Glubisz, A. RIPK4 Downregulation Reduces ABCG2 Expression, Increasing BRAF-Mutated Melanoma Cell Susceptibility to Cisplatin- and Doxorubicin-Induced Apoptosis. Biomolecules 2024, 14, 1573. https://doi.org/10.3390/biom14121573
Olajossy B, Wronski N, Madej E, Komperda J, Szczygieł M, Wolnicka-Glubisz A. RIPK4 Downregulation Reduces ABCG2 Expression, Increasing BRAF-Mutated Melanoma Cell Susceptibility to Cisplatin- and Doxorubicin-Induced Apoptosis. Biomolecules. 2024; 14(12):1573. https://doi.org/10.3390/biom14121573
Chicago/Turabian StyleOlajossy, Bartlomiej, Norbert Wronski, Ewelina Madej, Joanna Komperda, Małgorzata Szczygieł, and Agnieszka Wolnicka-Glubisz. 2024. "RIPK4 Downregulation Reduces ABCG2 Expression, Increasing BRAF-Mutated Melanoma Cell Susceptibility to Cisplatin- and Doxorubicin-Induced Apoptosis" Biomolecules 14, no. 12: 1573. https://doi.org/10.3390/biom14121573
APA StyleOlajossy, B., Wronski, N., Madej, E., Komperda, J., Szczygieł, M., & Wolnicka-Glubisz, A. (2024). RIPK4 Downregulation Reduces ABCG2 Expression, Increasing BRAF-Mutated Melanoma Cell Susceptibility to Cisplatin- and Doxorubicin-Induced Apoptosis. Biomolecules, 14(12), 1573. https://doi.org/10.3390/biom14121573