


Biomarkers in Peripartum Cardiomyopathy—What We Know and What Is Still to Be Found


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Epidemiology, Risk Factors, and Outcomes
1.2. Different Phenotypes and Courses of Peripartum Cardiomyopathy
2. Pathophysiology
2.1. Oxidative Stress and Inflammation, Endothelial and Microcirculatory Dysfunction
2.2. Heart Muscle Tissue
2.2.1. Cardiomyocytes
2.2.2. Fibrosis
2.2.3. Extracellular Matrix
2.2.4. Genetics
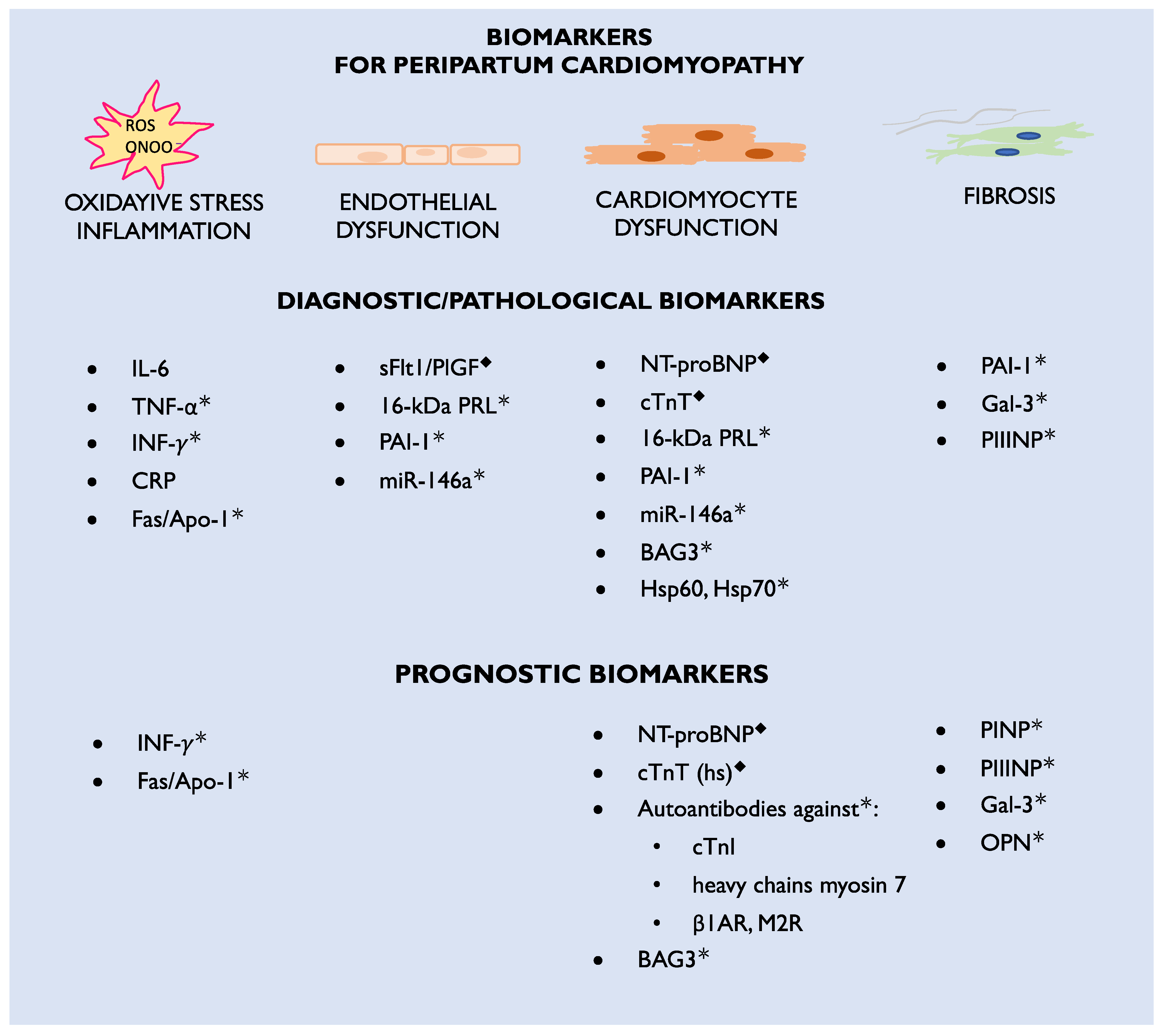
3. Biomarkers
3.1. Biomarkers Currently Used in Clinical Practice
3.1.1. NT-proBNP and Brain-Type Natriuretic Peptide (BNP)
3.1.2. Cardiac Troponin T (cTnT) and Cardiac Troponin I (cTnI)
3.1.3. Soluble Fms-like Tyrosine Kinase-1 and Placenta Growth Factor
3.1.4. 23-kDa PRL
3.2. Biomarker Candidates for Future Practice
3.2.1. 16-kDa PRL
3.2.2. Gal-3
3.2.3. PINP and PIIINP
3.3. Markers of Inflammation
3.3.1. Fas/Apo-1
3.3.2. C-Reactive Protein (CRP)
3.3.3. IL-6
3.3.4. Tumor Necrosis Factor Alpha (TNF-α)
3.3.5. Interferon Gamma (IFN-γ)
3.3.6. PAI-1
3.4. Autoantibodies to Cardiac Antigens
4. New Specific Biomarkers to Be Found
4.1. Fibrosis and Inflammation
4.2. MicroRNA
4.3. Heat Shock Proteins
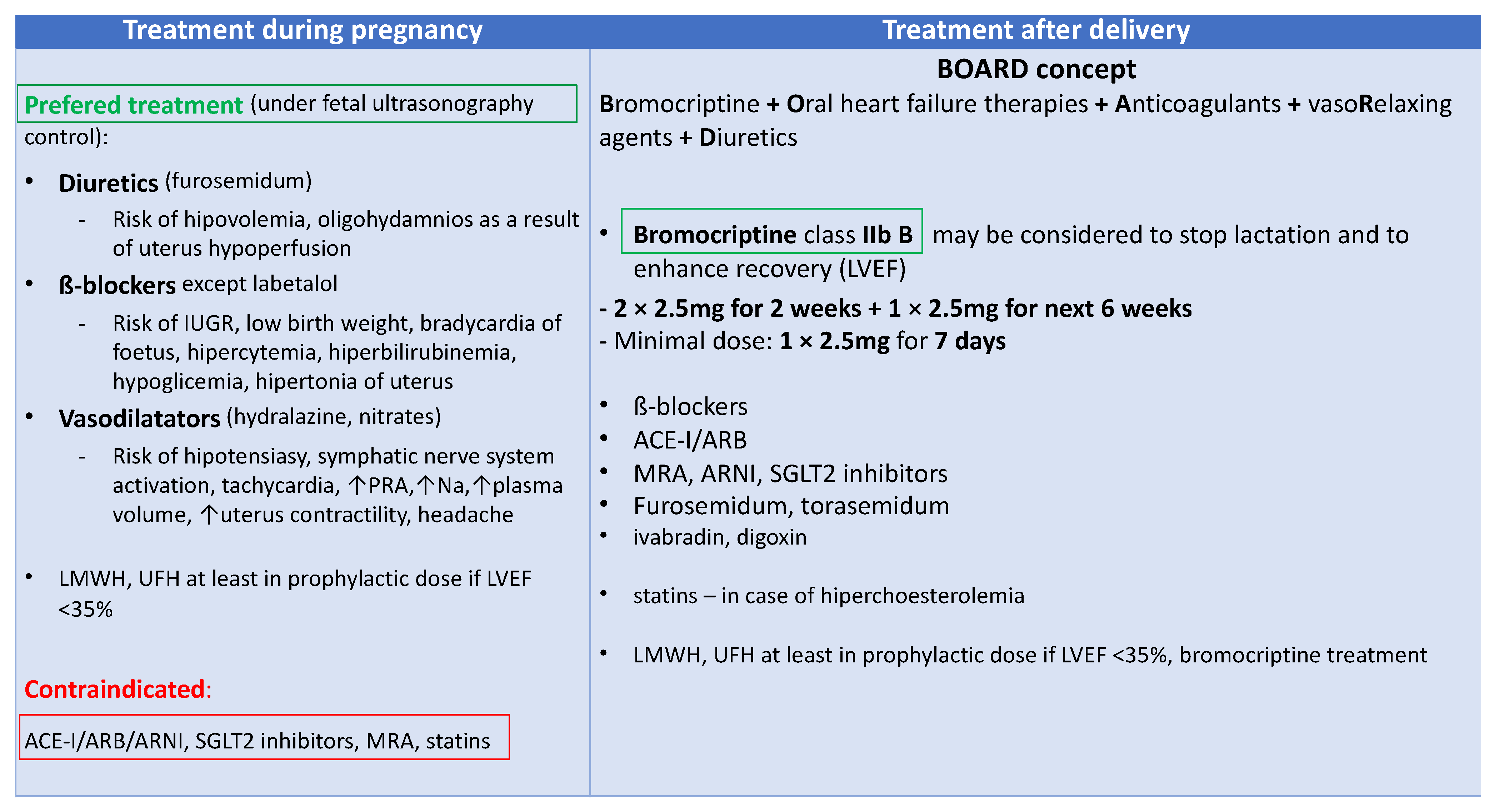
5. Therapy for Peripartum Cardiomyopathy
5.1. New Biomarker-Based Therapies
5.2. Biomarker-Guided Therapy
6. Conclusions
Take Home Messages
- Although rare, PPCM can be a life-threatening condition and may be difficult to diagnose. The etiology of PPCM is complex and remains unclear. The most important pathological pathways include antiangiogenic 16 kDa PRL, with bromocriptine being the most specific PPCM treatment to date.
- There is a need to explore new pathophysiological pathways concerning endothelial miRs, ECM fibrosis, and cardiac tissue proteostasis, as well as to identify new drug targets to improve patient outcomes.
- A broad range of drug molecules requires further testing in PPCM, particularly concerning the safety of the MPO inhibitor, which is the most advanced in clinical HF studies to date.
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Sliwa, K.; Hilfiker-Kleiner, D.; Petrie, M.C.; Mebazaa, A.; Pieske, B.; Buchmann, E.; Regitz-Zagrosek, V.; Schaufelberger, M.; Tavazzi, L.; van Veldhuisen, D.J.; et al. Current state of knowledge on aetiology, diagnosis, management and therapy of peripartum cardiomyopathy (PPCM): A position statement from the Heart Failure Association of the European Society of Cardiology Working group on PPCM. Eur. J. Heart Fail. 2010, 12, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Roos-Hesselink, J.W.; Bauersachs, J.; Blomström-Lundqvist, C.; Cífková, R.; De Bonis, M.; Iung, B.; Johnson, M.R.; Kintscher, U.; Kranke, P.; et al. 2018 ESC Guidelines on the management of cardiovascular diseases during pregnancy, the Task Force on the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 3165–3241. [Google Scholar] [CrossRef]
- Hibbard, J.U.; Lindheimer, M.; Lang, R.M. A modified definition for peripartum cardiomyopathy and prognosis based on echocardiography. Obstet. Gynecol. 1999, 94, 311–316. [Google Scholar]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.; et al. ESC Scientific Document Group 2023 ESC Guidelines for the management of cardiomyopathies, Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Sliwa, K.; Bauersachs, J.; Arany, Z.; Spracklen, T.F.; Hilfiker-Kleiner, D. Peripartum cardiomyopathy: From genetics to management. Eur. Heart J. 2021, 42, 3094–3102. [Google Scholar] [CrossRef]
- Sliwa, K.; Petrie, M.C.; van der Meer, P.; Mebazaa, A.; Hilfiker-Kleiner, D.; Jackson, A.M.; Maggioni, A.P.; Laroche, C.; Regitz-Zagrosek, V.; Schaufelberger, M.; et al. Clinical presentation, management, and 6-month outcomes in women with peripartum cardiomyopathy: An ESC EORP registry. Eur. Heart J. 2020, 41, 3787–3797. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- Pachariyanon, P.; Bogabathina, H.; Jaisingh, K.; Modi, M.; Modi, K. Long-Term Outcomes of Women with Peripartum Cardiomyopathy Having Subsequent Pregnancies. J. Am. Coll. Cardiol. 2023, 82, 16–26. [Google Scholar] [CrossRef]
- WHO International Programme on Chemical Safety Biomarkers in Risk Assessment, Validity and Validation. 2001. Available online: http://www.inchem.org/documents/ehc/ehc/ehc222.htm (accessed on 1 October 2023).
- Isezuo, S.A.; Abubakar, S.A. Epidemiologic profile of peripartum cardiomyopathy in a tertiary care hospital. Ethn. Dis. 2007, 17, 228–233. [Google Scholar]
- Fet, J.D.; Christie, L.G.; Carraway, R.D.; Murphy, J.G. Five-year prospective study of the incidence and prognosis of peripartum cardiomyopathy at a single institution. Mayo Clin. Proc. 2005, 80, 1602–1606. [Google Scholar] [CrossRef]
- Desai, D.; Moodley, J.; Naidoo, D. Peripartum Cardiomyopathy: Experiences at King Edward VIII Hospital, Durban, South Africa and a Review of the Literature. Trop. Dr. 1995, 25, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Nabbaale, J.; Okello, E.; Kibirige, D.; Ssekitoleko, I.; Isanga, J.; Karungi, P.; Sebatta, E.; Zhu, Z.W.; Nakimuli, A.; Omagino, J.; et al. Burden, predictors and short-term outcomes of peripartum cardiomyopathy in a black African cohort. PLoS ONE 2020, 15, e0240837. [Google Scholar] [CrossRef]
- Isogai, T.; Kamiya, C.A. Worldwide Incidence of Peripartum Cardiomyopathy and Overall Maternal Mortality. Int. Heart J. 2019, 60, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Arany, Z.; McNamara, D.M.; Goland, S.; Elkayam, U. Peripartum Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 207–221. [Google Scholar] [CrossRef]
- Kolte, D.; Khera, S.; Aronow, W.S.; Palaniswamy, C.; Mujib, M.; Ahn, C.; Jain, D.; Gass, A.; Ahmed, A.; Panza, J.A.; et al. Temporal Trends in Incidence and Outcomes of Peripartum Cardiomyopathy in the United States: A Nationwide Population-Based Study. J. Am. Heart. Assoc. 2014, 3, e001056. [Google Scholar] [CrossRef]
- Sliwa, K.; Mebazaa, A.; Hilfiker-Kleiner, D.; Petrie, M.C.; Maggioni, A.P.; Laroche, C.; Regitz-Zagrosek, V.; Schaufelberger, M.; Tavazzi, L.; van der Meer, P.; et al. Clinical characteristics of patients from the worldwide registry on peripartum cardiomyopathy (PPCM): EURObservational Research Programme in conjunction with the Heart Failure Association of the European Society of Cardiology Study Group on PPCM. Eur. J. Heart Fail. 2017, 19, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Petryka-Mazurkiewicz, J.; Kryczka, K.; Marona, M.; Kuriata, J.; Sitkowska-Rysiak, E.; Konopka, A.; Marczak, M.; Kołsut, P.; Kuśmierczyk, M.; Demkow, M.; et al. Cardiovascular magnetic resonance imaging of biventricular assist device-induced recovery from acute heart failure in peripartum cardiomyopathy. Kardiol. Pol. 2020, 78, 1284–1285. [Google Scholar] [CrossRef]
- Sugahara, M.; Kagiyama, N.; Hasselberg, N.E.; Blauwet, L.A.; Briller, J.; Cooper, L.; Fett, J.D.; Hsich, E.; Wells, G.; McNamara, D.; et al. Global Left Ventricular Strain at Presentation is Associated with Subsequent Recovery in Patients with Peripartum Cardiomyopathy. J. Am. Soc. Echocardiogr. 2019, 32, 1565–1573. [Google Scholar] [CrossRef]
- Fidziańska, A.; Walczak, E.; Glinka, Z.; Religa, G.; Sobieszczanska-Malek, M.; Bilinska, Z.T. Ultrastructural evidence of myocardial capillary remodeling in peripartum cardiomyopathy. Med. Sci. Monit. 2010, 16, CS62–CS66. [Google Scholar]
- Hilfiker-Kleiner, D.; Kaminski, K.; Podewski, E.; Bonda, T.; Schaefer, A.; Sliwa, K.; Forster, O.; Quint, A.; Landmesser, U.; Doerries, C.; et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 2007, 128, 589–600. [Google Scholar] [CrossRef]
- Halkein, J.; Tabruyn, S.P.; Ricke-Hoch, M.; Haghikia, A.; Nguyen, N.Q.; Scherr, M.; Castermans, K.; Malvaux, L.; Lambert, V.; Thiry, M.; et al. MicroRNA-146a is a Therapeutic Target and Biomarker for Peripartum Cardiomyopathy. J. Clin. Investig. 2013, 123, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Patten, I.S.; Rana, S.; Shahul, S.; Rowe, G.C.; Jang, C.; Liu, L.; Hacker, M.R.; Rhee, J.S.; Mitchell, J.; Mahmood, F.; et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 2012, 485, 333–338. [Google Scholar] [CrossRef]
- Lenke, L.; de la Escalera, G.M.; Clapp, C.; Bertsch, T.; Triebel, J. A Dysregulation of the Prolactin/Vasoinhibin Axis Appears to Contribute to Preeclampsia. Front. Endocrinol. 2020, 10, 893. [Google Scholar] [CrossRef]
- Ricke-Hoch, M.; Bultmann, I.; Stapel, B.; Condorelli, G.; Rinas, U.; Sliwa, K.; Scherr, M.; Hilfiker-Kleiner, D. Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovasc. Res. 2014, 101, 587–596. [Google Scholar] [CrossRef]
- Bollen, I.A.E.; Ehler, E.; Fleischanderl, K.; Bouwman, F.; Kempers, L.; Ricke-Hoch, M.; Hilfiker-Kleiner, D.; Dos Remedios, C.G.; Krüger, M.; Vink, A.; et al. Myofilament Remodeling and Function is More Impaired in Peripartum Cardiomyopathy Compared with Dilated Cardiomyopathy and Ischemic Heart Disease. Am. J. Pathol. 2017, 187, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Seno, A.; Takeda, Y.; Matsui, M.; Okuda, A.; Nakano, T.; Nakada, Y.; Kumazawa, T.; Nakagawa, H.; Nishida, T.; Onoue, K.; et al. Suppressed Production of Soluble Fms-Like Tyrosine Kinase-1 Contributes to Myocardial Remodeling and Heart Failure. Hypertension 2016, 68, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Petryka-Mazurkiewicz, J.; Kryczka, K.; Mazurkiewicz, Ł.; Miłosz-Wieczorek, B.; Śpiewak, M.; Marczak, M.; Henzel, J.; Grzybowski, J.; Demkow, M.; Dzielińska, Z. Cardiovascular Magnetic Resonance in Peripartum Cardiomyopathy: Comparison with Idiopathic Dilated Cardiomyopathy. Diagnostics 2021, 11, 1752. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Winkler, J.; Ramos, I. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef]
- Azibani, F.; Pfeffer, T.J.; Ricke-Hoch, M.; Dowling, W.; Pietzsch, S.; Briton, O.; Baard, J.; Abou Moulig, V.; König, T.; Berliner, D.; et al. Outcome in German and South African peripartum cardiomyopathy cohorts associates with medical therapy and fibrosis markers. ESC Heart Fail. 2020, 7, 512–522. [Google Scholar] [CrossRef]
- Nikolov, A.; Popovski, N. Extracellular Matrix in Heart Disease, Focus on Circulating Collagen Type I and III Derived Peptides as Biomarkers of Myocardial Fibrosis and Their Potential in the Prognosis of Heart Failure: A Concise Review. Metabolites 2022, 12, 297. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, F.J.; Ruiz-Laiglesia, F.J.; Samperiz-Legarre, P.; Lasierra-Diaz, P.; Flamarique-Pascual, A.; Morales-Rull, J.L.; Perez-Calvo, J.I. Propeptide of procollagen type I (PIP) and outcomes in decompensated heart failure. Eur. J. Intern. Med. 2007, 18, 129–134. [Google Scholar] [CrossRef]
- Spracklen, T.F.; Chakafana, G.; Schwartz, P.J.; Kotta, M.C.; Shaboodien, G.; Ntusi, N.A.B.; Sliwa, K. Genetics of Peripartum Cardiomyopathy: Current Knowledge, Future Directions and Clinical Implications. Genes 2021, 12, 103. [Google Scholar] [CrossRef]
- Kryczka, K.E.; Dzielińska, Z.; Franaszczyk, M.; Kryczka, K.E.; Dzielińska, Z.; Franaszczyk, M.; Wojtkowska, I.; Henzel, J.; Śpiewak, M.; Stępińska, J.; et al. Severe Course of Peripartum Cardiomyopathy and Subsequent Recovery in a Patient with a Novel TTN Gene-Truncating Mutation. Am. J. Case Rep. 2018, 19, 820–824. [Google Scholar] [CrossRef] [PubMed]
- Franaszczyk, M.; Chmielewski, P.; Truszkowska, G.; Stawinski, P.; Michalak, E.; Rydzanicz, M.; Sobieszczanska-Malek, M.; Pollak, A.; Szczygieł, J.; Kosinska, J.; et al. Titin Truncating Variants in Dilated Cardiomyopathy—Prevalence and Genotype-Phenotype Correlations. PLoS ONE 2017, 12, e0169007. [Google Scholar] [CrossRef] [PubMed]
- Felkin, L.E.; Walsh, R.; Ware, J.S.; Yacoub, M.H.; Birks, E.J.; Barton, P.J.; Cook, S.A. Recovery of Cardiac Function in Cardiomyopathy Caused by Titin Truncation. JAMA Cardiol. 2016, 1, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, E.J.; Takayama, H.; Ferrari, G. Stretch your heart-but not too far: The role of titin mutations in dilated cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2018, 156, 209–214. [Google Scholar] [CrossRef]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef]
- Horne, B.D.; Rasmusson, K.D.; Alharethi, R.; Budge, D.; Brunisholz, K.D.; Metz, T.; Carlquist, J.F.; Connolly, J.J.; Porter, T.F.; Lappé, D.L.; et al. Genome-wide significance and replication of the chromosome 12p11.22 locus near the PTHLH gene for peripartum cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 359–366. [Google Scholar] [CrossRef]
- Pfeffer, T.J.; Schlothauer, S.; Pietzsch, S.; Schaufelberger, M.; Auber, B.; Ricke-Hoch, M.; List, M.; Berliner, D.; Abou Moulig, V.; König, T.; et al. Increased cancer prevalence in peripartum cardiomyopathy. JACC Cardio Oncol. 2019, 1, 196–205. [Google Scholar] [CrossRef]
- Mebazaa, A.; Seronde, M.F.; Gayat, E.; Tibazarwa, K.; Anumba, D.O.C.; Akrout, N.; Sadoune, M.; Sarb, J.; Arrigo, M.; Motiejunaite, J.; et al. Imbalanced angiogenesis in peripartum cardiomyopathydiagnostic value of placenta growth factor. Circ. J. 2017, 81, 1654–1661. [Google Scholar] [CrossRef]
- Sliwa, K.; Blauwet, L.; Tibazarwa, K.; Libhaber, E.; Smedema, J.P.; Becker, A.; McMurray, J.; Yamac, H.; Labidi, S.; Struman, I.; et al. Evaluation of bromocriptine in the treatment of acute severe peripartum cardiomyopathy: A proof-of-concept pilot study. Circulation 2010, 121, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Sarma, A.; Aggarwal, N.; Briller, J.; Briller, J.E.; Davis, M.; Economy, K.E.; Hameed, A.B.; Januzzi, J.L.; Lindley, K.J.; Mattina, D.J.; et al. The Utilization and Interpretation of Cardiac Biomarkers During Pregnancy. JACC Adv. 2022, 1, 100064. [Google Scholar] [CrossRef]
- Weber, M.; Hamm, C. Role of B-type natriuretic peptide (BNP) and NT-proBNP in clinical routine. Heart 2006, 92, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Resnik, J.L.; Hong, C.; Resnik, R.; Kazanegra, R.; Beede, J.; Bhalla, V.; Maisel, A. Evaluation of B-type natriuretic peptide (BNP) levels in normal and preeclamptic women. Am. J. Obstet. Gynecol. 2005, 193, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Kale, E.; Yalinkaya, A.; Akdeniz, N.; Canoruç, N. The comparison of amino-terminal probrain natriuretic peptide levels in preeclampsia and normotensive pregnancy. J. Perinat. Med. 2005, 33, 121–124. [Google Scholar] [CrossRef]
- Dockree, S.; Brook, J.; Shine, B.; James, T.; Vatish, M. Pregnancy-specific Reference Intervals for BNP and NT-pro BNP-Changes in Natriuretic Peptides Related to Pregnancy. J. Endocr. Soc. 2021, 5, bvab091. [Google Scholar] [CrossRef]
- Imran, T.F.; Mohebali, D.; Lopez, D.; Goli, R.R.; DeFilippis, E.M.; Truong, S.; Bello, N.A.; Gaziano, J.M.; Djousse, L.; Coglianese, E.E.; et al. NT-proBNP and predictors of event free survival and left ventricular systolic function recovery in peripartum cardiomyopathy. Inter. J. Cardiol. 2022, 357, 48–54. [Google Scholar] [CrossRef]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar]
- Communal, C.; Colucci, W.S. The Control of Cardiomyocyte Apoptosis via the Beta-Adrenergic Signaling Pathways. Arch. Mal. Coeur. Vaiss. 2005, 98, 236–241. [Google Scholar]
- Ricke-Hoch, M.; Hoes, M.F.; Pfeffer, T.J.; Schlothauer, S.; Nonhoff, J.; Haidari, S.; Bomer, N.; Scherr, M.; Stapel, B.; Stelling, E.; et al. In peripartum cardiomyopathy plasminogen activator inhibitor-1 is a potential new biomarker with controversial roles. Cardiovasc. Res. 2019, 116, 1875–1886. [Google Scholar] [CrossRef]
- Hu, C.L.; Li, Y.B.; Zou, Y.G.; Zhang, J.M.; Chen, J.B.; Liu, J.; Tang, Y.H.; Tang, Q.Z.; Huang, C.X. Troponin T measurement can predict persistent left ventricular dysfunction in peripartum cardiomyopathy. Heart 2007, 93, 488–490. [Google Scholar] [CrossRef]
- Peng, B.S.; Frederick, B.; Hidalgo, M.D.; Ryan, J. Peripartum Cardiomyopathy: A Cause of Heart Failure in Young Women. Med. Forum 2017, 18, 15. [Google Scholar] [CrossRef][Green Version]
- Nikuei, P.; Rajaei, M.; Roozbeh, N.; Mohseni, F.; Poordarvishi, F.; Azad, M.; Haidari, S. Diagnostic accuracy of sFlt1/PlGF ratio as a marker for preeclampsia. BMC Pregnancy Childbirth 2020, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Powe, C.E.; Salahuddin, S.; Verlohren, S.; Perschel, F.H.; Levine, R.J.; Lim, K.H.; Wenger, J.B.; Thadhani, R.; Karumanchi, S.A. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation 2012, 125, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Kwiatkowska, E.; Torbe, A. The role of disordered angiogenesis tissue markers (sFlt-1; PlGF) in present day diagnosis of preeclampsia. Ginkol. Pol. 2019, 90, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Young, J.; Chanson, P.; Binart, N. New insights in prolactin, pathological implications. Nat. Rev. Endocrinol. 2015, 11, 265–275. [Google Scholar] [CrossRef]
- Brown, R.S.E.; Herbison, A.E.; Grattan, D.R. Effects of Prolactin and Lactation on A15 Dopamine Neurones in the Rostral Preoptic Area of Female Mice. J. Neuroendocrinol. 2015, 27, 708–717. [Google Scholar] [CrossRef]
- Forster, O.; Hilfiker-Kleiner, D.; Ansari, A.A.; Sundstrom, J.B.; Libhaber, E.; Tshani, W.; Becker, A.; Yip, A.; Klein, G.; Sliwa, K. Reversal of IFN-gamma; oxLDL and prolactin serum levels correlate with clinical improvement in patients with peripartum cardiomyopathy. Eur. J. Heart Fail. 2008, 10, 861–868. [Google Scholar] [CrossRef]
- Bauersachs, J.; Arrigo, M.; Hilfiker-Kleiner, D.; Veltmann, C.; Coats, A.J.; Crespo-Leiro, M.G.; De Boer, R.A.; van der Meer, P.; Maack, C.; Mouquet, F.; et al. Current management of patients with severe acute peripartum cardiomyopathy, practical guidance from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur. J. Heart Fail. 2016, 18, 1096–1105. [Google Scholar] [CrossRef]
- Lee, S.; Nishino, M.; Mazumdar, T.; Garcia, G.E.; Galfione, M.; Lee, F.L.; Lee, C.L.; Liang, A.; Kim, J.; Feng, L.; et al. 16-kDa Prolactin Down-Regulates Inducible Nitric Oxide Synthase Expression through Inhibition of the Signal Transducer and Activator of Transcription 1/IFN Regulatory Factor-1 Pathway. Cancer Res. 2005, 65, 7984–7992. [Google Scholar] [CrossRef][Green Version]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Sherwi, N.; Merali, S.; Wong, K. Personalizing biomarker strategies in heart failure with galectin-3. Future Cardiol. 2012, 8, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Groh, K.; Alharethi, R.; Ewald, G.; Givertz, M.; Felker, G.M.; Pisarcik, J.; Hanley-Yanez, K.; Halder, I.; McTiernan, C.; McNamara, D. Galectin-3 levels and outcomes in periprartum cardiomyopathy: Results from the multicenter IPAC Investigation. J. Am. Coll. Cardiol. 2016, 67, 1533. [Google Scholar] [CrossRef]
- Yu, L.; Ruifrok, W.P.; Meissner, M.; Bos, E.M.; van Goor, H.; Sanjabi, B.; van der Harst, P.; Pitt, B.; Goldstein, I.J.; Koerts, J.A.; et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ. Heart Fail. 2013, 6, 107–117. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses, cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, K.; Skudicky, D.; Bergmann, A. Peripartum cardiomyopathy, analysis of clinical outcome, left ventricular function, plasma levels of cytokines and Fas/APO-1. J. Am. Coll. Cardiol. 2000, 35, 701–705. [Google Scholar] [CrossRef]
- Sliwa, K.; Forster, O.; Libhaber, E.; Fett, J.D.; Sundstrom, J.B.; Hilfiker-Kleiner, D.; Ansari, A.A. Peripartum cardiomyopathy, inflammatory markers as predictors of outcome in 100 prospectively studied patients. Eur. Heart J. 2006, 27, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation, conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef]
- Ni, C.W.; Hsieh, H.J.; Chao, Y.J.; Wang, D.L. Interleukin-6-induced JAK2/STAT3 signaling pathway in endothelial cells is suppressed by hemodynamic flow. Am. J. Physiol. Cell Physiol. 2004, 287, C771–C780. [Google Scholar] [CrossRef]
- Lecour, S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J. Mol. Cell Cardiol. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- Mann, D.L. Stress-activated cytokines and the heart: From adaptation to maladaptation. Annu. Rev. Physiol. 2003, 65, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, K.; Skudicky, D.; Candy, G.; Bergemann, A.; Hopley, M.; Sareli, P. The addition of pentoxifylline to conventional therapy improves outcome in patients with peripartum cardiomyopathy. Eur. J. Heart Fail. 2002, 4, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Lasfar, A.; Cook, J.R.; Cohen Solal, K.A.; Reuhl, K.; Kotenko, S.V.; Langer, J.A.; Laskin, D.L. Critical role of the endogenous interferon ligand-receptors in type I and type II interferons response. Immunology 2014, 142, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chakravarty, S.D.; Ivashkiv, L.B. Regulation of interferon and toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008, 226, 41–56. [Google Scholar] [CrossRef]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen activator inhibitor-1 (PAI-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, D.Y.; Li, J.; Wu, Y.F.; Liu, X.L.; Yang, X.C. Autoantibodies against the myocardial β1-adrenergic and M2-muscarinic receptors in patients with congestive heart failure. Chin. Med. J. 2002, 115, 1127–1131. [Google Scholar]
- Magnusson, Y.; Marullo, S.; Hoyert, S.B.; Waagstein, F.; Andersson, B.; Vahlne, A.; Guillet, J.G. Mapping of a Functional Autoimmune Epitope on the Adrenergic Receptor in Patients with Idiopathic Dilated Cardiomyopathy. J. Clin. Investig. 1990, 86, 1658–1663. [Google Scholar] [CrossRef]
- Duan, X.; Liu, R.; Luo, X.-L.; Gao, X.-J.; Hu, F.-H.; Guo, C.; Wang, J.; Hu, X.-Y.; Chun, Y.-S.; Yuan, J.-S.; et al. The relationship between β1-adrenergic and M2-muscarinic receptor autoantibodies and hypertrophic cardiomyopathy. Exp. Physiol. 2020, 105, 522–530. [Google Scholar] [CrossRef]
- Jane-wit, D.; Altuntas, C.Z.; Johnson, J.M.; Yong, S.; Wickley, P.J.; Clark, P.; Wang, Q.; Popović, Z.B.; Penn, M.S.; Damron, D.S.; et al. β1-Adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation 2007, 116, 399–410. [Google Scholar] [CrossRef]
- Stavrakis, S.; Kem, D.C.; Patterson, E.; Lozano, P.; Huang, S.; Szabo, B.; Cunningham, M.W.; Lazzara, R.; Yu, X. Opposing cardiac effect of autoantibody activation of β-adrenergic and M2 muscarinic receptors in cardiac-related diseases. Int. J. Cardiol. 2011, 148, 331–336. [Google Scholar] [CrossRef]
- Vatner, D.E.; Sato, N.; Galper, J.B.; Vatner, S.F. Physiological and biochemical evidence for coordinate increases in muscarinic receptors and Gi during pacing-induced heart failure. Circulation 1996, 94, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Kaya, Z.; Schwab, J.; Westenfeld, R.; Ehlermann, P.; Bachelier, K.; Oettl, R.; von Kaisenberg, C.S.; Katus, H.A.; Bauersachs, J.; et al. Evidence of autoantibodies against cardiac troponin I and sarcomeric myosin in peripartum cardiomyopathy. Basic Res. Cardiol. 2015, 110, 60. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Chen, M.; Zhao, W.; Wang, X.; Wang, H.; Zhang, Z.; Zhang, J.; Xu, L.; Chen, J.; et al. The Correlation between Peripartum Cardiomyopathy and Autoantibodies against Cardiovascular Receptors. PLoS ONE 2014, 9, e86770. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.L.; Hung, H.F.; Shyu, K.G.; Yeh, J.H.; Chiu, H.C. Successful Treatment of Peripartum Cardiomyopathy with Plasmapheresis. Acta Cardiol. Sin. 2013, 29, 471–474. [Google Scholar]
- Lau, D.; Baldus, S. Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol. Ther. 2006, 111, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J. Biol. Chem. 2001, 276, 41279–41287. [Google Scholar] [CrossRef]
- Shabani, F.; McNeil, J.; Tippett, L. The oxidative inactivation of tissue inhibitor of metalloproteinase-1 (TIMP-1) by hypochlorous acid (HOCl) is suppressed by anti-rheumatic drugs. Free Radic. Res. 1998, 28, 115–123. [Google Scholar] [CrossRef]
- Reichlin, T.; Socrates, T.; Egli, P.; Potocki, M.; Breidthardt, T.; Arenja, N.; Meissner, J.; Noveanu, M.; Reiter, M.; Twerenbold, R.; et al. Use of myeloperoxidase for risk stratification in acute heart failure. Clin. Chem. 2010, 56, 944–951. [Google Scholar] [CrossRef]
- Serraino, G.F.; Jiritano, F.; Costa, D.; Ielapi, N.; Napolitano, D.; Mastroroberto, P.; Bracale, U.M.; Andreucci, M.; Serra, R. Metalloproteinases and Hypertrophic Cardiomyopathy: A Systematic Review. Biomolecules 2023, 13, 665. [Google Scholar] [CrossRef]
- Münch, J.; Avanesov, M.; Bannas, P.; Säring, D.; Krämer, E.; Mearini, G.; Carrier, L.; Suling, A.; Lund, G.; Patten, M. Serum Matrix Metalloproteinases as Quantitative Biomarkers for Myocardial Fibrosis and Sudden Cardiac Death Risk Stratification in Patients with Hypertrophic Cardiomyopathy. J. Card. Fail. 2016, 2, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, J.P.; Colan, S.D.; Lang, P.; Triedman, J.K.; Alexander, M.E.; Walsh, E.P.; Berul, C.I.; Cecchin, F. Circulating Matrix Metalloproteinases in Adolescents with Hypertrophic Cardiomyopathy and Ventricular Arrhythmia. Circ. Heart Fail. 2012, 5, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E.; Tobe, T.; Yamada, H.; Okamoto, N.; Zack, D.J.; Werb, Z.; Soloway, P.D.; Campochiaro, P.A. TIMP-1 promotes VEGF-induced neovascularization in the retina. Histol. Histopathol. 2001, 16, 87–97. [Google Scholar]
- Cui, Q.; Yu, Z.; Pan, Y.; Purisima, E.O.; Wang, E. MicroRNAs preferentially target the genes with high transcriptional regulation complexity. Biochem. Biophys. Res. Commun. 2007, 352, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Zhou, S.; Jin, J.; Wang, J.; Zhang, Z.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAs in cardiovascular diseases, potential biomarkers: Therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef]
- Kasprzyk-Pawelec, A.; Wojciechowska, A.; Kuc, M.; Zielinski, J.; Parulski, A.; Kusmierczyk, M.; Lutynska, A.; Kozar-Kaminska, K. microRNA expression profile in Smooth Muscle Cells isolated from thoracic aortic aneurysm samples. Adv. Med. Sci. 2019, 64, 331–337. [Google Scholar] [CrossRef]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of Flow-Sensitive microRNAs in Endothelial Dysfunction and Atherosclerosis. Mechanosensitive Athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef]
- Licholai, S.; Blaż, M.; Kapelak, B.; Sanak, M. Unbiased Profile of MicroRNA Expression in Ascending Aortic Aneurysm Tissue Appoints Molecular Pathways Contributing to the Pathology. Ann. Thorac. Surg. 2016, 102, 1245–1252. [Google Scholar] [CrossRef]
- Staszel, T.; Zapała, B.; Polus, A.; Sadakierska-Chudy, A.; Kieć-Wilk, B.; Stępień, E.; Wybrańska, I.; Chojnacka, M.; Dembińska-Kieć, A. Role of microRNAs in endothelial cell pathophysiology. Pol. Arch. Med. Wewn. 2011, 121, 361–367. [Google Scholar] [CrossRef]
- Chakafana, G.; Spracklen, T.F.; Kamuli, S.; Zininga, T.; Shonhai, A.; Ntusi, N.A.B.; Sliwa, K. Heat Shock Proteins, Potential Modulators and Candidate Biomarkers of Peripartum Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 633013. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Haghikia, A.; Berliner, D.; Vogel-Claussen, J.; Schwab, J.; Franke, A.; Schwarzkopf, M.; Ehlermann, P.; Pfister, R.; Michels, G.; et al. Bromocriptine for the treatment of peripartum cardiomyopathy: A multicentre randomized study. Eur. Heart J. 2017, 38, 2671–2679. [Google Scholar] [CrossRef]
- Wallace, B.; Peisl, A.; Seedorf, G.; Kim, C.; Bosco, J.; Kenniston, J.; Keefe, D.; Abman, S.H. Anti-sFlt-1 Therapy Preserves Lung Alveolar and Vascular Growth in Antenatal Models of Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2018, 197, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Anttila, V.; Saraste, A.; Knuuti, J.; Jaakkola, P.; Hedman, M.; Svedlund, S.; Lagerström-Fermér, M.; Kjaer, M.; Jeppsson, A.; Gan, L.M. Synthetic mRNA Encoding VEGF-A in Patients Undergoing Coronary Artery Bypass Grafting: Design of a Phase 2a Clinical Trial. Mol. Ther. Methods Clin. Dev. 2020, 18, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, T.J.; List, M.; Müller, J.H.; Jaakkola, P.; Hedman, M.; Svedlund, S.; Lagerström-Fermér, M.; Kjaer, M.; Jeppsson, A.; Gan, L.M. Perhexiline treatment improves toxic effects of β-adrenergic receptor stimulation in experimental peripartum cardiomyopathy. ESC Heart Fail. 2021, 8, 3375–3381. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; González, A.; Pizard, A.; Papageorgiou, A.P.; López-Andrés, N.; Jaisser, F.; Thum, T.; Zannad, F.; Díez, J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur. J. Heart Fail. 2015, 17, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.F.; Nigri, M.; Higuchi, M.L.; Pomerantzeff, P.M.; Spina, G.S.; Sampaio, R.O.; Tarasoutchi, F.; Grinberg, M.; Rochitte, C.E. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J. Am. Coll. Cardiol. 2010, 56, 278–287. [Google Scholar] [CrossRef]
- Aoki, T.; Fukumoto, Y.; Sugimura, K.; Oikawa, M.; Satoh, K.; Nakano, M.; Nakayama, M.; Shimokawa, H. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure—Comparison between preserved and reduced ejection fraction heart failure. Circ. J. 2011, 75, 2605–2613. [Google Scholar] [CrossRef]
- Siraki, A.G. The many roles of myeloperoxidase: From inflammation and immunity to biomarkers, drug metabolism and drug discovery. Redox Biol. 2021, 46, 102109. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Troughton, R.W.; Frampton, C.M.; Yandle, T.G.; Espiner, E.A.; Nicholls, M.G.; Richards, A.M. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000, 355, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, P.; Jondeau, G.; Funck, F.; Gueffet, P.; Le Helloco, A.; Donal, E.; Aupetit, J.F.; Aumont, M.C.; Galinier, M.; Eicher, J.C.; et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: The STARS-BNP Multicenter Study. J. Am. Coll. Cardiol. 2007, 49, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Lainchbury, J.G.; Troughton, R.W.; Strangman, K.M.; Frampton, C.M.; Pilbrow, A.; Yandle, T.G.; Hamid, A.K.; Nicholls, M.G.; Richards, A.M. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: Results from the BATTLESCARRED (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) trial. J. Am. Coll. Cardiol. 2009, 55, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Anstrom, K.J.; Adams, K.F.; Ezekowitz, J.A.; Fiuzat, M.; Houston-Miller, N.; Januzzi, J.L., Jr.; Mark, D.B.; Pina, I.L.; Passmore, G.; et al. Effect of natriuretic peptideguided therapy on hospitalization or cardiovascular mortality in high-risk patients with heart failure and reduced ejection fraction: A randomized clinical trial. JAMA 2017, 318, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, M.; Buser, P.; Rickli, H.; Gutmann, M.; Erne, P.; Rickenbacher, P.; Vuillomenet, A.; Jeker, U.; Dubach, P.; Beer, H.; et al. BNP-guided vs symptom-guided heart failure therapy: The Trial of Intensified vs Standard Medical Therapy in Elderly Patients with Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009, 301, 383–392. [Google Scholar] [CrossRef]
- Porapakkham, P.; Porapakkham, P.; Zimmet, H.; Billah, B.; Krum, H. B-type natriuretic peptide-guided heart failure therapy: A meta-analysis. Arch. Intern. Med. 2010, 170, 507–514. [Google Scholar] [CrossRef]
- Troughton, R.W.; Frampton, C.M.; Brunner-La Rocca, H.P.; Pfisterer, M.; Eurlings, L.W.; Erntell, H.; Persson, H.; O’Connor, C.M.; Moertl, D.; Karlstrom, P.; et al. Effect of B-type natriuretic peptide-guided treatment of chronic heart failure on total mortality and hospitalization: An individual patient meta-analysis. Eur. Heart J. 2014, 35, 1559–1567. [Google Scholar] [CrossRef]
- Kodogo, V.; Viljoen, C.; Hoevelmann, J.; Chakafana, G.; Tromp, J.; Farhan, H.A.; Goland, S.; van der Meer, P.; Karaye, K.; Kryczka, K.; et al. Proteomic Profiling in Patients with Peripartum Cardiomyopathy: A Biomarker Study of the ESC EORP PPCM Registry. JACC Heart Fail. 2023, 11, 1708–1725. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryczka, K.E.; Demkow, M.; Dzielińska, Z. Biomarkers in Peripartum Cardiomyopathy—What We Know and What Is Still to Be Found. Biomolecules 2024, 14, 103. https://doi.org/10.3390/biom14010103
Kryczka KE, Demkow M, Dzielińska Z. Biomarkers in Peripartum Cardiomyopathy—What We Know and What Is Still to Be Found. Biomolecules. 2024; 14(1):103. https://doi.org/10.3390/biom14010103
Chicago/Turabian StyleKryczka, Karolina E., Marcin Demkow, and Zofia Dzielińska. 2024. "Biomarkers in Peripartum Cardiomyopathy—What We Know and What Is Still to Be Found" Biomolecules 14, no. 1: 103. https://doi.org/10.3390/biom14010103
APA StyleKryczka, K. E., Demkow, M., & Dzielińska, Z. (2024). Biomarkers in Peripartum Cardiomyopathy—What We Know and What Is Still to Be Found. Biomolecules, 14(1), 103. https://doi.org/10.3390/biom14010103