
Sirtuin-5 Is Recruited to Hepatic Peroxisomes in Mice Fed Dodecanedioic Acid but Has Little Impact on the Peroxisomal Succinylome
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and DC12 Administration
2.2. Density Gradient Centrifugation
2.3. SIRT5 Co-Immunoprecipitation
2.4. Immunoblotting
2.5. Fatty Acid Oxidation Flux Assays
2.6. Urinary Dicarboxylic Acids
2.7. Proteomics and Succinylomics
2.8. Recombinant Protein Expression and Purification
2.9. Enzyme Activity Assays
3. Results
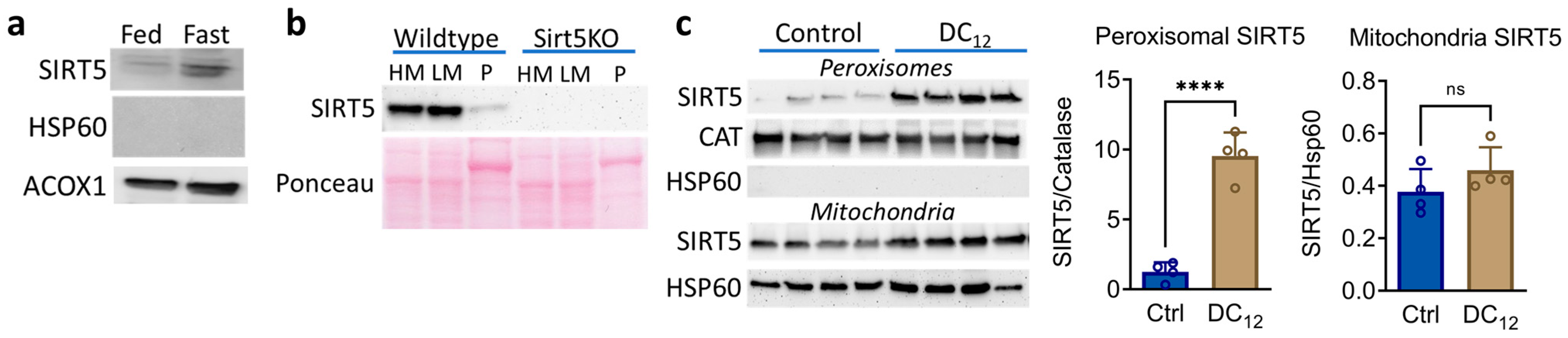
3.1. Dicarboxylic Acid Feeding Recruits SIRT5 to Liver Peroxisomes
3.2. SIRT5 Knockout Mice Have Impaired DC12 Catabolism
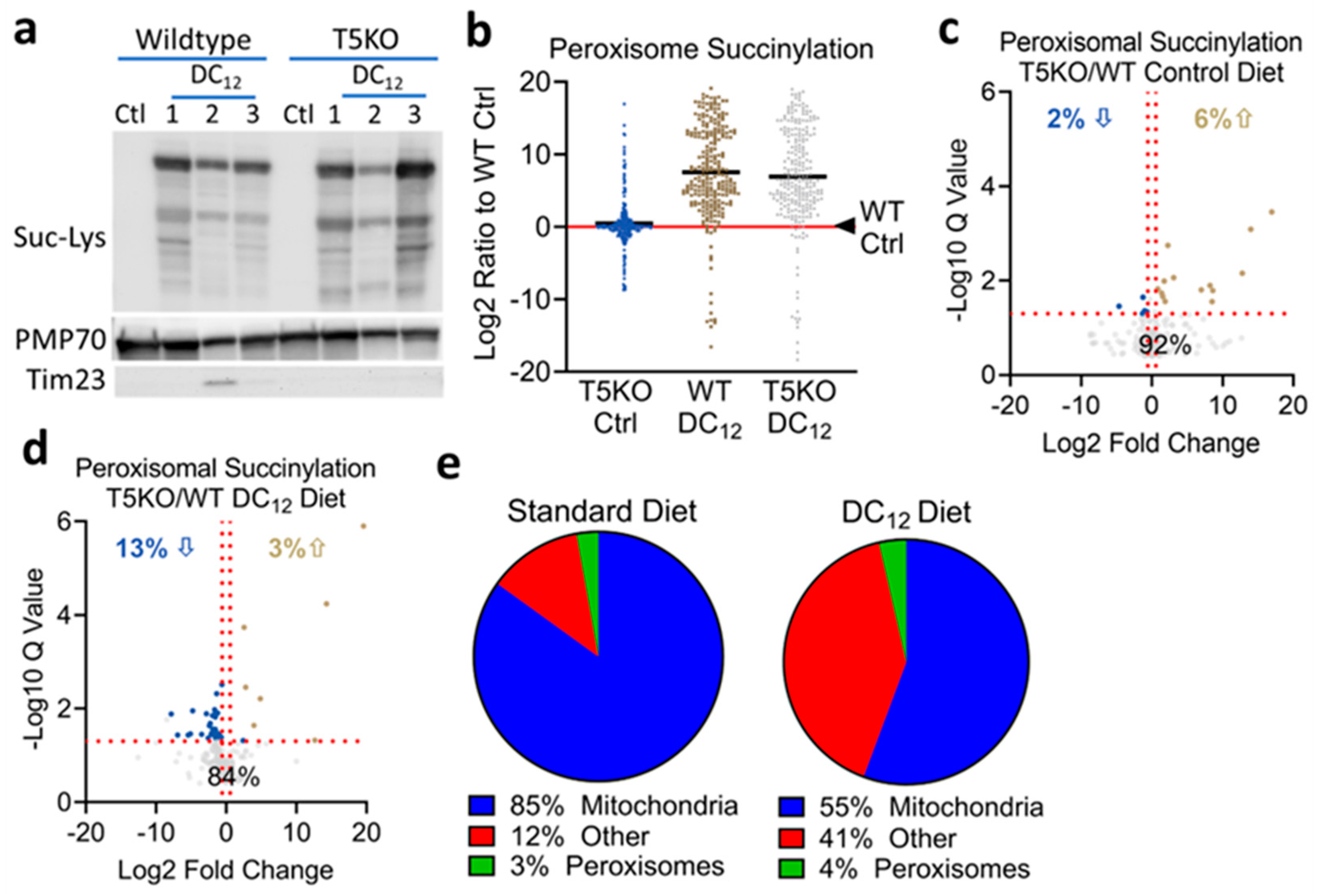
3.3. Knocking out SIRT5 Has Minimal Effects on the Peroxisomal Succinylome
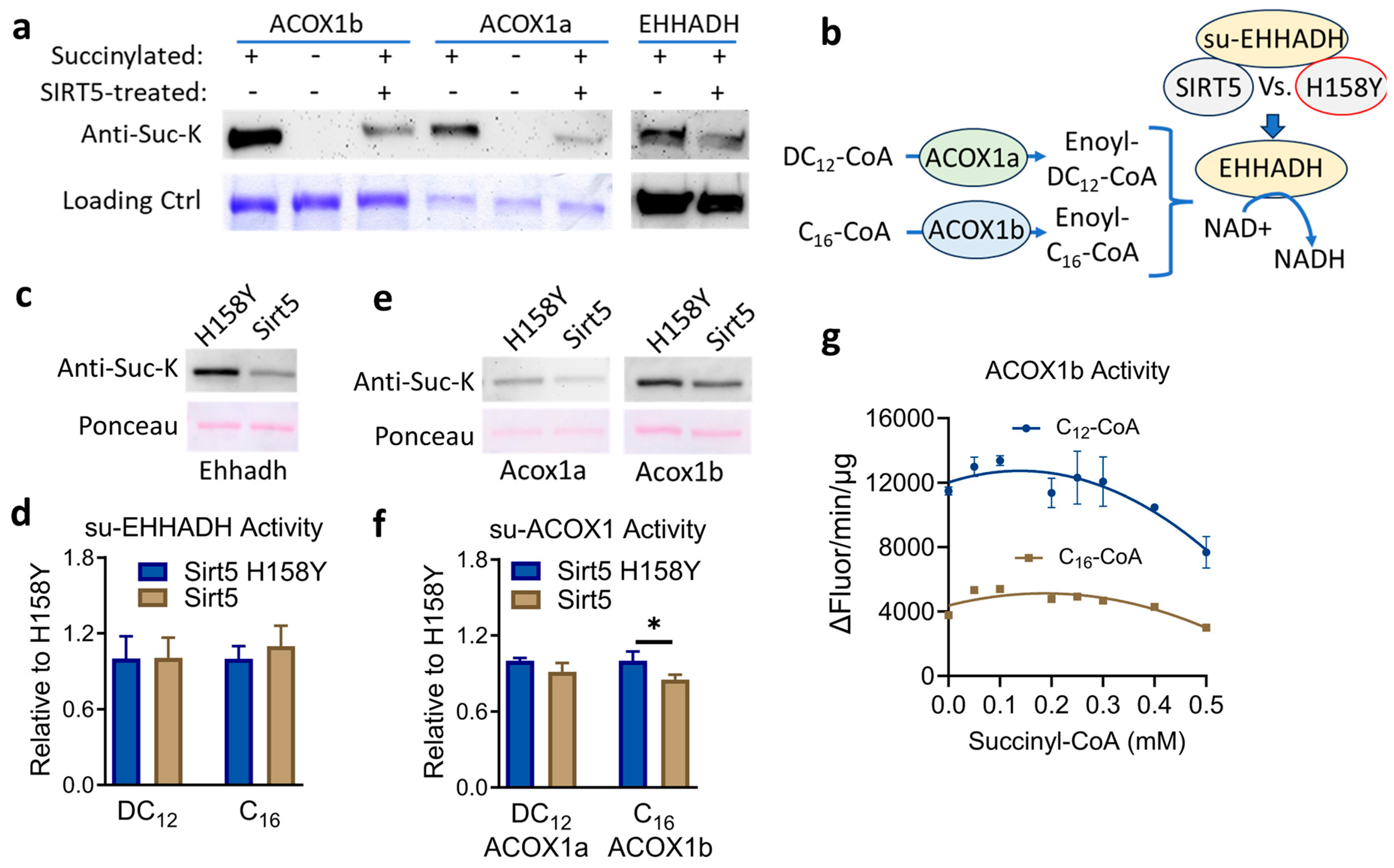
3.4. SIRT5 Desuccinylates Recombinant ACOX1 and EHHADH but the Impact on Enzyme Activity Is Minimal
4. Conclusions
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Zhao, Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol. Cell. Proteom. 2015, 14, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Goetzman, E.S.; Zhang, B.B.; Zhang, Y.; Bharathi, S.S.; Bons, J.; Rose, J.; Shah, S.; Solo, K.J.; Schmidt, A.V.; Richert, A.C.; et al. Dietary dicarboxylic acids provide a non-storable alternative fat source that protects mice against obesity. J. Clin. Investig. 2024, 134, e174186. [Google Scholar] [CrossRef]
- Ranea-Robles, P.; Houten, S.M. The biochemistry and physiology of long-chain dicarboxylic acid metabolism. Biochem. J. 2023, 480, 607–627. [Google Scholar] [CrossRef]
- Jin, Z.; Bian, F.; Tomcik, K.; Kelleher, J.K.; Zhang, G.F.; Brunengraber, H. Compartmentation of Metabolism of the C12-, C9-, and C5-n-dicarboxylates in Rat Liver, Investigated by Mass Isotopomer Analysis: ANAPLEROSIS FROM DODECANEDIOATE. J. Biol. Chem. 2015, 290, 18671–18677. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Baumgart, E.; Mannaerts, G.P. Mannaerts, Iodixanol (Optiprep), an improved density gradient medium for the iso-osmotic isolation of rat liver peroxisomes. Anal. Biochem. 1996, 237, 17–23. [Google Scholar] [CrossRef]
- Mi, J.; Garcia-Arcos, I.; Alvarez, R.; Cristobal, S. Age-related subproteomic analysis of mouse liver and kidney peroxisomes. Proteome Sci. 2007, 5, 19. [Google Scholar] [CrossRef]
- Schmidt, A.V.; Bharathi, S.S.; Solo, K.J.; Bons, J.; Rose, J.P.; Schilling, B.; Goetzman, E.S. Sirt2 Regulates Liver Metabolism in a Sex-Specific Manner. Biomolecules 2024, 14, 1160. [Google Scholar] [CrossRef] [PubMed]
- Mihalik, S.J.; Rainville, A.M.; Watkins, P.A. Phytanic acid alpha-oxidation in rat liver peroxisomes. Production of alpha-hydroxyphytanoyl-CoA and formate is enhanced by dioxygenase cofactors. Eur. J. Biochem. 1995, 232, 545–551. [Google Scholar] [PubMed]
- Huynh, F.K.; Green, M.F.; Koves, T.R.; Hirschey, M.D. Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 2014, 542, 391–405. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Veerkamp, J.H.; van Moerkerk, T.B.; Glatz, J.F.; Zuurveld, J.G.; Jacobs, A.E.; Wagenmakers, A.J. 14CO2 production is no adequate measure of [14C]fatty acid oxidation. Biochem. Med. Metab. Biol. 1986, 35, 248–259. [Google Scholar] [CrossRef]
- Schlüter, A.; Real-Chicharro, A.; Gabaldón, T.; Sánchez-Jiménez, F.; Pujol, A. PeroxisomeDB 2.0: E. Nucleic Acids Res. 2010, 38, D800–D805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef]
- Zhang, Y.; Goetzman, E. The enzyme activity of mitochondrial trifunctional protein is not altered by lysine acetylation or lysine succinylation. PLoS ONE 2021, 16, e0256619. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Uppala, R.; Verdin, E.; Gibson, B.W.; Goetzman, E.S. SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain acyl-CoA dehydrogenase. PLoS ONE 2015, 10, e0122297. [Google Scholar] [CrossRef]
- He, A.Y.; Liu, C.Z.; Chen, L.Q.; Ning, L.J.; Zhang, M.L.; Li, E.C.; Du, Z.Y. Identification, characterization and nutritional regulation of two isoforms of acyl-coenzyme A oxidase 1 gene in Nile tilapia (Oreochromis niloticus). Gene 2014, 545, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Oaxaca-Castillo, D.; Andreoletti, P.; Vluggens, A.; Yu, S.; van Veldhoven, P.P.; Reddy, J.K.; Cherkaoui-Malki, M. Biochemical characterization of two functional human liver acyl-CoA oxidase isoforms 1a and 1b encoded by a single gene. Biochem. Biophys. Res. Commun. 2007, 360, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Morais, S.; Knoll-Gellida, A.; André, M.; Barthe, C.; Babin, P.J. Conserved expression of alternative splicing variants of peroxisomal acyl-CoA oxidase 1 in vertebrates and developmental and nutritional regulation in fish. Physiol. Genom. 2007, 28, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.W.; Jeske, J.; Schmidt, N.; Erdmann, R. ATP-Dependent Steps in Peroxisomal Protein Import. Annu. Rev. Biochem. 2024, 93, 233–259. [Google Scholar] [CrossRef] [PubMed]
- Kunze, M. The type-2 peroxisomal targeting signal. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118609. [Google Scholar] [CrossRef]
- Kunze, M. Predicting Peroxisomal Targeting Signals to Elucidate the Peroxisomal Proteome of Mammals. Subcell. Biochem. 2018, 89, 157–199. [Google Scholar]
- Hershberger, K.A.; Abraham, D.M.; Liu, J.; Locasale, J.W.; Grimsrud, P.A.; Hirschey, M.D. Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J. Biol. Chem. 2018, 293, 10630–10645. [Google Scholar] [CrossRef]
- Alleyn, M.; Breitzig, M.; Lockey, R.; Kolliputi, N. The dawn of succinylation: A posttranslational modification. Am. J. Physiol. Cell Physiol. 2018, 314, C228–C232. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292, 10239–10249. [Google Scholar] [CrossRef]
- Ranea-Robles, P.; Violante, S.; Argmann, C.; Dodatko, T.; Bhattacharya, D.; Chen, H.; Yu, C.; Friedman, S.L.; Puchowicz, M.; Houten, S.M. Murine deficiency of peroxisomal L-bifunctional protein (EHHADH) causes medium-chain 3-hydroxydicarboxylic aciduria and perturbs hepatic cholesterol homeostasis. Cell. Mol. Life Sci. 2021, 78, 5631–5646. [Google Scholar] [CrossRef]
- Ding, J.; Loizides-Mangold, U.; Rando, G.; Zoete, V.; Michielin, O.; Reddy, J.K.; Wahli, W.; Riezman, H.; Thorens, B. The peroxisomal enzyme L-PBE is required to prevent the dietary toxicity of medium-chain fatty acids. Cell Rep. 2013, 5, 248–258. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | % Coverage |
|---|---|---|
| Uricase (UOX) | Oxidizes uric acid to 5-hydroxyisourate | 86 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | Steps 2 and 3 of β-oxidation | 74 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | Steps 2 and 3 of β-oxidation | 65 |
| Acyl-CoA dehydrogenase 11 (ACAD11) | Step 1 of very long-chain β-oxidation | 50 |
| Peroxisomal 2,4-dienoyl-CoA reductase (DECR2) | Reduces double bonds, β-oxidation | 49 |
| Peroxisomal acyl-coenzyme A oxidase 1 (ACOX1) | Step 1 of β-oxidation | 40 |
| Catalase (CAT) | Degrades H2O2 to H2O and O2 | 33 |
| ATP-binding cassette sub-family D member 3 (ABCD3) | Fatty acid transporter | 23 |
| Acyl-coenzyme A thioesterase 4 (ACOT4) | Cleaves CoA from succinyl-CoA | 21 |
| Non-specific lipid-transfer protein (SCP2) | Step 4 of β-oxidation | 21 |
| Peroxisomal sarcosine oxidase (PIPOX) | Metabolizes sarcosine & pipecolic acid | 16 |
| Lon protease homolog 2, peroxisomal (LONP2) | Degrades misfolded proteins | 14 |
| 3-ketoacyl-CoA thiolase B (ACAA1b) | Step 4 of β-oxidation | 13 |
| Very long-chain acyl-CoA synthetase (SLC27A2) | Activates fatty acids into acyl-CoAs | 12 |
| Peroxisomal coenzyme A diphosphatase (NUDT7) | Hydrolyzes CoA moiety of acyl-CoAs | 10 |
| Protein | Lysine Residue | Log2 FC KO/WT |
|---|---|---|
| Increased Succinylation (q < 0.05) | ||
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K481 | 16.9 |
| Enoyl-CoA isomerase (ECH1) | K320 | 14.0 |
| Enoyl-CoA isomerase (ECH1) | K76 | 12.8 |
| Acyl-coenzyme A diphosphatase (NUDT19) | K351 | 8.6 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K390 | 8.5 |
| Alpha-methylacyl-CoA racemase (AMACR) | K204 | 8.2 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K295 | 7.0 |
| Prostaglandin reductase-3 (ZADH2) | K214 | 3.1 |
| Dehydrogenase/reductase SDR family member 4 (DHRS4) | K106 | 2.3 |
| Catalase (CAT) | K522 | 1.9 |
| Peroxisomal 2,4-dienoyl-CoA reductase (DECR2) | K230 | 1.7 |
| ATP-binding cassette sub-family D member 3 (ABCD3) | K6 | 1.5 |
| Alanine--glyoxylate aminotransferase (AGXT) | K322 | 1.4 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K355 | 0.8 |
| Decreased Succinylation (q < 0.05) | ||
| Dehydrogenase/reductase SDR family member 4 (DHRS4) | K99 | −0.7 |
| Catalase (CAT) | K98 | −1.0 |
| Peroxisomal trans-2-enoyl-CoA reductase (PECR) | K83 | −1.3 |
| Enoyl-CoA isomerase (ECH1) | K230 | −1.3 |
| 2-Hydroxyacid oxidase 1 (HAO1) | K249 | −4.6 |
| Protein | Lysine Residue | Log2 FC KO/WT |
|---|---|---|
| Increased Succinylation (q < 0.05) | ||
| Peroxisomal ATPase (PEX1) | K1205 | 14.3 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K50/K57 | 12.6 |
| Alanine--glyoxylate aminotransferase (AGXT) | K411/K314 | 4.9 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K295 | 4.0 |
| Catalase (CAT) | K477 | 2.8 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K348/K355 | 2.6 |
| Acyl-coenzyme A diphosphatase (NUDT19) | K351 | 2.4 |
| Decreased Succinylation (q < 0.05) | ||
| Uricase (UOX) | K27 | −0.6 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K579 | −0.7 |
| Acyl-CoA dehydrogenase-11 (ACAD11) | K422 | −0.9 |
| Uricase (UOX) | K118 | −1.0 |
| Uricase (UOX) | K80 | −1.1 |
| Bile acid-CoA: amino acid N-acyltransferase (BAAT) | K346 | −1.2 |
| Peroxisomal sarcosine oxidase (PIPOX) | K104 | −1.3 |
| ATP-binding cassette sub-family D member 3 (ABCD3) | K260 | −1.4 |
| Bile acid-CoA: amino acid N-acyltransferase (BAAT) | K406 | −1.4 |
| Non-specific lipid-transfer protein (SCP2) | K26 | −1.5 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K527 | −1.6 |
| 2-Hydroxyacid oxidase 1 (HAO1) | K353 | −1.6 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K81 | −1.6 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K459 | −1.6 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K57 | −1.8 |
| Dehydrogenase/reductase SDR family member 4 (DHRS4) | K99 | −1.8 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K65/K68 | −1.8 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K87 | −1.8 |
| Acyl-CoA dehydrogenase-11 (ACAD11) | K90 | −1.9 |
| Peroxisomal 2,4-dienoyl-CoA reductase (DECR2) | K70/K71 | −1.9 |
| Peroxisomal trans-2-enoyl-CoA reductase (PECR) | K32 | −1.9 |
| Peroxisomal sarcosine oxidase (PIPOX) | K153 | −2.1 |
| Peroxisomal sarcosine oxidase (PIPOX) | K98 | −2.1 |
| Alpha-methylacyl-CoA racemase (AMACR) | K57 | −2.3 |
| Bifunctional epoxide hydrolase 2 (EPHX2) | K482 | −2.4 |
| 2-hydroxyacyl-CoA lyase 1 (HACL1) | K21 | −2.5 |
| Enoyl-CoA isomerase (ECH1) | K230 | −2.8 |
| Peroxisomal coenzyme A diphosphatase (NUDT7) | K20 | −3.5 |
| 2-hydroxyacyl-CoA lyase 1 (HACL1) | K238 | −4.8 |
| 2-Hydroxyacid oxidase 1 (HAO1) | K309 | −5.1 |
| Acyl-CoA oxidase-2 (ACOX2) | K678 | −5.4 |
| Peroxisomal trans-2-enoyl-CoA reductase (PECR) | K17 | −6.9 |
| Acyl-CoA oxidase-2 (ACOX2) | K303 | −7.8 |
| NAD-capped RNA hydrolase (NUDT12) | K282 | −8.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, B.B.; Bharathi, S.S.; Bons, J.; Rose, J.P.; Shah, S.; Dobrowolski, S.F.; Sims-Lucas, S.; Schilling, B.; Goetzman, E.S. Sirtuin-5 Is Recruited to Hepatic Peroxisomes in Mice Fed Dodecanedioic Acid but Has Little Impact on the Peroxisomal Succinylome. Biomolecules 2024, 14, 1508. https://doi.org/10.3390/biom14121508
Zhang Y, Zhang BB, Bharathi SS, Bons J, Rose JP, Shah S, Dobrowolski SF, Sims-Lucas S, Schilling B, Goetzman ES. Sirtuin-5 Is Recruited to Hepatic Peroxisomes in Mice Fed Dodecanedioic Acid but Has Little Impact on the Peroxisomal Succinylome. Biomolecules. 2024; 14(12):1508. https://doi.org/10.3390/biom14121508
Chicago/Turabian StyleZhang, Yuxun, Bob B. Zhang, Sivakama S. Bharathi, Joanna Bons, Jacob P. Rose, Samah Shah, Steven F. Dobrowolski, Sunder Sims-Lucas, Birgit Schilling, and Eric S. Goetzman. 2024. "Sirtuin-5 Is Recruited to Hepatic Peroxisomes in Mice Fed Dodecanedioic Acid but Has Little Impact on the Peroxisomal Succinylome" Biomolecules 14, no. 12: 1508. https://doi.org/10.3390/biom14121508
APA StyleZhang, Y., Zhang, B. B., Bharathi, S. S., Bons, J., Rose, J. P., Shah, S., Dobrowolski, S. F., Sims-Lucas, S., Schilling, B., & Goetzman, E. S. (2024). Sirtuin-5 Is Recruited to Hepatic Peroxisomes in Mice Fed Dodecanedioic Acid but Has Little Impact on the Peroxisomal Succinylome. Biomolecules, 14(12), 1508. https://doi.org/10.3390/biom14121508