
Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson’s Disease
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Risk Factors Associated with PD
1.1.1. Aging
1.1.2. Gender
1.1.3. Ethnicity
1.1.4. Genetics
1.1.5. Environmental Factors
2. Genetic Basis of PD
2.1. α-Synuclein
2.2. LRRK2
2.3. PINK1
2.4. Parkin
2.5. DJ-1
2.6. Vacuolar Protein Sorting 35 (VPS35)
2.7. Glucocerebrosidase 1 (GBA1)
3. Cellular and Molecular Mechanism Underlying the Pathogenesis of Neurodegeneration in PD
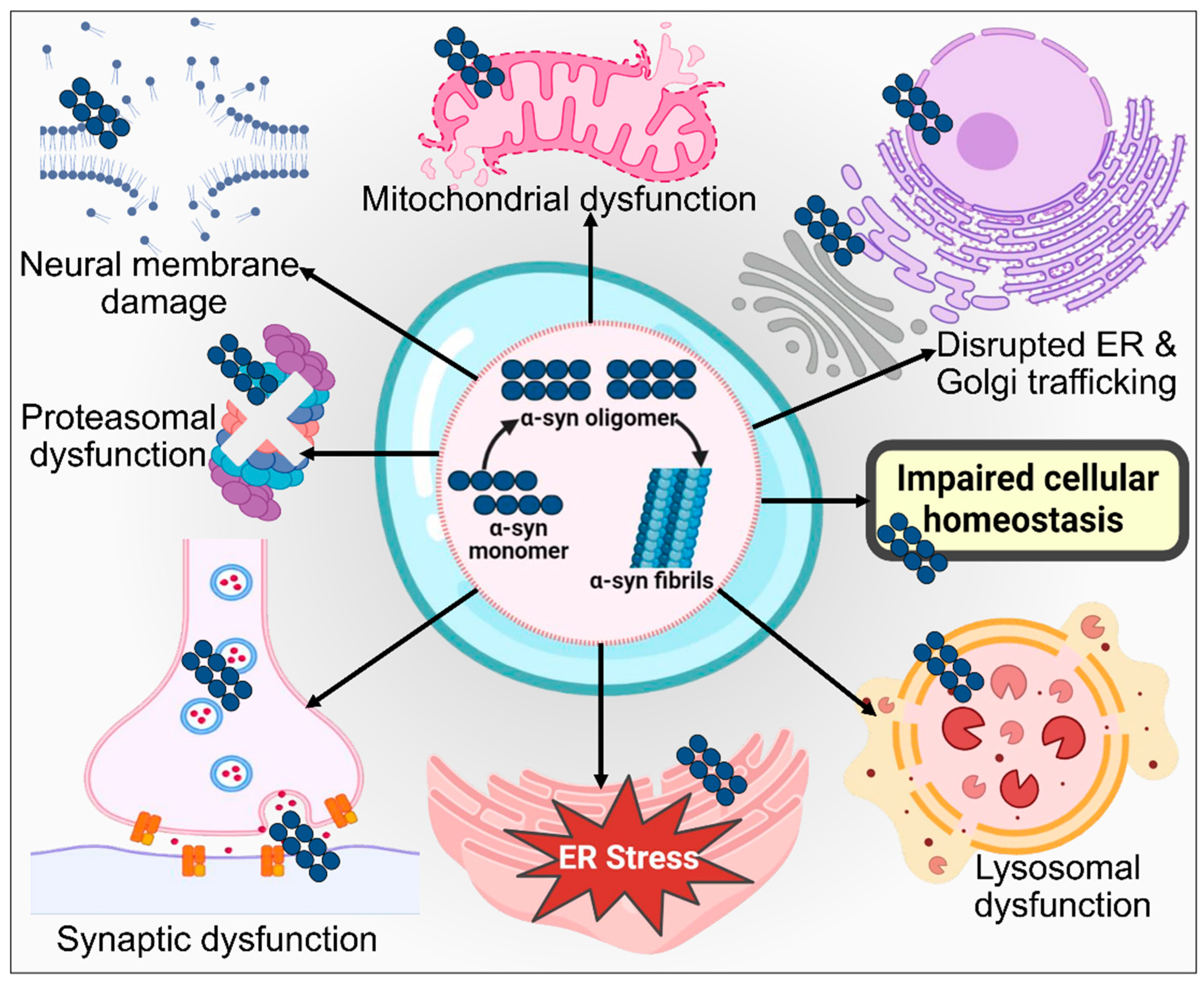
3.1. Protein Misfolding and Aggregation
3.2. ER Stress
3.3. Calcium Homeostasis
3.4. Dopamine Metabolism and Toxicity
3.5. Mitochondrial Dysfunction
3.6. Oxidative Stress
3.7. Nitrosative Stress
3.8. Apoptosis
3.9. Neuroinflammation
3.10. Immune System Deregulation
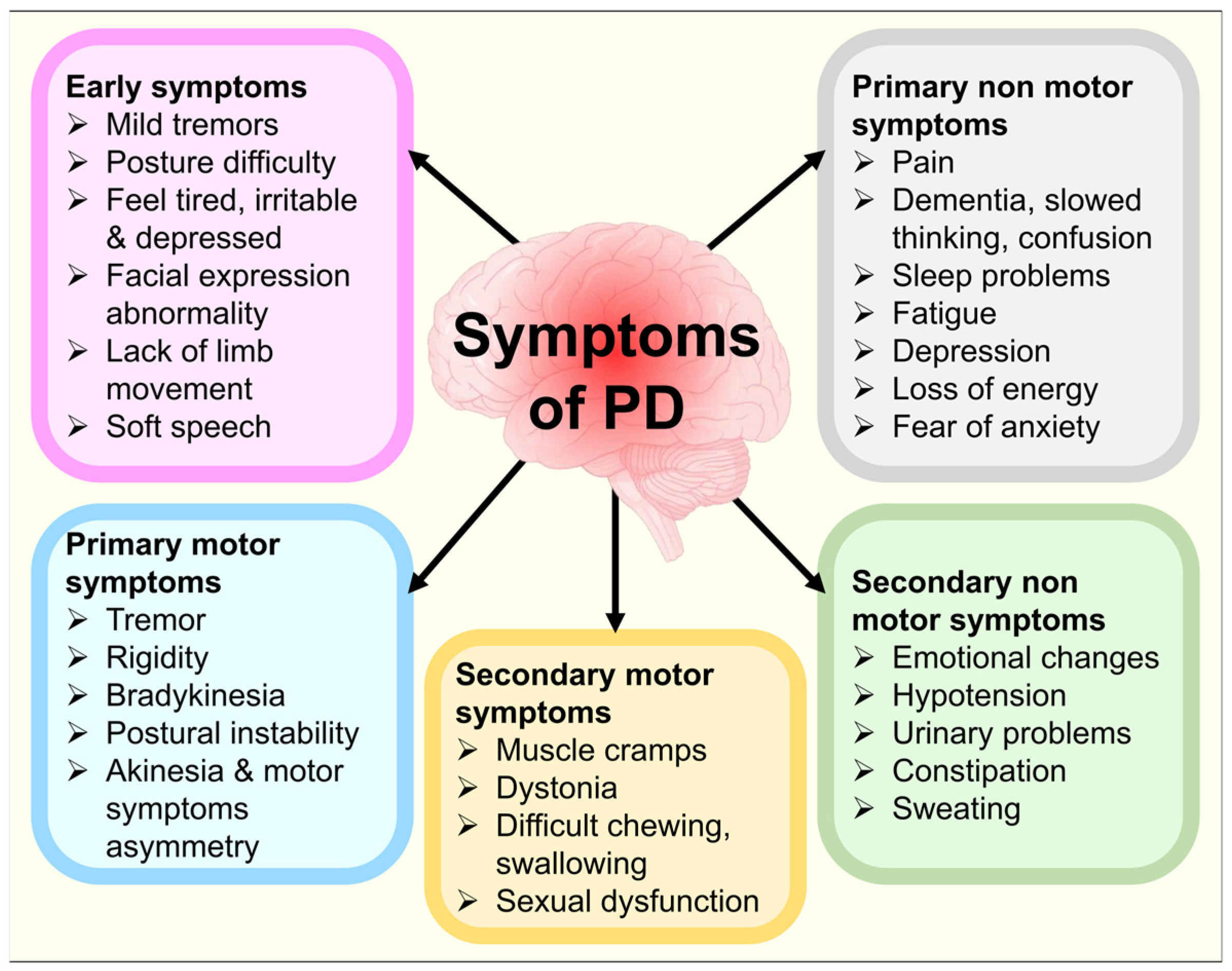
3.11. Non-Motor Pathologies in PD
3.12. Non-Dopaminergic Pathologies in PD
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Ray Chaudhuri, K.; Weintraub, D. Parkinson Disease-Associated Cognitive Impairment. Nat. Rev. Dis. Prim. 2021, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s Disease: An Introspection of Its Journey towards Precision Medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Urso, D.; Savica, R. Descriptive Epidemiology of Neurodegenerative Diseases: What Are the Critical Questions? Neuroepidemiology 2022, 56, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Delamarre, A.; Meissner, W.G. Epidemiology, Environmental Risk Factors and Genetics of Parkinson’s Disease. Press. Medicale 2017, 46, 175–181. [Google Scholar] [CrossRef]
- Pang, S.Y.Y.; Ho, P.W.L.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The Interplay of Aging, Genetics and Environmental Factors in the Pathogenesis of Parkinson’s Disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Dawson, V.L.; Dawson, T.M. Recent Advances in the Genetics of Parkinson’s Disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 301–325. [Google Scholar] [CrossRef]
- Davie, C.A. A Review of Parkinson’s Disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef]
- Rizek, P.; Kumar, N.; Jog, M.S. An Update on the Diagnosis and Treatment of Parkinson Disease. Can. Med. Assoc. J. 2016, 188, 1157–1165. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. The Role of Environmental Exposures in Neurodegeneration and Neurodegenerative Diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H. Non-Dopaminergic Treatments for Motor Control in Parkinson’s Disease. Drugs 2013, 73, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Kumaresan, M.; Khan, S. Spectrum of Non-Motor Symptoms in Parkinson’s Disease. Cureus 2021, 13, e13275. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.; Lim, J.; Choi, H.J. Recent Advances in the Pathology of Prodromal Non-Motor Symptoms Olfactory Deficit and Depression in Parkinson’s Disease: Clues to Early Diagnosis and Effective Treatment. Arch. Pharm. Res. 2021, 44, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Dahbour, S.S.; Al Murr, M.J.; Oweis, L.H.; Al Antary, N.T.; Mohsen, M.; Al Fegi, S. Non-Motor Manifestation of Parkinson’s Disease: A Cross-Sectional Study in a Teaching Hospital in Jordan. Egypt. J. Neurol. Psychiatry Neurosurg. 2022, 58, 148. [Google Scholar] [CrossRef]
- Gupta, S.; Shukla, S. Non-Motor Symptoms in Parkinson’s Disease: Opening New Avenues in Treatment. Curr. Res. Behav. Sci. 2021, 2, 100049. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s Disease: Etiopathogenesis and Treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Stephan, M.A.; Meier, B.; Zaugg, S.W.; Kaelin-Lang, A. Motor Sequence Learning Performance in Parkinson’s Disease Patients Depends on the Stage of Disease. Brain Cogn. 2011, 75, 135–140. [Google Scholar] [CrossRef]
- Sanjari Moghaddam, H.; Valitabar, Z.; Ashraf-Ganjouei, A.; Mojtahed Zadeh, M.; Ghazi Sherbaf, F.; Aarabi, M.H. Cerebrospinal Fluid C-Reactive Protein in Parkinson’s Disease: Associations with Motor and Non-Motor Symptoms. NeuroMol. Med. 2018, 20, 376–385. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson Disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and Neurodegenerative Diseases. A Systematic Review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Politi, C.; Ciccacci, C.; Novelli, G.; Borgiani, P. Genetics and Treatment Response in Parkinson’s Disease: An Update on Pharmacogenetic Studies. NeuroMol. Med. 2018, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Korczyn, A.D. Are Dementia with Lewy Bodies and Parkinson’s Disease Dementia the Same Disease? BMC Med. 2018, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Gökçal, E.; Gür, V.E.; Selvitop, R.; Babacan Yildiz, G.; Asil, T. Motor and Non-Motor Symptoms in Parkinson’s Disease: Effects on Quality of Life. Noropsikiyatri Ars. 2017, 54, 143–148. [Google Scholar]
- Schapira, A.H.V. Glucocerebrosidase and Parkinson Disease: Recent Advances. Mol. Cell. Neurosci. 2015, 66, 37–42. [Google Scholar] [CrossRef]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Chen, C.M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Moradi Vastegani, S.; Nasrolahi, A.; Ghaderi, S.; Belali, R.; Rashno, M.; Farzaneh, M.; Khoshnam, S.E. Mitochondrial Dysfunction and Parkinson’s Disease: Pathogenesis and Therapeutic Strategies. Neurochem. Res. 2023, 48, 2285–2308. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Chakravarthy, V.S. Influence of Energy Deficiency on the Subcellular Processes of Substantia Nigra Pars Compacta Cell for Understanding Parkinsonian Neurodegeneration. Sci. Rep. 2021, 11, 1754. [Google Scholar] [CrossRef]
- Błaszczyk, J.W. The Emerging Role of Energy Metabolism and Neuroprotective Strategies in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 2016–2019. [Google Scholar] [CrossRef] [PubMed]
- Himmelberg, M.M.; West, R.J.H.; Elliott, C.J.H.; Wade, A.R. Abnormal Visual Gain Control and Excitotoxicity in Early-Onset Parkinson’s Disease Drosophila Models. J. Neurophysiol. 2018, 119, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-Induced Excitotoxicity in Parkinson’s Disease: The Role of Glial Cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The Cell Biology of Parkinson’s Disease. J. Cell Biol. 2021, 220, e202012095. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Huang, G.; Ma, L.; Tong, Y.; Chen, P.; Shen, J. Cell Autonomous Role of Leucine-Rich Repeat Kinase in Protection of Dopaminergic Neuron Survival ELife Assessment. bioRxiv 2023, 2, 1–57. [Google Scholar]
- Meng, L.; Liu, C.; Li, Y.; Chen, G.; Xiong, M.; Yu, T.; Pan, L.; Zhang, X.; Zhou, L.; Guo, T.; et al. The Yeast Prion Protein Sup35 Initiates α-Synuclein Pathology in Mouse Models of Parkinson’s Disease. Sci. Adv. 2023, 9, eadj1092. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-like Mechanisms in Parkinson’s Disease. Front. Neurosci. 2019, 13, 552. [Google Scholar] [CrossRef]
- Lehtonen, Š.; Sonninen, T.M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-Onset Impairment of the Ubiquitin-Proteasome System in Dopaminergic Neurons Caused by α-Synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Rosemary, A.; Hamilton, M.; Pascal, C.; Sabina, L.; James, S.; Caroline, A.G.C. Onset and progression factors in Parkinson’s disease: A systematic review. Neurotoxicology 2017, 61, 132–141. [Google Scholar]
- Chauhan, A.; Jeans, A.F. Is Parkinson’s Disease Truly a Prion-like Disorder? An Appraisal of Current Evidence. Neurol. Res. Int. 2015, 2015, 345285. [Google Scholar] [CrossRef] [PubMed]
- Berg, D. Biomarkers for the Early Detection of Parkinson’s and Alzheimer’s Disease. Neurodegener. Dis. 2008, 5, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-Synuclein Biology in Lewy Body Diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Marques, O.; Outeiro, T.F. Alpha-Synuclein: From Secretion to Dysfunction and Death. Cell Death Dis. 2012, 3, e350. [Google Scholar] [CrossRef] [PubMed]
- Bourdenx, M.; Bezard, E.; Dehay, B. Lysosomes and α-Synuclein Form a Dangerous Duet Leading to Neuronal Cell Death. Front. Neuroanat. 2014, 8, 83. [Google Scholar] [CrossRef]
- Arena, G.; Sharma, K.; Agyeah, G.; Krüger, R.; Grünewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson’s Disease: A Self-Sustained Loop. Curr. Neurol. Neurosci. Rep. 2022, 22, 427–440. [Google Scholar] [CrossRef]
- Horsager, J.; Knudsen, K.; Sommerauer, M. Clinical and Imaging Evidence of Brain-First and Body-First Parkinson’s Disease. Neurobiol. Dis. 2022, 164, 105626. [Google Scholar] [CrossRef]
- Klann, E.M.; Dissanayake, U.; Gurrala, A.; Farrer, M.; Shukla, A.W.; Ramirez-Zamora, A.; Mai, V.; Vedam-Mai, V. The Gut–Brain Axis and Its Relation to Parkinson’s Disease: A Review. Front. Aging Neurosci. 2022, 13, 782082. [Google Scholar] [CrossRef]
- Li, Q.; Meng, L.B.; Chen, L.J.; Shi, X.; Tu, L.; Zhou, Q.; Yu, J.L.; Liao, X.; Zeng, Y.; Yuan, Q.Y. The Role of the Microbiota-Gut-Brain Axis and Intestinal Microbiome Dysregulation in Parkinson’s Disease. Front. Neurol. 2023, 14, 1185375. [Google Scholar] [CrossRef]
- Li, Z.; Liang, H.; Hu, Y.; Lu, L.; Zheng, C.; Fan, Y.; Wu, B.; Zou, T.; Luo, X.; Zhang, X.; et al. Gut Bacterial Profiles in Parkinson’s Disease: A Systematic Review. CNS Neurosci. Ther. 2023, 29, 140–157. [Google Scholar] [CrossRef]
- Han, Y.; Wang, B.; Gao, H.; He, C.; Hua, R.; Liang, C.; Zhang, S.; Wang, Y.; Xin, S.; Xu, J. Vagus Nerve and Underlying Impact on the Gut Microbiota-Brain Axis in Behavior and Neurodegenerative Diseases. J. Inflamm. Res. 2022, 15, 6213–6230. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, S.; Xu, C.; Tang, L.; Liang, Y.; Zhao, Y.; Zhu, G. Interactions between Gut Microbiota and Parkinson’s Disease: The Role of Microbiota-Derived Amino Acid Metabolism. Front. Aging Neurosci. 2022, 14, 976316. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhao, D.; Ali Shah, S.Z.; Wu, W.; Lai, M.; Zhang, X.; Li, J.; Guan, Z.; Zhao, H.; Li, W.; et al. The Role of the Gut Microbiota in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2019, 10, 1155. [Google Scholar] [CrossRef] [PubMed]
- Manfredsson, F.P.; Luk, K.C.; Benskey, M.J.; Gezer, A.; Garcia, J.; Kuhn, N.C.; Sandoval, I.M.; Patterson, J.R.; O’Mara, A.; Yonkers, R.; et al. Induction of Alpha-Synuclein Pathology in the Enteric Nervous System of the Rat and Non-Human Primate Results in Gastrointestinal Dysmotility and Transient CNS Pathology. Neurobiol. Dis. 2018, 112, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Imbriani, P.; Bonsi, P.; Martella, G.; Peppe, A. Beyond the Microbiota: Understanding the Role of the Enteric Nervous System in Parkinson’s Disease from Mice to Human. Biomedicines 2023, 11, 1560. [Google Scholar] [CrossRef] [PubMed]
- Schaffernicht, G.; Shang, Q.; Stievenard, A.; Bötzel, K.; Dening, Y.; Kempe, R.; Toussaint, M.; Gündel, D.; Kranz, M.; Reichmann, H.; et al. Pathophysiological Changes in the Enteric Nervous System of Rotenone-Exposed Mice as Early Radiological Markers for Parkinson’s Disease. Front. Neurol. 2021, 12, 642604. [Google Scholar] [CrossRef]
- Chalazonitis, A.; Rao, M. Enteric Nervous System Manifestations of Neurodegenerative Disease. Brain Res. 2018, 1693, 207–213. [Google Scholar] [CrossRef]
- Keszthelyi, D. Targeting the Enteric Nervous System to Treat Constipation in Parkinson’s Disease. Gastroenterology 2023, 164, 1017–1018. [Google Scholar] [CrossRef]
- Bindas, A.J.; Nichols, K.N.; Roth, N.J.; Brady, R.; Koppes, A.N.; Koppes, R.A. Aggregation of Alpha-Synuclein in Enteric Neurons Does not Impact Function In Vitro. Sci. Rep. 2022, 12, 22211. [Google Scholar] [CrossRef]
- Hashish, S.; Salama, M. The Role of an Altered Gut Microbiome in Parkinson’s Disease: A Narrative Review. Appl. Microbiol. 2023, 3, 429–447. [Google Scholar] [CrossRef]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-Analysis of the Parkinson’s Disease Gut Microbiome Suggests Alterations Linked to Intestinal Inflammation. NPJ Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Salim, S.; Ahmad, F.; Banu, A.; Mohammad, F. Gut Microbiome and Parkinson’s Disease: Perspective on Pathogenesis and Treatment. J. Adv. Res. 2023, 50, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Müller-Nedebock, A.C.; Dekker, M.C.J.; Farrer, M.J.; Hattori, N.; Lim, S.Y.; Mellick, G.D.; Rektorová, I.; Salama, M.; Schuh, A.F.S.; Stoessl, A.J.; et al. Different Pieces of the Same Puzzle: A Multifaceted Perspective on the Complex Biological Basis of Parkinson’s Disease. NPJ Park. Dis. 2023, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, M.M. Medical Management of Parkinson’s Disease. Pharm. J. 2008, 33, 590–606. [Google Scholar]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s Disease: Why Is Advancing Age the Biggest Risk Factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative Diseases: An Overview of Environmental Risk Factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef]
- Myhre, O.; Utkilen, H.; Duale, N.; Brunborg, G.; Hofer, T. Metal Dyshomeostasis and Inflammation in Alzheimer’s and Parkinson’s Diseases: Possible Impact of Environmental Exposures. Oxid. Med. Cell. Longev. 2013, 2013, 726954. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial Dysfunction in Aging: Much Progress but Many Unresolved Questions. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1347–1353. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial Abnormalities in Parkinson’s Disease and Alzheimer’s Disease: Can Mitochondria Be Targeted Therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The Prevalence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and Etiology of Parkinson’s Disease: A Review of the Evidence. Eur. J. Epidemiol. 2011, 26, S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Wright Willis, A.; Evanoff, B.A.; Lian, M.; Criswell, S.R.; Racette, B.A. Geographic and Ethnic Variation in Parkinson Disease: A Population-Based Study of Us Medicare Beneficiaries. Neuroepidemiology 2010, 34, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Marder, K.; Tang, M.; Mejia, H.; Alfaro, B.; Cbte, B.A.L.; Louis, E. Risk of Parkinson’s Disease among First-Degree Relatives: A Community-Based Study. Neurology 1996, 47, 155–160. [Google Scholar] [CrossRef]
- Shino, M.Y.; McGuire, V.; Van Den Eeden, S.K.; Tanner, C.M.; Popat, R.; Leimpeter, A.; Bernstein, A.L.; Nelson, L.M. Familial Aggregation of Parkinson’s Disease in a Multiethnic Community-Based Case-Control Study. Mov. Disord. 2010, 25, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Rohde, M.; Buschbaum, S.; Severin, D. Throughput Optimized A-Si/Μc-Si Tandem Solar Cells on Sputter-Etched ZnO Substrates. Sol. Energy Mater. Sol. Cells 2012, 98, 363–369. [Google Scholar] [CrossRef]
- Greggio, E.; Cookson, M.R. Leucine-Rich Repeat Kinase 2 Mutations and Parkinson’s Disease: Three Questions. ASN Neuro 2009, 1, 13–24. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Karri, V.; Ramos, D.; Martinez, J.B.; Odena, A.; Oliveira, E.; Coort, S.L.; Evelo, C.T.; Mariman, E.C.M.; Schuhmacher, M.; Kumar, V. Differential Protein Expression of Hippocampal Cells Associated with Heavy Metals (Pb, As, and MeHg) Neurotoxicity: Deepening into the Molecular Mechanism of Neurodegenerative Diseases. J. Proteom. 2018, 187, 106–125. [Google Scholar] [CrossRef] [PubMed]
- Aizenman, E.; Mastroberardino, P.G. Metals and Neurodegeneration. Neurobiol. Dis. 2015, 81, 1–3. [Google Scholar] [CrossRef]
- Duan, W.; Liu, C.; Zhou, J.; Yu, Q.; Duan, Y.; Zhang, T.; Li, Y.; Fu, G.; Sun, Y.; Tian, J.; et al. Upregulation of Mitochondrial Calcium Uniporter Contributes to Paraquat-Induced Neuropathology Linked to Parkinson’s Disease via Imbalanced OPA1 Processing. J. Hazard. Mater. 2023, 453, 131369. [Google Scholar] [CrossRef]
- Ball, N.; Teo, W.P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Goldman, S.M.; Tanner, C.M.; Oakes, D.; Bhudhikanok, G.S.; Gupta, A.; Langston, J.W. Head Injury and Parkinson’s Disease Risk in Twins. Ann. Neurol. 2006, 60, 65–72. [Google Scholar] [CrossRef]
- Yan, D.; Yang, Y.; Lang, J.; Wang, X.; Huang, Y.; Meng, J.; Wu, J.; Zeng, X.; Li, H.; Ma, H.; et al. SIRT1/FOXO3-Mediated Autophagy Signaling Involved in Manganese-Induced Neuroinflammation in Microglia. Ecotoxicol. Environ. Saf. 2023, 256, 114872. [Google Scholar] [CrossRef]
- Coon, S.; Stark, A.; Peterson, E.; Gloi, A.; Kortsha, G.; Pounds, J.; Chettle, D.; Gorell, J. Whole-Body Lifetime Occupational Lead Exposure and Risk of Parkinson’s Disease. Environ. Health Perspect. 2006, 114, 1872–1876. [Google Scholar] [CrossRef]
- Gorell, J.M.; Rybicki, B.A.; Johnson, C.C.; Peterson, E.L. Occupational Metal Exposures and the Risk of Parkinson’s Disease. Neuroepidemiology 1999, 18, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, J.; Leonard, S.S.; Rao, K.M.K. Cadmium Inhibits the Electron Transfer Chain and Induces Reactive Oxygen Species. Free Radic. Biol. Med. 2004, 36, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Cholanians, A.B.; Phan, A.V.; Ditzel, E.J.; Camenisch, T.D.; Lau, S.S.; Monks, T.J. Arsenic Induces Accumulation of α-Synuclein: Implications for Synucleinopathies and Neurodegeneration. Toxicol. Sci. 2016, 153, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and Mitochondria: Recent Advances and Current Views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the Mechanism of Pathogenesis of Parkinson’s Disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Romero, A.; Montpeyó, M.; Martinez-Vicente, M. The Emerging Role of the Lysosome in Parkinson’s Disease. Cells 2020, 9, 2399. [Google Scholar] [CrossRef]
- Thi Phuong Thao, D. Ubiquitin Carboxyl-Terminal Hydrolase L1 in Parkinson’s Disease. In Ubiquitin Proteasome System—Current Insights into Mechanism Cellular Regulation and Disease Relation; Matthew Summers, A., Ed.; IntechOpen: London, UK, 2019; pp. 1–12. [Google Scholar]
- Cookson, M.R. Parkinsonism Due to Mutations in PINK1, Parkin, and DJ-1 and Oxidative Stress and Mitochondrial Pathways. Cold Spring Harb. Perspect. Med. 2012, 2, a009415. [Google Scholar] [CrossRef]
- Pan, L.; Li, C.; Meng, L.; Tian, Y.; He, M.; Yuan, X.; Zhang, G.; Zhang, Z.; Xiong, J.; Chen, G.; et al. Tau Accelerates α-Synuclein Aggregation and Spreading in Parkinson’s Disease. Brain 2022, 145, 3454–3471. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef]
- Sarchione, A.; Marchand, A.; Taymans, J.M.; Chartier-Harlin, M.C. Alpha-Synuclein and Lipids: The Elephant in the Room? Cells 2021, 10, 2452. [Google Scholar] [CrossRef] [PubMed]
- Stojkovska, I.; Krainc, D.; Mazzulli, J.R. Molecular Mechanisms of α-Synuclein and GBA1 in Parkinson’s Disease. Cell Tisuue 2018, 373, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Klein, C. The Many Faces of Alpha-Synuclein Mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Kalomoiri, M.; Majb, N.; Xanthou, G.; El-agnaf, O.M.A. Phosphorylated Exogenous Alpha-Synuclein Fibrils Exacerbate Pathology and Induce Neuronal Dysfunction in Mice. Sci. Rep. 2017, 7, 16533. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Arawaka, S.; Hara, S.; Fukushima, S.; Koga, K.; Koyama, S.; Kato, T. Authentically Phosphorylated-Synuclein at Ser129 Accelerates Neurodegeneration in a Rat Model of Familial Parkinson’s Disease. J. Neurosci. 2011, 31, 16884–16894. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef]
- Qi, X.; Li, J.; Zhu, Y.; Yu, L.; Wang, P. Abnormal Modification of Alpha-Synuclein and Its Mechanism in Parkinson’s Disease. Chin. J. Tissue Eng. Res. 2024, 28, 1301–1306. [Google Scholar]
- Mondal, A.; Dolui, S.; Dhabal, S.; Kundu, S.; Das, L. Structure Specific Neuro-Toxicity of α-Synuclein Oligomer. Int. J. Biol. Macromol. 2023, 253, 126683. [Google Scholar] [CrossRef]
- Bisi, N.; Feni, L.; Peqini, K.; Pérez-peña, H.; Ongeri, S. α-Synuclein: An All-Inclusive Trip Around Its Structure, in Fl Uencing Factors and Applied Techniques. Front. Chem. 2021, 9, 666585. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-Synuclein Structure and Parkinson’s Disease—Lessons and Emerging Principles. Mol. Neurodegener. 2019, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Su, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms Underlying Extensive Ser129-Phosphorylation in α-Synuclein Aggregates. Acta Neuropathol. Commun. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Ara, J.; Uemura, N.; Lougee, M.G.; Meymand, E.S.; Zhang, B.; Petersson, E.J.; Trojanowski, J.Q.; Lee, V.M.Y. Alpha-Synuclein from Patient Lewy Bodies Exhibits Distinct Pathological Activity That Can Be Propagated In Vitro. Acta Neuropathol. Commun. 2021, 9, 188. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Chartier, S.; Duyckaerts, C. Is Lewy Pathology in the Human Nervous System Chiefly an Indicator of Neuronal Protection or of Toxicity? Cell Tissue Res. 2018, 373, 149–160. [Google Scholar] [CrossRef]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic Investigation of α-Synuclein Multiplication and Parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of Genetic Synucleinopathies with Parkinsonism: Review of the Literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-Synuclein Induces the Unfolded Protein Response in Parkinson’s Disease SNCA Triplication IPSC-Derived Neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Ito, S.; Matsui, H.; Onodera, O. Phosphorylation of α-Synuclein at T64 Results in Distinct Oligomers and Exerts Toxicity in Models of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2214652120. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein Phosphorylation in Neurodegeneration: Friend or Foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J. Da The Roles of Post-Translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C. Hierarchical Chemical Determination of Amyloid Polymorphs in Neurodegenerative Disease. Nat. Chem. Biol. 2021, 17, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C. Structural Diversity of Amyloid Fibrils and Advances in Their Structure Determination. Biochemistry 2020, 59, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Kai, J.; Meng, F.; Wang, C.; Min, H.; Jian, Y.; Zhang, N.; Gu, L.; Zhang, H. α-Synuclein Induced the Occurrence of RBD via Interaction with OX1R and Modulated Its Degradation. Neuromol. Med. 2023, 25, 286–300. [Google Scholar]
- Maor, G.; Dubreuil, R.R.; Feany, M.B. α-Synuclein Promotes Neuronal Dysfunction and Death by Disrupting the Binding of Ankyrin to β-Spectrin. J. Neurosci. 2023, 43, 1614–1626. [Google Scholar] [CrossRef]
- Ratan, Y.; Rajput, A.; Maleysm, S.; Pareek, A.; Jain, V.; Pareek, A.; Kaur, R.; Singh, G. An Insight into Cellular and Molecular Mechanisms Underlying the Pathogenesis of Neurodegeneration in Alzheimer’s Disease. Biomedicines 2023, 11, 1398. [Google Scholar] [CrossRef]
- Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.M.; Saad, M.; Simón-Sánchez, J.; Schulte, C.; Lesage, S.; Sveinbjörnsdóttir, S.; et al. Imputation of Sequence Variants for Identification of Genetic Risks for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet 2011, 377, 641–649. [Google Scholar]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein Aggregates in the Pathogenesis, Prognosis, and Therapeutics for Neurodegenerative Diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, T.; Credle, J.; Rodriguez, O.; Wills, J.; Oaks, A.W.; Masliah, E.; Sidhu, A. Hyperphosphorylated Tau in an α-Synuclein-Overexpressing Transgenic Model of Parkinson’s Disease. Eur. J. Neurosci. 2011, 33, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Jackson, G.R. Interactions between Tau and α-Synuclein Augment Neurotoxicity in a Drosophila Model of Parkinson’s Disease. Hum. Mol. Genet. 2014, 23, 3008–3023. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-Synuclein-Induced Mitochondrial Dysfunction Is Mediated via a Sirtuin 3-Dependent Pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s Disease-Associated Mutant VPS35 Causes Mitochondrial Dysfunction by Recycling DLP1 Complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Akther, M.; Azam, S.; Kim, I.-S.; Lin, Y.; Lee, Y.-H.; Choi, D.-K. Targeting Alpha-synuclein Aggregation and Its Role in Mitochondrial Dysfunction in Parkinson’s Diasease. Br. J. Pharmacol. 2021, 179, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Pupyshev, A.B.; Korolenko, T.A.; Akopyan, A.A.; Amstislavskaya, T.G.; Tikhonova, M.A. Suppression of Autophagy in the Brain of Transgenic Mice with Overexpression of A53T-Mutant α-Synuclein as an Early Event at Synucleinopathy Progression. Neurosci. Lett. 2018, 672, 140–144. [Google Scholar] [CrossRef]
- Zhang, Z.; Chu, S.F.; Wang, S.S.; Jiang, Y.N.; Gao, Y.; Yang, P.F.; Ai, Q.D.; Chen, N.H. RTP801 is a critical factor in the neurodegeneration process of A53T α-synuclein in a mouse model of Parkinson’s disease under chronic restraint stress. Br. J. Pharmacol. 2018, 175, 590–605. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome Impairment by α-Synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of α-Synuclein-Containing Erythrocyte-Derived Extracellular Vesicles across the Blood-Brain Barrier via Adsorptive Mediated Transcytosis: Another Mechanism for Initiation and Progression of Parkinson’s Disease? Acta Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef]
- Fang, F.; Yang, W.; Florio, J.B.; Rockenstein, E.; Spencer, B.; Orain, X.M.; Dong, S.X.; Li, H.; Chen, X.; Sung, K.; et al. Synuclein Impairs Trafficking and Signaling of BDNF in a Mouse Model of Parkinson’s Disease. Sci. Rep. 2017, 7, 3868. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB Neurotrophic Activities Are Blocked by α-Synuclein, Triggering Dopaminergic Cell Death in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Sekar, S.; Barathidasan, R.; Manivasagam, T.; Thenmozhi, A.J.; Sevanan, M.; Chidambaram, S.B.; Essa, M.M.; Guillemin, G.J.; Sakharkar, M.K. Naringenin Decreases α-Synuclein Expression and Neuroinflammation in MPTP-Induced Parkinson’s Disease Model in Mice. Neurotox. Res. 2018, 33, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E.A. ShRNA Targeting α-Synuclein Prevents Neurodegeneration in a Parkinson’s Disease Model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef] [PubMed]
- Drolet, R.E.; Sanders, J.M.; Kern, J.T. Leucine-Rich Repeat Kinase 2 (LRRK2) Cellular Biology: A Review of Recent Advances in Identifying Physiological Substrates and Cellular Functions. J. Neurogenet. 2011, 25, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.; Massano, J. An Updated Review of Parkinson’s Disease Genetics and Clinicopathological Correlations. Acta Neurol. Scand. 2017, 135, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Mills, R.D.; Mulhern, T.D.; Cheng, H.C.; Culvenor, J.G. Analysis of LRRK2 Accessory Repeat Domains: Prediction of Repeat Length, Number and Sites of Parkinson’s Disease Mutations. Biochem. Soc. Trans. 2012, 40, 1086–1089. [Google Scholar] [CrossRef]
- Cookson, M.R. The Role of Leucine-Rich Repeat Kinase 2 (LRRK2) in Parkinson’s Disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef]
- Correia Guedes, L.; Ferreira, J.J.; Rosa, M.M.; Coelho, M.; Bonifati, V.; Sampaio, C. Worldwide Frequency of G2019S LRRK2 Mutation in Parkinson’s Disease: A Systematic Review. Park. Relat. Disord. 2010, 16, 237–242. [Google Scholar] [CrossRef]
- Benamer, H.T.S.; De Silva, R. LRRK2 G2019S in the North African Population: A Review. Eur. Neurol. 2010, 63, 321–325. [Google Scholar] [CrossRef]
- Thaler, A.; Ash, E.; Gan-Or, Z.; Orr-Urtreger, A.; Giladi, N. The LRRK2 G2019S Mutation as the Cause of Parkinson’s Disease in Ashkenazi Jews. J. Neural Transm. 2009, 116, 1473–1482. [Google Scholar] [CrossRef]
- Hu, Z.X.; Peng, D.T.; Cai, M.; Pu, J.L.; Lei, X.G.; Yin, X.Z.; Ou-Yang, Z.Y.; Luo, W.; Zhang, B.R. A Study of Six Point Mutation Analysis of LRRK2 Gene in Chinese Mainland Patients with Parkinson’s Disease. Neurol. Sci. 2011, 32, 741–742. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Y.; Wu, Q.; Hong, H.; Zhou, H.; Chen, J.; Wang, H.; Xian, W.; Li, J.; Liu, Z.; et al. Confirmation of LRRK2 S1647T Variant as a Risk Factor for Parkinson’s Disease in Southern China. Eur. J. Neurol. 2011, 18, 538–540. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S.; Liu, Y.; Hong, H.; Wang, H.; Zheng, Y.; Zhou, H.; Chen, J.; Xian, W.; He, Y.; et al. LRRK2 R1398H Polymorphism Is Associated with Decreased Risk of Parkinson’s Disease in a Han Chinese Population. Park. Relat. Disord. 2011, 17, 291–292. [Google Scholar] [CrossRef]
- Zhou, Y.; Luo, X.; Li, F.; Tian, X.; Zhu, L.; Yang, Y.; Ren, Y.; Pang, H. Association of Parkinson’s Disease with Six Single Nucleotide Polymorphisms Located in Four PARK Genes in the Northern Han Chinese Population. J. Clin. Neurosci. 2012, 19, 1011–1015. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, J.Y.; Kim, H.J.; Kim, J.S.; Shin, E.S.; Cho, J.H.; Park, S.S.; Jeon, B.S. The LRRK2 G2385R Variant Is a Risk Factor for Sporadic Parkinson’s Disease in the Korean Population. Park. Relat. Disord. 2010, 16, 85–88. [Google Scholar] [CrossRef]
- Papapetropoulos, S.; Adi, N.; Shehadeh, L.; Bishopric, N.; Singer, C.; Argyriou, A.A.; Chroni, E. Is the G2019S LRRK2 Mutation Common in All Southern European Populations? J. Clin. Neurosci. 2008, 15, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic Proteomic Analysis of LRRK2-Mediated Rab GTPase Phosphorylation Establishes a Connection to Ciliogenesis. Elife 2017, 6, e31012. [Google Scholar] [CrossRef] [PubMed]
- Kang, U.-B.; Marto, J.A. Leucine-Rich Repeat Kinase 2 (LRRK2) and Parkinsons Disease. Adv. Neurobiol. 2017, 14, 1–23. [Google Scholar]
- Mills, R.D.; Mulhern, T.D.; Liu, F.; Culvenor, J.G.; Cheng, H.C. Prediction of the Repeat Domain Structures and Impact of Parkinsonism-Associated Variations on Structure and Function of All Functional Domains of Leucine-Rich Repeat Kinase 2 (LRRK2). Hum. Mutat. 2014, 35, 395–412. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Taymans, J.M.; Mutez, E.; Drouyer, M.; Sibran, W.; Chartier-Harlin, M.C. LRRK2 Detection in Human Biofluids: Potential Use as a Parkinson’s Disease Biomarker? Biochem. Soc. Trans. 2017, 45, 207–212. [Google Scholar] [CrossRef]
- Galper, J.; Kim, W.S.; Dzamko, N. LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease. Biomolecules 2022, 12, 1597. [Google Scholar] [CrossRef]
- Bouhouche, A.; Tibar, H.; Ben El Haj, R.; El Bayad, K.; Razine, R.; Tazrout, S.; Skalli, A.; Bouslam, N.; Elouardi, L.; Benomar, A.; et al. LRRK2 G2019S Mutation: Prevalence and Clinical Features in Moroccans with Parkinson’s Disease. Parkinsons. Dis. 2017, 2017, 2412486. [Google Scholar] [CrossRef]
- Zhang, X.; Kortholt, A. LRRK2 Structure-Based Activation Mechanism and Pathogenesis. Biomolecules 2023, 13, 612. [Google Scholar] [CrossRef]
- Watanabe, R.; Buschauer, R.; Boassa, D.; Taylor, S.; Villa, E.; Audagnotto, M.; Lasker, K.; Lu, T. The In Situ Structure of Parkinson’s Disease-Linked LRRK2 Ll. Cell 2020, 182, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.L.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019s-LRRK2 Expression Augments α-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.P.T.; Daniel, G.; Valdés, P.; Islam, M.S.; Schneider, B.L.; Moore, D.J. G2019S LRRK2 Enhances the Neuronal Transmission of Tau in the Mouse Brain. Hum. Mol. Genet. 2018, 27, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Mo, J.S.; Kim, M.Y.; Ann, E.J.; Ahn, J.S.; Jo, E.H.; Lee, H.J.; Lee, Y.C.; Seol, W.; Yarmoluk, S.M.; et al. LRRK2 Functions as a Scaffolding Kinase of ASK1-Mediated Neuronal Cell Death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-Rich Repeat Kinase 2 (LRRK2) Interacts with Parkin, and Mutant LRRK2 Induces Neuronal Degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef]
- Han, K.A.; Shin, W.H.; Jung, S.; Seol, W.; Seo, H.; Ko, C.; Chung, K.C. Leucine-Rich Repeat Kinase 2 Exacerbates Neuronal Cytotoxicity through Phosphorylation of Histone Deacetylase 3 and Histone Deacetylation. Hum. Mol. Genet. 2017, 26, 1–18. [Google Scholar] [CrossRef]
- Rassu, M.; Del Giudice, M.G.; Sanna, S.; Taymans, J.M.; Morari, M.; Brugnoli, A.; Frassineti, M.; Masala, A.; Esposito, S.; Galioto, M.; et al. Role of LRRK2 in the Regulation of Dopamine Receptor Trafficking. PLoS ONE 2017, 12, e0179082. [Google Scholar] [CrossRef]
- Pan, P.Y.; Li, X.; Wang, J.; Powell, J.; Wang, Q.; Zhang, Y.; Chen, Z.; Wicinski, B.; Hof, P.; Ryan, T.A.; et al. Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J. Neurosci. 2017, 37, 11366–11376. [Google Scholar] [CrossRef]
- Howlett, E.H.; Jensen, N.; Belmonte, F.; Zafar, F.; Hu, X.; Kluss, J.; Schüle, B.; Kaufman, B.A.; Greenamyre, J.T.; Sanders, L.H. LRRK2 G2019S-Induced Mitochondrial DNA Damage Is LRRK2 Kinase Dependent and Inhibition Restores MtDNA Integrity in Parkinson’s Disease. Hum. Mol. Genet. 2017, 26, 4340–4351. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 Regulates Mitochondrial Dynamics and Function through Direct Interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Mortiboys, H.; Aasly, J.; Bandmann, O. Ursocholanic Acid Rescues Mitochondrial Function in Common Forms of Familial Parkinson’s Disease. Brain 2013, 136, 3038–3050. [Google Scholar] [CrossRef]
- Rüb, C.; Wilkening, A.; Voos, W. Mitochondrial Quality Control by the PINK1/Parkin System. Cell Tissue Res. 2017, 367, 111–123. [Google Scholar] [CrossRef]
- Bekris, L.M.; Mata, I.F.; Zabetian, C.P. The Genetics of Parkinson Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guardia-Laguarta, C.; Yin, J.; Erdjument-Bromage, H.; Martin, B.; James, M.; Jiang, X.; Przedborski, S. The Ubiquitination of PINK1 Is Restricted to Its Mature 52-KDa Form. Cell Rep. 2017, 20, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.J.; Yoo, H.; Woo, D.; Lee, D.H.; Park, K.; Chung, J. PINK1 and Parkin Regulate IP 3 R-Mediated ER Calcium Release. Nat. Commun. 2023, 14, 5202. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Muqit, M.M.K. PTEN-Induced Kinase 1 (PINK1) and Parkin: Unlocking a Mitochondrial Quality Control Pathway Linked to Parkinson’s Disease. Curr. Opin. Neurobiol. 2022, 72, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Mcwilliams, T.G.; Muqit, M.M.K. PINK1 and Parkin: Emerging Themes in Mitochondrial Homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-Mediated Phosphorylation of LETM1 Regulates Mitochondrial Calcium Transport and Protects Neurons against Mitochondrial Stress. Nat. Commun. 2017, 8, 1399. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. Elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S Increases Risk of Parkinson’s Disease via a Dominant-Negative Mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the Mitochondrial Unfolded Protein Response Promotes Longevity and Dopamine Neuron Survival in Parkinson’s Disease Models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef]
- Ando, M.; Fiesel, F.C.; Hudec, R.; Caulfield, T.R.; Ogaki, K.; Górka-Skoczylas, P.; Koziorowski, D.; Friedman, A.; Chen, L.; Dawson, V.L.; et al. The PINK1 p.I368N Mutation Affects Protein Stability and Ubiquitin Kinase Activity. Mol. Neurodegener. 2017, 12, 32. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, N.; Li, L.; Chen, S.; Wang, T. PINK1-Dependent Phosphorylation of PINK1 and Parkin Is Essential for Mitochondrial Quality Control. Cell Death Dis. 2016, 7, e2501. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tamjar, J.; Waddell, A.D.; Woodroof, H.I.; Raimi, O.G.; Shaw, A.M.; Peggie, M.; Muqit, M.M.K.; van Aalten, D.M.F. Structure of PINK1 and Mechanisms of Parkinson’s Disease-Associated Mutations. Elife 2017, 6, e29985. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, J.; Jana, A.; Ghosh, E.; Banerjee, T.K.; Chakraborty, D.P.; Rao, V.R. Evaluation of PARKIN Gene Variants in West Bengal Parkinson’s Disease Patients. J. Hum. Genet. 2015, 60, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin-Induced Mitophagy in the Pathogenesis of Parkinson Disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive Oxygen Species Trigger Parkin/PINK1 Pathway–Dependent Mitophagy by Inducing Mitochondrial Recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef]
- Kathrin Lutz, A.; Exner, N.; Fett, M.E.; Schleke, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of Parkin or PINK1 Function Increases Drp1-Dependent Mitochondrial Fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef]
- Lee, Y.; Stevens, D.A.; Kang, S.; Dawson, V.L.; Shin, J.; Dawson, T.M. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. CellReports 2017, 18, 918–932. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. DJ-1 (PARK7), a Novel Gene for Autosomal Recessive, Early Onset Parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent Findings on the Physiological Function of DJ-1: Beyond Parkinson’s Disease. Neurobiol. Dis. 2017, 108, 65–72. [Google Scholar] [CrossRef]
- Malgieri, G.; Eliezer, D. Structural Effects of Parkinson’s Disease Linked DJ-1 Mutations. Protein Sci. 2008, 17, 855–868. [Google Scholar] [CrossRef]
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhães, M. DJ-1 Linked Parkinsonism (PARK7) is Associated with Lewy Body Pathology. Brain 2016, 139, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xiong, H.; Sun, P.; Zhang, Y.; Wang, D.; Hu, Z.; Zhu, Z.; Ma, H.; Pan, Q.; Xia, J.H.; et al. Association of PINK1 and DJ-1 Confers Digenic Inheritance of Early-Onset Parkinson’s Disease. Hum. Mol. Genet. 2006, 15, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Patel, S.H.; Han, S. DJ-1, a Parkinson’s Disease Related Protein, Aggregates under Denaturing Conditions and Co-Aggregates with α-Synuclein through Hydrophobic Interaction. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1759–1769. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Gonçalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 Interactions with a-Synuclein Attenuate Aggregation and Cellular Toxicity in Models of Parkinson’s Disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Li, Y.; Wei, Q.; Lin, R.; Kang, R.; Ruan, Y.; Lin, Z. Nuclear DJ-1 Regulates DNA Damage Repair via the Regulation of PARP1 Activity. Int. J. Mol. Sci. 2023, 24, 8651. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, S.; Albanese, A.; Moore, D.J. VPS35-Based Approach: A Potential Innovative Treatment in Parkinson’s Disease. Front. Neurol. 2019, 10, 1272. [Google Scholar] [CrossRef]
- Mukherjee, U.A.; Ong, S.B.; Ong, S.G.; Hausenloy, D.J. Parkinson’s Disease Proteins: Novel Mitochondrial Targets for Cardioprotection. Pharmacol. Ther. 2015, 156, 34–43. [Google Scholar] [CrossRef]
- Ishikawa, S.; Taira, T.; Niki, T.; Takahashi-Niki, K.; Maita, C.; Maita, H.; Ariga, H.; Iguchi-Ariga, S.M.M. Oxidative Status of DJ-1-Dependent Activation of Dopamine Synthesis through Interaction of Tyrosine Hydroxylase and 4-Dihydroxy-L-Phenylalanine (L-DOPA) Decarboxylase with DJ-1. J. Biol. Chem. 2009, 284, 28832–28844. [Google Scholar] [CrossRef]
- Im, J.Y.; Lee, K.W.; Woo, J.M.; Junn, E.; Mouradian, M.M. Dj-1 Induces Thioredoxin 1 Expression through the Nrf2 Pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef]
- Giaime, E.; Sunyach, C.; Druon, C.; Scarzello, S.; Robert, G.; Grosso, S.; Auberger, P.; Goldberg, M.S.; Shen, J.; Heutink, P.; et al. Loss of Function of DJ-1 Triggered by Parkinson’s Disease-Associated Mutation Is Due to Proteolytic Resistance to Caspase-6. Cell Death Differ. 2010, 17, 158–169. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Giasson, B.I. L10p and P158DEL DJ-1 Mutations Cause Protein Instability, Aggregation, and Dimerization Impairments. J. Neurosci. Res. 2010, 88, 3111–3124. [Google Scholar] [CrossRef] [PubMed]
- Batelli, S.; Invernizzi, R.W.; Negro, A.; Calcagno, E.; Rodilossi, S.; Forloni, G.; Albani, D. The Parkinson’s Disease-Related Protein Dj-1 Protects Dopaminergic Neurons in Vivo and Cultured Cells from Alpha-Synuclein and 6-Hydroxydopamine Toxicity. Neurodegener. Dis. 2015, 15, 13–23. [Google Scholar] [CrossRef]
- Vidyadhara, D.J.; Lee, J.E.; Chandra, S.S. Role of the Endolysosomal System in Parkinson’s Disease. J. Neurochem. 2019, 150, 487–506. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P.J. Retromer: A Master Conductor of Endosome Sorting. Cold Spring Harb. Perspect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.E.; Healy, M.D.; Collins, B.M. Towards a Molecular Understanding of Endosomal Trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef]
- Kovtun, O.; Leneva, N.; Bykov, Y.S.; Ariotti, N.; Teasdale, R.D.; Schaffer, M.; Engel, B.D.; Owen, D.J.; Briggs, J.A.G.; Collins, B.M. Structure of the Membrane-Assembled Retromer Coat Determined by Cryo-Electron Tomography. Nature 2018, 561, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Gershlick, D.C.; Vidaurrazaga, A.; Rojas, A.L.; Bonifacino, J.S.; Hierro, A. Structural Mechanism for Cargo Recognition by the Retromer Complex. Cell 2016, 167, 1623–1635. [Google Scholar] [CrossRef]
- Brodin, L.; Shupliakov, O. Retromer in Synaptic Function and Pathology. Front. Synaptic Neurosci. 2018, 10, 37. [Google Scholar] [CrossRef]
- Kendall, A.K.; Xie, B.; Xu, P.; Wang, J.; Burcham, R.; Frazier, M.N.; Binshtein, E.; Wei, H.; Graham, T.R.; Nakagawa, T.; et al. Mammalian Retromer Is an Adaptable Scaffold for Cargo Sorting from Endosomes. Structure 2020, 28, 393–405. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chang, Y.Y.; Lan, M.Y.; Chen, P.L.; Lin, C.H. Identification of VPS35 p.D620N Mutation-Related Parkinson’s Disease in a Taiwanese Family with Successful Bilateral Subthalamic Nucleus Deep Brain Stimulation: A Case Report and Literature Review. BMC Neurol. 2017, 17, 191. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, W.; Hoppel, C.; Liu, J.; Zhu, X. Parkinson’s Disease-Associated Pathogenic VPS35 Mutation Causes Complex I Deficits. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Follett, J.; Bugarcic, A.; Yang, Z.; Ariotti, N.; Norwood, S.J.; Collins, B.M.; Parton, R.G.; Teasdale, R.D. Parkinson Disease-Linked VPS35 R524W Mutation Impairs the Endosomal Association of Retromer and Induces α-Synuclein Aggregation. J. Biol. Chem. 2016, 291, 18283–18298. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Toh, J.; Ho, P.; Tio, M.; Zhao, Y.; Tan, E.K. In Vivo Evidence of Pathogenicity of VPS35 Mutations in the Drosophila. Mol. Brain 2014, 7, 73. [Google Scholar] [CrossRef][Green Version]
- Bae, E.J.; Yang, N.Y.; Lee, C.; Lee, H.J.; Kim, S.; Sardi, S.P.; Lee, S.J. Loss of Glucocerebrosidase 1 Activity Causes Lysosomal Dysfunction and α-Synuclein Aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and Its Relevance to Parkinson Disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T. Aroldo Rossi Lysosomal Hydrolases in Cerebrospinal Fluid from Subjects with Parkinson’s Disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase Deficiency in Substantia Nigra of Parkinson Disease Brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced Glucocerebrosidase Is Associated with Increased α-Synuclein in Sporadic Parkinson’s Disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Granek, Z.; Barczuk, J.; Siwecka, N. GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance. Int. J. Mol. Sci. 2023, 24, 2044. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive Decline of Glucocerebrosidase in Aging and Parkinson’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal Fluid β-Glucocerebrosidase Activity Is Reduced in Parkinson’s Disease Patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H.V. A Clinical and Family History Study of Parkinson’s Disease in Heterozygous Glucocerebrosidase Mutation Carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 Deficiency Negatively Affects Physiological α-Synuclein Tetramers and Related Multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef]
- García-Sanz, P.; Orgaz, L.; Fuentes, J.M.; Vicario, C.; Moratalla, R. Cholesterol and Multilamellar Bodies: Lysosomal Dysfunction in GBA-Parkinson Disease. Autophagy 2018, 14, 717–718. [Google Scholar] [CrossRef]
- García-Sanz, P.; Orgaz, L.; Bueno-Gil, G.; Espadas, I.; Rodríguez-Traver, E.; Kulisevsky, J.; Gutierrez, A.; Dávila, J.C.; González-Polo, R.A.; Fuentes, J.M.; et al. N370S-GBA1 Mutation Causes Lysosomal Cholesterol Accumulation in Parkinson’s Disease. Mov. Disord. 2017, 32, 1409–1422. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Rusconi, R.; Deangeli, G.; Di Monte, D.A.; Spillantini, M.G.; Schapira, A.H.V. The L444P GBA1 Mutation Enhances Alpha-Synuclein Induced Loss of Nigral Dopaminergic Neurons in Mice. Brain 2017, 140, 2706–2721. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular Chaperone Dysfunction in Neurodegenerative Diseases and Effects of Curcumin. Biomed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Flint Beal, M. Parkinson’s Disease. Hum. Mol. Genet. 2007, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Delenclos, M.; Baine, A.M.T.; De Ture, M.; Murray, M.E.; Dickson, D.W.; McLean, P.J. Transmission of Soluble and Insoluble α-Synuclein to Mice. J. Neuropathol. Exp. Neurol. 2015, 74, 1158–1169. [Google Scholar] [PubMed]
- Alenina, N.; Klempin, F. The Role of Serotonin in Adult Hippocampal Neurogenesis. Behav. Brain Res. 2014, 277, 49–57. [Google Scholar] [CrossRef]
- Kohl, Z.; Ben Abdallah, N.; Vogelgsang, J.; Tischer, L.; Deusser, J.; Amato, D.; Anderson, S.; Müller, C.P.; Riess, O.; Masliah, E.; et al. Severely Impaired Hippocampal Neurogenesis Associates with an Early Serotonergic Deficit in a BAC α-Synuclein Transgenic Rat Model of Parkinson’s Disease. Neurobiol. Dis. 2016, 85, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Motaln, H.; Rogelj, B. The Role of C-Abl Tyrosine Kinase in Brain and Its Pathologies. Cells 2023, 12, 2041. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, S.; Ge, P.; Lee, S.H.; Kim, D.; Karuppagounder, S.S.; Kumar, M.; Mao, X.; Shin, J.H.; Lee, Y.; Pletnikova, O.; et al. Activation of Tyrosine Kinase C-Abl Contributes to α-Synuclein-Induced Neurodegeneration. J. Clin. Investig. 2016, 126, 2970–2988. [Google Scholar] [CrossRef]
- Wu, W.; Sung, C.C.; Yu, P.; Li, J.; Chung, K.K.K. S-Nitrosylation of G Protein-Coupled Receptor Kinase 6 and Casein Kinase 2 Alpha Modulates Their Kinase Activity toward Alpha-Synuclein Phosphorylation in an Animal Model of Parkinson’s Disease. PLoS ONE 2020, 15, e0232019. [Google Scholar]
- Bergeron, M.; Motter, R.; Tanaka, P.; Fauss, D.; Babcock, M.; Chiou, S.S.; Nelson, S.; San Pablo, F.; Anderson, J.P. In Vivo Modulation of Polo-like Kinases Supports a Key Role for PLK2 in Ser129 α-Synuclein Phosphorylation in Mouse Brain. Neuroscience 2014, 256, 72–82. [Google Scholar] [CrossRef]
- Sano, K.; Iwasaki, Y.; Yamashita, Y.; Irie, K.; Hosokawa, M.; Satoh, K.; Mishima, K. Tyrosine 136 Phosphorylation of α-Synuclein Aggregates in the Lewy Body Dementia Brain: Involvement of Serine 129 Phosphorylation by Casein Kinase 2. Acta Neuropathol. Commun. 2021, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and α-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- van der Vlag, M.; Havekes, R.; Heckman, P.R.A. The Contribution of Parkin PINK1 and DJ-1 Genes to Selective Neuronal Degeneration in Parkinson’s Disease. Eur. J. Neurosci. 2019, 52, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S. Di DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef]
- Zhou, J.; Broe, M.; Huang, Y.; Anderson, J.P.; Gai, W.P.; Milward, E.A.; Porritt, M.; Howells, D.; Hughes, A.J.; Wang, X.; et al. Changes in the Solubility and Phosphorylation of α-Synuclein over the Course of Parkinson’s Disease. Acta Neuropathol. 2011, 121, 695–704. [Google Scholar] [CrossRef]
- Pacheco, C.R.; Morales, C.N.; Ramírez, A.E.; Muñoz, F.J.; Gallegos, S.S.; Caviedes, P.A.; Aguayo, L.G.; Opazo, C.M. Extracellular α-Synuclein Alters Synaptic Transmission in Brain Neurons by Perforating the Neuronal Plasma Membrane. J. Neurochem. 2015, 132, 731–741. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal Cell-to-Cell Transmission of Alpha Synuclein Oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Li, X.; Yang, W.; Yu, S. Blood Plasma of Patients with Parkinson’s Disease Increases Alpha-Synuclein Aggregation and Neurotoxicity. Parkinsons. Dis. 2016, 2016, 7596482. [Google Scholar] [CrossRef]
- Lichtenberg, M.; Mansilla, A.; Zecchini, V.R.; Fleming, A.; Rubinsztein, D.C. The Parkinson’s Disease Protein LRRK2 Impairs Proteasome Substrate Clearance without Affecting Proteasome Catalytic Activity. Cell Death Dis. 2011, 2, e196. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.; Kim, K.S.; Seol, W.; Choi, H.J. LRRK2 Interferes with Aggresome Formation for Autophagic Clearance. Mol. Cell. Neurosci. 2016, 75, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, T.; Thomas, J.M.; Pei, Z.; Jiang, H.; Engelender, S.; Ross, C.A.; Smith, W.W. Synphilin-1 Attenuates Mutant LRRK2-Induced Neurodegeneration in Parkinson’s Disease Models. Hum. Mol. Genet. 2016, 25, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-Induced ER Stress in Mouse Dopaminergic Neurons Involves Downregulation of TRPC1 and Inhibition of AKT/MTOR Signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Cellular Models of Parkinson’s Disease. J. Neurosci. 2002, 22, 10690–10698. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic Reticulum Stress is Important for the Manifestations of α-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Fouillet, A.; Levet, C.; Virgone, A.; Robin, M.; Dourlen, P.; Rieusset, J.; Belaidi, E.; Ovize, M.; Touret, M.; Nataf, S.; et al. ER Stress Inhibits Neuronal Death by Promoting Autophagy. Autophagy 2012, 8, 915–926. [Google Scholar] [CrossRef]
- Nakka, V.P.; Gusain, A.; Raghubir, R. Endoplasmic Reticulum Stress Plays Critical Role in Brain Damage after Cerebral Ischemia/Reperfusion in Rats. Neurotox. Res. 2010, 17, 189–202. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Düssmann, H.; Prehn, J.H.M. ER Stress Signaling Has an Activating Transcription Factor 6 (ATF6)-Dependent “off-Switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Delgado, L.; Labrador-Espinosa, M.Á.; Macías-García, D.; Jesús, S.; Benítez Zamora, B.; Fernández-Rodríguez, P.; Adarmes-Gómez, A.D.; Reina Castillo, M.I.; Castro-Labrador, S.; Silva-Rodríguez, J.; et al. Peripheral Inflammation Is Associated with Dopaminergic Degeneration in Parkinson’s Disease. Mov. Disord. 2023, 38, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, Y.; Wang, X.; Li, J.Y.; Zhang, C.; Yang, Y.; Zhang, J. The Cervical Lymph Node Contributes to Peripheral Inflammation Related to Parkinson’s Disease. J. Neuroinflamm. 2023, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of Dopaminergic Neuron Survival by the Unfolded Protein Response Transcription Factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-Mediated ER Stress Triggers Neurodegeneration in PINK1/Parkin Models of Parkinson’s Disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.B.; Jeon, B.S. Protective Role of Heat Shock and Heat Shock Protein 70 in Lactacystin-Induced Cell Death Both in the Rat Substantia Nigra and PC12 Cells. Brain Res. 2006, 1087, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wolfer, D.P.; Lipp, H.P.; Büeler, H. Hsp70 Gene Transfer by Adeno-Associated Virusi Inhibits MPTP-Induced Nigrostriatal Degeneration in the Mouse Model of Parkinson Disease. Mol. Ther. 2005, 11, 80–88. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose Regulated Protein 78 Diminishes α-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef]
- Scheper, W.; Hoozemans, J.J.M. The Unfolded Protein Response in Neurodegenerative Diseases: A Neuropathological Perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef]
- Cóppola-Segovia, V.; Cavarsan, C.; Maia, F.G.; Ferraz, A.C.; Nakao, L.S.; Lima, M.M.; Zanata, S.M. ER Stress Induced by Tunicamycin Triggers α-Synuclein Oligomerization, Dopaminergic Neurons Death and Locomotor Impairment: A New Model of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 5798–5806. [Google Scholar] [CrossRef]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium Homeostasis and ER Stress in Control of Autophagy in Cancer Cells. Biomed Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [PubMed]
- Rcom-H’cheo-Gauthier, A.; Goodwin, J.; Pountney, D.L. Interactions between Calcium and Alpha-Synuclein in Neurodegeneration. Biomolecules 2014, 4, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Sehrawat, A.; Mastana, S.S.; Kandimalla, R.; Sharma, P.K.; Bhatti, G.K.; Bhatti, J.S. Targeting Calcium Homeostasis and Impaired Inter-Organelle Crosstalk as a Potential Therapeutic Approach in Parkinson’s Disease. Life Sci. 2023, 330, 121995. [Google Scholar] [CrossRef] [PubMed]
- Zaichick, S.V.; Mcgrath, K.M.; Caraveo, G. The Role of Ca2+ Signaling in Parkinson’s Disease. Dis. Model. Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin Determines Toxic versus Beneficial Responses to α-Synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, 3544–3552. [Google Scholar] [CrossRef]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s Disease Is Associated with Altered Expression of Ca V1 Channels and Calcium-Binding Proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [PubMed]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-Terminal Calcium Binding of α-Synuclein Modulates Synaptic Vesicle Interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Minakaki, G.; Keramioti, M.V.; Xylaki, M.; Balafas, E.; Chrysanthou-piterou, M.; Kloukina, I. GABA Transmission via ATP-Dependent K+ Channels Regulates Alpha-Synuclein Secretion in Mouse Striatum. Brain 2016, 139, 871–890. [Google Scholar] [CrossRef]
- Lieberman, O.J.; Choi, S.J.; Kanter, E.; Saverchenko, A.; Micah, D.; Fiore, G.M.; Wu, M.; Kondapalli, J.; Zampese, E.; Surmeier, D.J. Alpha-Synuclein-Dependent Calcium Entry Underlies Differential Sensitivity of Cultured SN and VTA Dopaminergic Neurons to a Parkinsonian Neurotoxin. eNeuro 2017, 4, 1–22. [Google Scholar] [CrossRef]
- Hotka, M.; Cagalinec, M.; Hilber, K.; Hool, L.; Boehm, S.; Kubista, H. L-Type Ca2+ Channel—Mediated Ca2+ Influx Adjusts Neuronal Mitochondrial Function to Physiological and Pathophysiological Conditions. Sci. Signal. 2020, 13, eaaw6923. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Magalhaes, J.; Beavan, M.S.; Mcneill, A.; Gegg, M.E.; Cleeter, M.W.J.; Bloor-young, D.; Churchill, G.C.; Duchen, M.R.; Schapira, A.H.; et al. Cell Calcium Endoplasmic Reticulum and Lysosomal Ca2+ Stores Are Remodelled in GBA1-Linked Parkinson Disease Patient Fibroblasts. Cell Calcium 2016, 59, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired Dopamine Metabolism in Parkinson’s Disease Pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, M.F.; Den Ouden, H.E.M.; Aarts, E.; Timmer, M.H.M.; Bloem, B.R.; Toni, I.; Helmich, R.C. Dopamine Controls Parkinson’s Tremor by Inhibiting the Cerebellar Thalamus. Brain 2017, 140, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine Oxidation Mediates Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Masoud, S.T.; Vecchio, L.M.; Bergeron, Y.; Hossain, M.M.; Nguyen, L.T.; Bermejo, M.K.; Kile, B.; Sotnikova, T.D.; Siesser, W.B.; Gainetdinov, R.R.; et al. Neurobiology of Disease Increased Expression of the Dopamine Transporter Leads to Loss of Dopamine Neurons, Oxidative Stress and L-DOPA Reversible Motor Deficits. Neurobiol. Dis. 2015, 74, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ding, Y.; Cagniard, B.; Van Laar, A.D.; Mortimer, A.; Chi, W.; Hastings, T.G.; Kang, U.J.; Zhuang, X. Unregulated Cytosolic Dopamine Causes Neurodegeneration Associated with Oxidative Stress in Mice. Neurobiol. Dis. 2008, 28, 425–433. [Google Scholar] [CrossRef]
- Herrera, A.; Muñoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Kerk, S.Y.; Xiong, G.G.; Lim, T.M. Dopamine Auto-Oxidation Aggravates Non-Apoptotic Cell Death Induced by over-Expression of Human A53T Mutant Alpha-Synuclein in Dopaminergic PC12 Cells. J. Neurochem. 2009, 108, 601–610. [Google Scholar] [CrossRef]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine Induces Soluble α-Synuclein Oligomers and Nigrostriatal Degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef]
- Panneton, W.M.; Kumar, V.B.; Gan, Q.; Burke, W.J.; Galvin, J.E. The Neurotoxicity of DOPAL: Behavioral and Stereological Evidence for Its Role in Parkinson Disease Pathogenesis. PLoS ONE 2010, 5, e15251. [Google Scholar] [CrossRef]
- Vanle, B.C.; Florang, V.R.; Murry, D.J.; Aguirre, A.L.; Doorn, J.A. Inactivation of Glyceraldehyde-3-Phosphate Dehydrogenase by the Dopamine Metabolite, 3,4-Dihydroxyphenylacetaldehyde. Biochem. Biophys. Res. Commun. 2017, 492, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Vaarmann, A.; Yao, Z.; Duchen, M.R.; Wood, N.W.; Abramov, A.Y. Dopamine Induced Neurodegeneration in a PINK1 Model of Parkinson’s Disease. PLoS ONE 2012, 7, e37564. [Google Scholar] [CrossRef]
- Su, P.; Liu, F. A Peptide Disrupting the D2R-DAT Interaction Protects against Dopamine Neurotoxicity. Exp. Neurol. 2017, 295, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on Experimental Methods to Assess Mitochondrial Dysfunction in Cellular Models of Neurodegenerative Diseases. Cell Death Differ. 2017, 25, 542–572. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Yang, T.; Gu, Y.; Sun, X.-H. Mitochondrial Dysfunction in Parkinson’s Disease: From Mechanistic Insights to Therapy. Front. Aging Neurosci. 2022, 14, 885500. [Google Scholar] [CrossRef]
- Li, J.L.; Lin, T.Y.; Chen, P.L.; Guo, T.N.; Huang, S.Y.; Chen, C.H.; Lin, C.H.; Chan, C.C. Mitochondrial Function and Parkinson’s Disease: From the Perspective of the Electron Transport Chain. Front. Mol. Neurosci. 2021, 14, 797833. [Google Scholar] [CrossRef]
- Norberg, E.; Orrenius, S.; Zhivotovsky, B. Mitochondrial Regulation of Cell Death: Processing of Apoptosis-Inducing Factor (AIF). Biochem. Biophys. Res. Commun. 2010, 396, 95–100. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, Prefibrillar α-Synuclein Oligomers Promote Complex I-Dependent, Ca2+-Induced Mitochondrial Dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Zhang, S.; Eitan, E.; Wu, T.Y.; Mattson, M.P. Intercellular Transfer of Pathogenic α-Synuclein by Extracellular Vesicles Is Induced by the Lipid Peroxidation Product 4-Hydroxynonenal. Neurobiol. Aging 2018, 61, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.; Seong, Y.; Heon, G.; Yoo, S. HtrA2 Regulates α-Synuclein-Mediated Mitochondrial Reactive Oxygen Species Production in the Mitochondria of Microglia. Biochem. Biophys. Res. Commun. 2023, 638, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Islam, M.S.; Rahman, M.M.; Hong, S.T. Neuroprotective Function of Omi to α-Synuclein-Induced Neurotoxicity. Neurobiol. Dis. 2020, 136, 104706. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.J.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase Inhibition Causes Mitochondrial Dysfunction and Free Radical Damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Storch, A.; Ludolph, A.C.; Schwarz, J. Dopamine Transporter: Involvement in Selective Dopaminergic Neurotoxicity and Degeneration. J. Neural Transm. 2004, 111, 1267–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khaldy, H.; Escames, G.; León, J.; Bikjdaouene, L.; Acuña-Castroviejo, D. Synergistic Effects of Melatonin and Deprenyl against MPTP-Induced Mitochondrial Damage and DA Depletion. Neurobiol. Aging 2003, 24, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Yana, M.H.; Wang, X.; Zhu, X. Mitochondrial Defects and Oxidative Stress in Alzheimer Disease and Parkinson Disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Sterky, F.H.; Hoffman, A.F.; Milenkovic, D.; Bao, B.; Paganelli, A.; Edgar, D.; Wibom, R.; Lupica, C.R.; Olson, L.; Larsson, N.G. Altered Dopamine Metabolism and Increased Vulnerability to MPTP in Mice with Partial Deficiency of Mitochondrial Complex I in Dopamine Neurons. Hum. Mol. Genet. 2012, 21, 1078–1089. [Google Scholar] [CrossRef]
- Lee, S.; Oh, S.T.; Jeong, H.J.; Pak, S.C.; Park, H.J.; Kim, J.; Cho, H.S.; Jeon, S. MPTP-Induced Vulnerability of Dopamine Neurons in A53T α-Synuclein Overexpressed Mice with the Potential Involvement of DJ-1 Downregulation. Korean J. Physiol. Pharmacol. 2017, 21, 625–632. [Google Scholar] [CrossRef]
- Liu, S.; Cui, B.; Dai, Z.; Shi, P.; Wang, Z.; Gu, Y. Long Non-Coding RNA HOTAIR Promotes Parkinsons Disease Induced by MPTP through up-Regulating the Expression of LRRK2. Curr. Neurovasc. Res. 2016, 13, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple Pathways of Cytochrome c Release from Mitochondria in Apoptosis. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dikshit, M. Apoptotic Neuronal Death in Parkinson’s Disease: Involvement of Nitric Oxide. Brain Res. Rev. 2007, 54, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Zhao, Y.; Skipper, L.; Tan, M.G.; Di Fonzo, A.; Sun, L.; Fook-Chong, S.; Tang, S.; Chua, E.; Yuen, Y.; et al. The LRRK2 Gly2385Arg Variant Is Associated with Parkinson’s Disease: Genetic and Functional Evidence. Hum. Genet. 2007, 120, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Wu, Y.R.; Lee, M.C.; Funayama, M.; Chen, M.L.; Soto, A.I.; Mata, I.F.; Lee-Chen, G.J.; Chiung, M.C.; Tang, M.; et al. Analysis of LRRK2 R1628P as a Risk Factor for Parkinson’s Disease. Ann. Neurol. 2008, 64, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Chang, L.; Hou, L.; Wang, Z.; Li, D.; Ren, Z.; Gu, T.; Wang, J.; Chen, G. Serum Metabolomics Analysis Revealed Metabolic Disorders in Parkinson’s Disease. Medicine 2023, 102, e33715. [Google Scholar] [CrossRef] [PubMed]
- Jacquemyn, J.; Kuenen, S.; Swerts, J.; Pavie, B.; Vijayan, V.; Kilic, A.; Chabot, D. Parkinsonism Mutations in DNAJC6 Cause Lipid Defects and Neurodegeneration That Are Rescued by Synj1. NPJ Park. Dis. 2023, 9, 19. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative Stress and Parkinson’s Disease. Handb. Clin. Neurol. 2007, 83, 507–520. [Google Scholar]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Lotharius, J.; Brundin, P. Impaired Dopamine Storage Resulting from α-Synuclein Mutations May Contribute to the Pathogenesis of Parkinson’s Disease. Hum. Mol. Genet. 2002, 11, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Ryan, S.D. Nitrosative Stress in Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.H.K.; Chung, K.K.K. Oxidative and Nitrosative Stress in Parkinson’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Neuropathology, Biochemistry, and Biophysics of α-Synuclein Aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-Nitrosylation of Parkin Regulates Ubiquitination and Compromises Parkin’s Protective Function. Science 2004, 304, 1328–1331. [Google Scholar] [CrossRef]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative Stress Factors in Parkinson’s Disease. Neural Regen. Res. 2021, 16, 1383–1391. [Google Scholar] [CrossRef]
- Tompkins, M.M.; Basgall, E.J.; Zamrini, E.; Hill, W.D. Apoptotic-like Changes in Lewy-Body-Associated Disorders and Normal Aging in Substantia Nigral Neurons. Am. J. Pathol. 1997, 150, 119–131. [Google Scholar]
- Gao, K.; Liu, M.; Cao, J.; Yao, M.; Lu, Y.; Li, J.; Zhu, X.; Yang, Z.; Wen, A. Protective Effects of Lycium Barbarum Polysaccharide on 6-OHDA-Induced Apoptosis in PC12 Cells through the ROS-NO Pathway. Molecules 2015, 20, 293–308. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Singapore, 2018; pp. 3–26. [Google Scholar]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase Activities and Tumor Necrosis Factor Receptor R1 (P55) Level Are Elevated in the Substantia Nigra from Parkinsonian Brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A Vulnerability Factor and Final Effector in Apoptotic Death of Dopaminergic Neurons in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef]
- Viswanath, V.; Wu, Y.; Boonplueang, R.; Chen, S.; Stevenson, F.F.; Yantiri, F.; Yang, L.; Flint Beal, M.; Andersen, J.K. Caspase-9 Activation Results in Downstream Caspase-8 Activation and Bid Cleavage in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinson’s Disease. J. Neurosci. 2001, 21, 9519–9528. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A. Caspases: Key Mediators of Apoptosis. Chem. Biol. 1998, 5, R97–R103. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W. Caspase Structure, Proteolytic Substrates, and Function during Apoptotic Cell Death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The Biochemistry of Apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) Apoptosis Systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, G.; Starkov, A.; Polster, B.M.; Chinopoulos, C. Mitochondrial Mechanisms of Neural Cell Death and Neuroprotective Interventions in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2003, 991, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; Van Gurp, M.; Van Loo, G.; Vandenabeele, P. Toxic Proteins Released from Mitochondria in Cell Death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, Q.; Chen, R.; Gan, Z.; Huang, X.; Wang, P.; Cao, X.; Zhao, N.; Yang, Z.; Yan, J. Iron-Inhibited Autophagy via Transcription Factor ZFP27 in Parkinson’s Disease. J. Cell. Mol. Med. 2023, 22, 3614–3627. [Google Scholar] [CrossRef]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial Association of Alpha-Synuclein Causes Oxidative Stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef]
- Martínez-Fábregas, J.; Díaz-Moreno, I.; González-Arzola, K.; Janochas, S.; Navarro, J.A.; Hervás, M.; Bernhardt, R.; Velázquez-Campoy, A.; Díaz-Quintana, A.; De La Rosa, M.A. Structural and Functional Analysis of Novel Human Cytochrome c Targets in Apoptosis. Mol. Cell. Proteom. 2014, 13, 1439–1456. [Google Scholar] [CrossRef]
- Greenlund, L.J.S.; Korsmeyer, S.J.; Johnson, E.M. Role of BCL-2 in the Survival and Function of Developing and Mature Sympathetic Neurons. Neuron 1995, 15, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kameda, M.; Yasuhara, T.; Tajiri, N.; Kikuchi, Y.; Liang, H.B.; Tayra, J.T.; Shinko, A.; Wakamori, T.; Agari, T.; et al. GDNF-Pretreatment Enhances the Survival of Neural Stem Cells Following Transplantation in a Rat Model of Parkinson’s Disease. Neurosci. Res. 2011, 71, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Galleguillos, B.M.; Kunert, L.; Brüggemann, N.; Prasuhn, J. Neuroinflammation and Mitochondrial Dysfunction in Parkinson’s Disease: Connecting Neuroimaging with Pathophysiology. Antioxidants 2023, 12, 1411. [Google Scholar] [CrossRef]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson Disease and the Immune System—Associations, Mechanisms and Therapeutics. Neurology 2020, 16, 303–318. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.; Quan, N.; Jonathan, P. Godbout Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-Synuclein: Pathology, Mitochondrial Dysfunction and Neuroinflammation in Parkinson’s Disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-Released Oligomeric α-Synuclein Is an Endogenous Agonist of TLR2 for Paracrine Activation of Microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-Synuclein Induces Toll-like Receptor 4 Dependent Inflammatory Responses in Astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef]
- Madsen, L.; Labrecque, N.; Engberg, J.; Dierich, A.; Svejgaard, A.; Benoist, C.; Mathis, D.; Fugger, L. Mice Lacking All Conventional MHC Class II Genes. Proc. Natl. Acad. Sci. USA 1999, 96, 10338–10343. [Google Scholar] [CrossRef] [PubMed]
- Harm, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII Is Required for α-Synuclein-Induced Activation of Microglia, CD4 T Cell Proliferation, and Dopaminergic Neurodegeneration. J. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.Y.; Chao, Y.X.; Rötzschke, O.; Tan, E.K. New Insights into Immune-Mediated Mechanisms in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 9302. [Google Scholar] [CrossRef] [PubMed]
- van de Warrenburg, B.P.C.; Church, A.J.; Martino, D.; Candler, P.M.; Bhatia, K.P.; Giovannoni, G.; Quinn, N.P. Antineuronal Antibodies in Parkinson’s Disease. Mov. Disord. 2008, 23, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor Necrosis Factor-α (TNF-α) Increases Both in the Brain and in the Cerebrospinal Fluid from Parkinsonian Patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Blum-Degena, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1β and Interleukin-6 Are Elevated in the Cerebrospinal Fluid of Alzheimer’s and de Novo Parkinson’s Disease Patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum Interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ Concentrations Are Elevated in Patients with Atypical and Idiopathic Parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Akiyama, H.; McGeer, P.L. Microglial Response to 6-Hydroxydopamine-Induced Substantia Nigra Lesions. Brain Res. 1989, 489, 247–253. [Google Scholar] [CrossRef]
- McGeer, P.L.; Schwab, C.; Parent, A.; Doudet, D. Presence of Reactive Microglia in Monkey Substantia Nigra Years after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Administration. Ann. Neurol. 2003, 54, 599–604. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective Microglial Activation in the Rat Rotenone Model of Parkinson’s Disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Cookson, M.R. Alpha-Synuclein Triggers T-Cell Response. Is Parkinson’s Disease an Autoimmune Disorder? Mov. Disord. 2017, 32, 1327. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic Changes in Presynaptic and Axonal Transport Proteins Combined with Striatal Neuroinflammation Precede Dopaminergic Neuronal Loss in a Rat Model of AAV α-Synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Ejlerskov, P.; Hultberg, J.G.; Wang, J.Y.; Carlsson, R.; Ambjørn, M.; Kuss, M.; Liu, Y.; Porcu, G.; Kolkova, K.; Friis Rundsten, C.; et al. Lack of Neuronal IFN-β-IFNAR Causes Lewy Body-and Parkinson’s Disease-like Dementia. Cell 2015, 163, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I Expression Renders Catecholaminergic Neurons Susceptible to T-Cell-Mediated Degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Jimenez-Ferrer, I.; Jewett, M.; Tontanahal, A.; Romero-Ramos, M.; Swanberg, M. Allelic Difference in Mhc2ta Confers Altered Microglial Activation and Susceptibility to α-Synuclein-Induced Dopaminergic Neurodegeneration. Neurobiol. Dis. 2017, 106, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Thome, A.D.; Yan, Z.; Schonhoff, A.M.; Williams, G.P.; Li, X.; Liu, Y.; Qin, H.; Benveniste, E.N.; Standaert, D.G. Peripheral Monocyte Entry Is Required for Alpha-Synuclein Induced Inflammation and Neurodegeneration in a Model of Parkinson Disease. Exp. Neurol. 2018, 300, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of Pathology in PINK1-Deficient Mouse Brain from Splicing via Ubiquitination, ER Stress, and Mitophagy Changes to Neuroinflammation. J. Neuroinflamm. 2017, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Mutez, E.; Nkiliza, A.; Belarbi, K.; de Broucker, A.; Vanbesien-Mailliot, C.; Bleuse, S.; Duflot, A.; Comptdaer, T.; Semaille, P.; Blervaque, R.; et al. Involvement of the Immune System, Endocytosis and EIF2 Signaling in Both Genetically Determined and Sporadic Forms of Parkinson’s Disease. Neurobiol. Dis. 2014, 63, 165–170. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.-W.; Cooper, S.C.; Broxmeyer, H.E.; Cannon, J.R.; Kim, C.H. Parkinson Disease–Associated LRRK2 G2019S Transgene Disrupts Marrow Myelopoiesis and Peripheral Th17 Response. J. Leukoc. Biol. 2017, 102, 1093–1102. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-Motor Features of Parkinson Disease. Nat. Publ. Gr. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.S.; Twisk, J.W.R.; Raijmakers, P.G.H.M.; Doty, R.L.; Berendse, H.W. Hyposmia as a Marker of (Non-) Motor Disease Severity in Parkinson’s Disease. J. Neural Transm. 2019, 126, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zhao, Y.; He, Y.; Zhou, Y.; Yang, J.; Zhou, X. Olfactory Dysfunction Predicts Disease Progression in Parkinson’s Disease: A Longitudinal Study. Front. Neurosci. 2020, 14, 569777. [Google Scholar] [CrossRef]
- Fang, T.; Chang, M.; Yang, C.; Chen, Y. The Association of Olfactory Dysfunction With Depression, Cognition, and Disease Severity in Parkinson’s Disease. Front. Neurol. 2021, 12, 779712. [Google Scholar] [CrossRef]
- Paper, O. Visual Attention and Saccadic Oculomotor Control in Parkinson’s Disease. Eur. Neurol. 2015, 73, 283–293. [Google Scholar] [CrossRef]
- Ekker, M.S.; Janssen, S.; Seppi, K.; Poewe, W.; De Vries, N.M.; Theelen, T.; Nonnekes, J.; Bloem, B.R. Ocular and visual disorders in Parkinson’s disease: Common but frequently overlooked. Park. Relat. Disord. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Archibald, N.K.; Clarke, M.P.; Mosimann, U.P.; Burn, D.J. Visual Symptoms in Parkinson’s Disease and Parkinson’s Disease Dementia. Mov. Disord. 2011, 26, 2387–2395. [Google Scholar] [CrossRef]
- Halperin, O.; Karni, R.; Israeli-Korn, S.; Hassin-Baer, S.; Zaidel, A. Overconfidence in Visual Perception in Parkinson’s Disease. Eur. J. Neurosci. 2021, 53, 2027–2039. [Google Scholar] [CrossRef]
- Ray, S.; Agarwal, P. Depression and Anxiety in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 93–104. [Google Scholar] [CrossRef]
- Khedr, E.M.; Abdelrahman, A.A.; Elserogy, Y.; Zaki, A.F.; Gamea, A. Depression and Anxiety among Patients with Parkinson’s Disease: Frequency, Risk Factors, and Impact on Quality of Life. Egypt. J. Neurol. Psychiatry Neurosurg. 2020, 56, 116. [Google Scholar] [CrossRef]
- Metta, V.; Leta, V.; Rukmini, K.; Prashanth, M.L.K.; Goyal, V.; Borgohain, R.; Chung, G.; Ray, F.K. Gastrointestinal Dysfunction in Parkinson’s Disease: Molecular Pathology and Implications of Gut Microbiome, Probiotics, and Fecal Microbiota Transplantation. J. Neurol. 2022, 269, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, T.; Schäfer, K.; Claus, I.; Del Tredici, K.; Jost, W.H. Gastrointestinal Involvement in Parkinson’s Disease: Pathophysiology, Diagnosis, and Management. NPJ Park. Dis. 2022, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Sarika, S. Noradrenergic Pathways of Locus Coeruleus in Parkinson’s and Alzheimer’s Pathology. Int. J. Neurosci. 2020, 130, 251–261. [Google Scholar]
- Cui, K.; Yang, F.; Tufan, T.; Raza, M.U.; Zhan, Y.; Fan, Y.; Zeng, F.; Brown, R.W.; Price, J.B.; Jones, T.C.; et al. Restoration of Noradrenergic Function in Parkinson’s Disease Model Mice. Am. Soc. Neurochem. 2021, 13, 17590914211009730. [Google Scholar] [CrossRef] [PubMed]
- Paredes-rodriguez, E.; Vegas-suarez, S.; Morera-herreras, T. The Noradrenergic System in Parkinson’s Disease. Front. Pharmacol. 2020, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Kinnerup, M.B.; Sommerauer, M.; Damholdt, M.F.; Schaldemose, J.L.; Ismail, R.; Terkelsen, A.J.; Stær, K.; Hansen, A.; Fedorova, T.D.; Knudsen, K.; et al. Neurobiology of Disease Preserved Noradrenergic Function in Parkinson’s Disease Patients with Rest Tremor. Neurobiol. Dis. 2021, 152, 105295. [Google Scholar] [CrossRef] [PubMed]
- Butkovich, L.M.; Houser, M.C.; Tansey, M.G. α-Synuclein and Noradrenergic Modulation of Immune Cells in Parkinson’s Disease Pathogenesis. Front. Neurosci. 2018, 12, 626. [Google Scholar] [CrossRef]
- Schindlbeck, K.A.; Eidelberg, D. Serotonergic Pathology and Braak’s Staging Hypothesis in Parkinson’s Disease New Terminology for a Common TDP-43 Proteinopathy. Lancet Neurol. 2019, 18, 713–714. [Google Scholar] [CrossRef]
- Miquel-rio, L.; Sarri, U.; Pavia-collado, R.; Meana, J.J. The Role of α-Synuclein in the Regulation of Serotonin System: Physiological and Pathological Features. Biomedicines 2023, 11, 541. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Koros, C.; Yousaf, T.; Picillo, M.; Polychronis, S.; Simitsi, A.; Giordano, B.; Chappell, Z.; et al. Serotonergic Pathology and Disease Burden in the Premotor and Motor Phase of A53T α-Synuclein Parkinsonism: A Cross-Sectional Study. Lancet Neurol. 2019, 18, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K. Roles of Glutamate Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef] [PubMed]
- Chegão, A.; Guarda, M.; Alexandre, B.M.; Shvachiy, L.; Temido-ferreira, M.; Marques-morgado, I.; Gomes, B.F.; Matthiesen, R.; Lopes, L.V.; Florindo, P.R.; et al. Glycation Modulates Glutamatergic Signaling and Exacerbates Parkinson’s Disease-like Phenotypes. NPJ Park. Dis. 2022, 51, 51. [Google Scholar] [CrossRef]
- Yadav, D.; Kumar, P. Parkinson’s Disease: An Overview and Role of Glutamate and Its Receptors. In Proceedings of the 2021 5th International Conference on Information Systems and Computer Networks (ISCON), Mathura, India, 22–23 October 2021; IEEE: NewYork, NY, USA, 2021; pp. 1–5. [Google Scholar]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratan, Y.; Rajput, A.; Pareek, A.; Pareek, A.; Jain, V.; Sonia, S.; Farooqui, Z.; Kaur, R.; Singh, G. Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson’s Disease. Biomolecules 2024, 14, 73. https://doi.org/10.3390/biom14010073
Ratan Y, Rajput A, Pareek A, Pareek A, Jain V, Sonia S, Farooqui Z, Kaur R, Singh G. Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson’s Disease. Biomolecules. 2024; 14(1):73. https://doi.org/10.3390/biom14010073
Chicago/Turabian StyleRatan, Yashumati, Aishwarya Rajput, Ashutosh Pareek, Aaushi Pareek, Vivek Jain, Sonia Sonia, Zeba Farooqui, Ranjeet Kaur, and Gurjit Singh. 2024. "Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson’s Disease" Biomolecules 14, no. 1: 73. https://doi.org/10.3390/biom14010073
APA StyleRatan, Y., Rajput, A., Pareek, A., Pareek, A., Jain, V., Sonia, S., Farooqui, Z., Kaur, R., & Singh, G. (2024). Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson’s Disease. Biomolecules, 14(1), 73. https://doi.org/10.3390/biom14010073