
Repurposing of Tibolone in Alzheimer’s Disease

{kind=link}
Abstract
1. Introduction
2. Mitochondrial Dysfunction in Alzheimer’s Pathology
2.1. Changes in Mitochondrial Transport by TOM Proteins
2.2. Impairment of Mitochondrial Porins in AD
2.3. Impaired Mitochondrial Respiration in AD via ATP Synthase
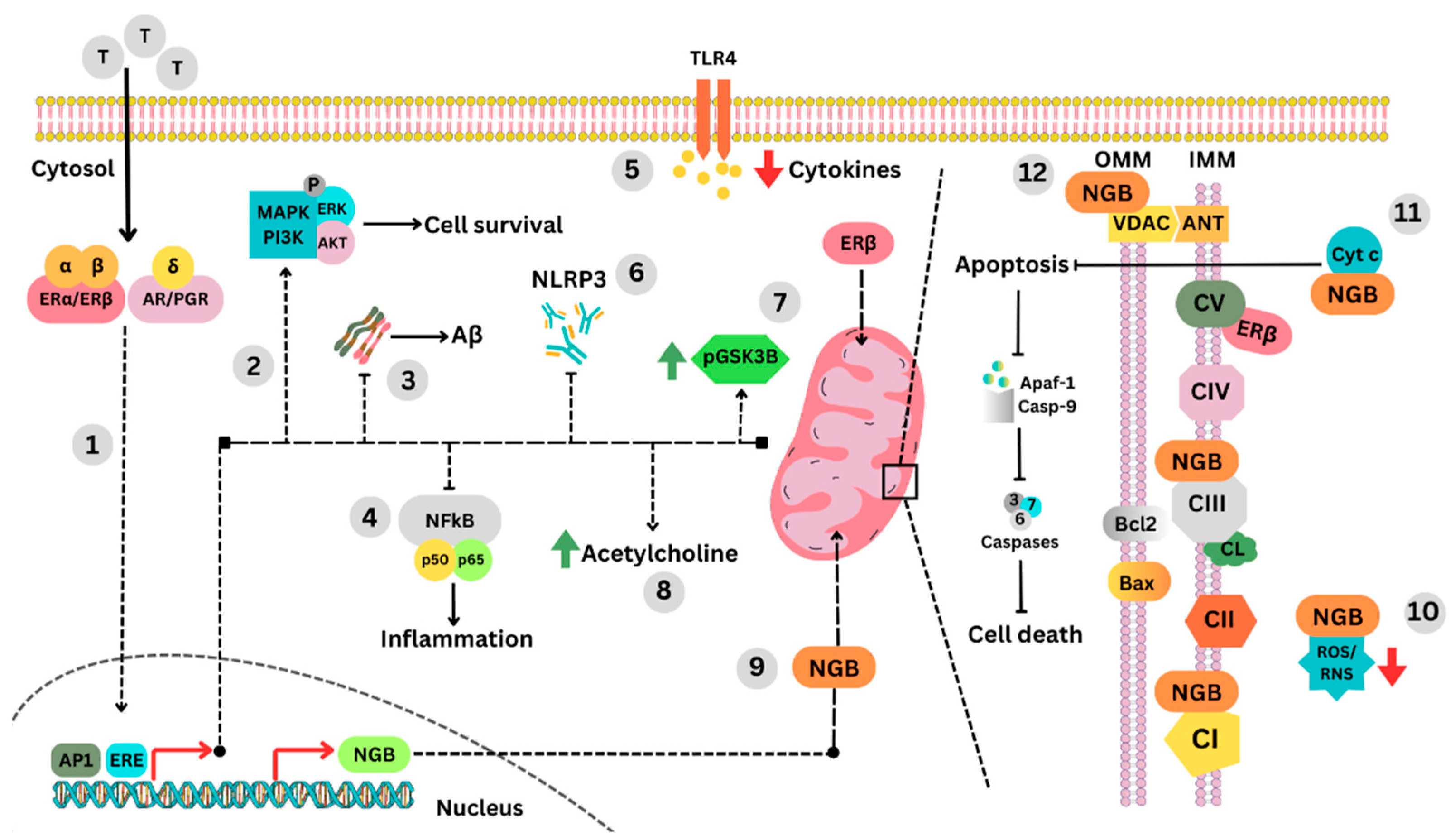
3. Molecular Signature of Tibolone’s Actions in the Brain
3.1. Neuroglobin Upregulation as a Protective Mechanism
3.2. Regulation of Apoptosis, VDAC, and Mitochondrial Trafficking
4. Regulation of Aβ and Tau Hyperphosphorylation
5. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Georges, J.; Miller, O.; Bintener, C. Estimating the prevalence of dementia in Europe. In Dementia in Europe Yearbook 2019; Alzheimer Europe: Luxembourg, 2020. [Google Scholar] [CrossRef]
- Mukadam, N.; Marston, L.; Lewis, G.; Mathur, R.; Rait, G.; Livingston, G. Incidence, age at diagnosis and survival with dementia across ethnic groups in England: A longitudinal study using electronic health records. Alzheimer’s Dement. 2023, 19, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.; Teixeira-Santos, A.C.; Caramelli, P.; Leist, A.K. Prevalence of dementia in Latin America and Caribbean countries: Systematic review and meta-analyses exploring age, sex, rurality, and education as possible determinants. Ageing Res. Rev. 2022, 81, 101703. [Google Scholar] [CrossRef] [PubMed]
- Custodio, N.; Wheelock, A.; Thumala, D.; Slachevsky, A. Dementia in Latin America: Epidemiological Evidence and Implications for Public Policy. Front. Aging Neurosci. 2017, 9, 221. [Google Scholar] [CrossRef]
- Lobo, A.; Launer, L.J.; Fratiglioni, L.; Andersen, K.; Di Carlo, A.; Breteler, M.M.; Copeland, J.R.; Dartigues, J.F.; Jagger, C.; Martinez-Lage, J.; et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54 (Suppl. 5), S4–S9. [Google Scholar]
- Fratiglioni, L.; Viitanen, M.; von Strauss, E.; Tontodonati, V.; Herlitz, A.; Winblad, B. Very Old Women at Highest Risk of Dementia and Alzheimer’s Disease: Incidence Data from the Kungsholmen Project, Stockholm. Neurology 1997, 48, 132–138. [Google Scholar] [CrossRef]
- Coughlan, G.T.; Betthauser, T.J.; Boyle, R.; Koscik, R.L.; Klinger, H.M.; Chibnik, L.B.; Jonaitis, E.M.; Yau, W.-Y.W.; Wenzel, A.; Christian, B.T.; et al. Association of Age at Menopause and Hormone Therapy Use with Tau and β-Amyloid Positron Emission Tomography. JAMA Neurol. 2023, 80, 462–473. [Google Scholar] [CrossRef]
- Imtiaz, B.; Tuppurainen, M.; Tiihonen, M.; Kivipelto, M.; Soininen, H.; Hartikainen, S.; Tolppanen, A.-M. Oophorectomy, Hysterectomy, and Risk of Alzheimer’s Disease: A Nationwide Case-Control Study. J. Alzheimer’s Dis. 2014, 42, 575–581. [Google Scholar] [CrossRef]
- Bove, R.; Secor, E.; Chibnik, L.B.; Barnes, L.L.; Schneider, J.A.; Bennett, D.A.; De Jager, P.L. Age at surgical menopause influences cognitive decline and Alzheimer pathology in older women. Neurology 2014, 82, 222–229. [Google Scholar] [CrossRef]
- Corbo, R.M.; Gambina, G.; Broggio, E.; Scacchi, R. Influence of Variation in the Follicle-Stimulating Hormone Receptor Gene (FSHR) and Age at Menopause on the Development of Alzheimer’s Disease in Women. Dement. Geriatr. Cogn. Disord. 2011, 32, 63–69. [Google Scholar] [CrossRef]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef]
- Vest, R.S.; Pike, C.J. Gender, sex steroid hormones, and Alzheimer’s disease. Horm. Behav. 2013, 63, 301–307. [Google Scholar] [CrossRef]
- Buckley, R.F.; Mormino, E.C.; Rabin, J.S.; Hohman, T.J.; Landau, S.; Hanseeuw, B.; Jacobs, H.; Papp, K.V.; Amariglio, R.E.; Properzi, M.J.; et al. Sex Differences in the Association of Global Amyloid and Regional Tau Deposition Measured by Positron Emission Tomography in Clinically Normal Older Adults. JAMA Neurol. 2019, 76, 542–551. [Google Scholar] [CrossRef]
- Shults, C.L.; Pinceti, E.; Giffin-Rao, Y.S.; Pak, T.R. Aging and Loss of Circulating 17β-Estradiol Alters the Alternative Splicing of ERβ in the Female Rat Brain. Endocrinology 2015, 156, 4187–4199. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Yuri, K. Estrogen-dependent changes in estrogen receptor-β mRNA expression in middle-aged female rat brain. Brain Res. 2014, 1543, 49–57. [Google Scholar] [CrossRef]
- Yamaguchi-Shima, N.; Yuri, K. Age-related changes in the expression of ER-β mRNA in the female rat brain. Brain Res. 2007, 1155, 34–41. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Patel, H.H.; Brand, F.J., 3rd; Perez-Pinzon, M.A.; Bramlett, H.M.; Raval, A.P. Estrogen receptor beta signaling alters cellular inflammasomes activity after global cerebral ischemia in reproductively senescence female rats. J. Neurochem. 2016, 136, 492–496. [Google Scholar] [CrossRef]
- González-Giraldo, Y.; Forero, D.A.; Echeverria, V.; Garcia-Segura, L.M.; Barreto, G.E. Tibolone attenuates inflammatory response by palmitic acid and preserves mitochondrial membrane potential in astrocytic cells through estrogen receptor beta. Mol. Cell. Endocrinol. 2019, 486, 65–78. [Google Scholar] [CrossRef]
- Hidalgo-Lanussa, O.; Ávila-Rodriguez, M.; Baez-Jurado, E.; Zamudio, J.; Echeverria, V.; Garcia-Segura, L.M.; Barreto, G.E. Tibolone Reduces Oxidative Damage and Inflammation in Microglia Stimulated with Palmitic Acid through Mechanisms Involving Estrogen Receptor Beta. Mol. Neurobiol. 2018, 55, 5462–5477. [Google Scholar] [CrossRef]
- Avila-Rodriguez, M.; Garcia-Segura, L.M.; Hidalgo-Lanussa, O.; Baez, E.; Gonzalez, J.; Barreto, G.E. Tibolone protects astrocytic cells from glucose deprivation through a mechanism involving estrogen receptor beta and the upregulation of neuroglobin expression. Mol. Cell. Endocrinol. 2016, 433, 35–46. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Varsavsky, I.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging. Neurology 2017, 89, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lee, J.H.; Pang, D.; Temkin, A.; Park, N.; Janicki, S.C.; Zigman, W.B.; Silverman, W.; Tycko, B.; Schupf, N. Estrogen Receptor-Beta Variants Are Associated with Increased Risk of Alzheimer’s Disease in Women with Down Syndrome. Dement. Geriatr. Cogn. Disord. 2011, 32, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Lindquist, K.; Sen, S.; Cauley, J.; Ferrell, R.; Penninx, B.; Harris, T.; Li, R.; Cummings, S.R. Estrogen receptor genotype and risk of cognitive impairment in elders: Findings from the Health ABC study. Neurobiol. Aging 2009, 30, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Luckhaus, C.; Spiegler, C.; Ibach, B.; Fischer, P.; Wichart, I.; Sterba, N.; Gatterer, G.; Rainer, M.; Jungwirth, S.; Huber, K.; et al. Estrogen Receptor β Gene (ESRβ) 3′-UTR Variants in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Pirskanen, M.; Hiltunen, M.; Mannermaa, A.; Helisalmi, S.; Lehtovirta, M.; Hänninen, T.; Soininen, H. Estrogen receptor beta gene variants are associated with increased risk of Alzheimer’s disease in women. Eur. J. Hum. Genet. 2005, 13, 1000–1006. [Google Scholar] [CrossRef]
- Cao, X.; Chen, Y.; Sang, X.; Xu, S.; Xie, Z.; Zhu, Z.; Wang, P.; Bi, J.; Xu, L. Impact prediction of translocation of the mitochondrial outer membrane 70 as biomarker in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1013943. [Google Scholar] [CrossRef]
- Leal, N.S.; Dentoni, G.; Schreiner, B.; Naia, L.; Piras, A.; Graff, C.; Cattaneo, A.; Meli, G.; Hamasaki, M.; Nilsson, P.; et al. Amyloid β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models. Cells 2020, 9, 2552. [Google Scholar] [CrossRef]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Hatanpää, K.; I Rapoport, S.; Brady, D.R. Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res. Mol. Brain Res. 1997, 44, 99–104. [Google Scholar] [CrossRef]
- Yang, S.-H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.-M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor β. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef]
- Álvarez-Delgado, C.; Mendoza-Rodríguez, C.A.; Picazo, O.; Cerbón, M. Different expression of α and β mitochondrial estrogen receptors in the aging rat brain: Interaction with respiratory complex V. Exp. Gerontol. 2010, 45, 580–585. [Google Scholar] [CrossRef]
- Barreto, G.E.; McGovern, A.J.; Garcia-Segura, L.M. Role of Neuroglobin in the Neuroprotective Actions of Estradiol and Estrogenic Compounds. Cells 2021, 10, 1907. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Aslani, S.; Barreto, G.E.; Báez-Jurado, E.; Kiaie, N.; Jamialahmadi, T.; Sahebkar, A. The potential of mitochondrial modulation by neuroglobin in treatment of neurological disorders. Free Radic. Biol. Med. 2021, 162, 471–477. [Google Scholar] [CrossRef]
- Cabezas, R.; Baez-Jurado, E.; Hidalgo-Lanussa, O.; Echeverria, V.; Ashrad, G.M.; Sahebkar, A.; Barreto, G.E. Growth Factors and Neuroglobin in Astrocyte Protection against Neurodegeneration and Oxidative Stress. Mol. Neurobiol. 2019, 56, 2339–2351. [Google Scholar] [CrossRef]
- Del Río, J.P.; Molina, S.; Hidalgo-Lanussa, O.; Garcia-Segura, L.M.; Barreto, G.E. Tibolone as Hormonal Therapy and Neuroprotective Agent. Trends Endocrinol. Metab. 2020, 31, 742–759. [Google Scholar] [CrossRef]
- Verheul, H.; Kloosterboer, H. Metabolism of exogenous sex steroids and effect on brain functions with a focus on tibolone. J. Steroid Biochem. Mol. Biol. 2006, 102, 195–204. [Google Scholar] [CrossRef]
- Kloosterboer, H.J. Tissue-selectivity: The mechanism of action of tibolone. Maturitas 2004, 48 (Suppl. 1), 30–40. [Google Scholar] [CrossRef]
- Sung, Y.-F.; Tsai, C.-T.; Kuo, C.-Y.; Lee, J.-T.; Chou, C.-H.; Chen, Y.-C.; Chou, Y.-C.; Sun, C.-A. Use of Hormone Replacement Therapy and Risk of Dementia: A Nationwide Cohort Study. Neurology 2022, 99, e1835–e1842. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Quesenberry, C.P., Jr.; Zhou, J.; Yaffe, K. Timing of hormone therapy and dementia: The critical window theory revisited. Ann. Neurol. 2011, 69, 163–169. [Google Scholar] [CrossRef]
- Rocca, W.A.; Grossardt, B.R.; Shuster, L.T. Oophorectomy, menopause, estrogen treatment, and cognitive aging: Clinical evidence for a window of opportunity. Brain Res. 2011, 1379, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Henderson, V.W.; Benke, K.S.; Green, R.C.; A Cupples, L.; A Farrer, L. Postmenopausal hormone therapy and Alzheimer’s disease risk: Interaction with age. J. Neurol. Neurosurg. Psychiatry 2005, 76, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Mills, Z.B.; Faull, R.L.M.; Kwakowsky, A. Is Hormone Replacement Therapy a Risk Factor or a Therapeutic Option for Alzheimer’s Disease? Int. J. Mol. Sci. 2023, 24, 3205. [Google Scholar] [CrossRef] [PubMed]
- Banarase, T.A.; Sammeta, S.S.; Wankhede, N.L.; Mangrulkar, S.V.; Rahangdale, S.R.; Aglawe, M.M.; Taksande, B.G.; Upaganlawar, A.B.; Umekar, M.J.; Kale, M.B. Mitophagy regulation in aging and neurodegenerative disease. Biophys. Rev. 2023, 15, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.-S.; Jiang, Q.-W.; Chen, S.-D. Sex difference in biological change and mechanism of Alzheimer’s disease: From macro- to micro-landscape. Ageing Res. Rev. 2023, 87, 101918. [Google Scholar] [CrossRef]
- Kalani, K.; Chaturvedi, P.; Chaturvedi, P.; Verma, V.K.; Lal, N.; Awasthi, S.K.; Kalani, A. Mitochondrial mechanisms in Alzheimer’s disease: Quest for therapeutics. Drug Discov. Today 2023, 28, 103547. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: A Current Overview. J. Alzheimer’s Dis. 2023, 92, 751–768. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]
- Hill, K.; Model, K.; Ryan, M.T.; Dietmeier, K.; Martin, F.; Wagner, R.; Pfanner, N. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nature 1998, 395, 516–521. [Google Scholar] [CrossRef]
- Petersen, C.A.H.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef]
- Weidberg, H.; Amon, A. MitoCPR—A surveillance pathway that protects mitochondria in response to protein import stress. Science 2018, 360, eaan4146. [Google Scholar] [CrossRef]
- Chai, Y.L.; Xing, H.; Chong, J.R.; Francis, P.T.; Ballard, C.G.; Chen, C.P.; Lai, M.K. Mitochondrial Translocase of the Outer Membrane Alterations May Underlie Dysfunctional Oxidative Phosphorylation in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 61, 793–801. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.-P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef]
- Lemeshko, V.V. VDAC as a voltage-dependent mitochondrial gatekeeper under physiological conditions. Biochim. Biophys. Acta Biomembr. 2023, 1865, 184175. [Google Scholar] [CrossRef]
- Galganska, H.; Karachitos, A.; Wojtkowska, M.; Stobienia, O.; Budzinska, M.; Kmita, H. Communication between mitochondria and nucleus: Putative role for VDAC in reduction/oxidation mechanism. Biochim. Biophys. Acta 2010, 1797, 1276–1280. [Google Scholar] [CrossRef]
- Benz, R. Permeation of hydrophilic solutes through mitochondrial outer membranes: Review on mitochondrial porins. Biochim. Biophys. Acta 1994, 1197, 167–196. [Google Scholar] [CrossRef]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent Anion Channel 1-based Peptides Interact with Bcl-2 to Prevent Antiapoptotic Activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Antonsson, B.; Suzuki, M.; Youle, R.J.; Colombini, M.; Bezrukov, S.M. Bid, but Not Bax, Regulates VDAC Channels. J. Biol. Chem. 2004, 279, 13575–13583. [Google Scholar] [CrossRef]
- Naghdi, S.; Várnai, P.; Hajnóczky, G. Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis. Proc. Natl. Acad. Sci. USA 2015, 112, E5590–E5599. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer’s disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Menz, R.; Walker, J.E.; Leslie, A.G. Structure of Bovine Mitochondrial F1-ATPase with Nucleotide Bound to All Three Catalytic Sites: Implications for the Mechanism of Rotary Catalysis. Cell 2001, 106, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ueno, H.; Mitome, N.; Suzuki, J.; Yoshida, M. F0 of ATP Synthase Is a Rotary Proton Channel. Obligatory coupling of proton translocation with rotation of c-subunit ring. J. Biol. Chem. 2002, 277, 13281–13285. [Google Scholar] [CrossRef]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef]
- Sergeant, N.; Wattez, A.; Galvn-Valencia, M.; Ghestem, A.; David, J.-P.; Lemoine, J.; Sautire, P.-E.; Dachary, J.; Mazat, J.-P.; Michalski, J.-C.; et al. Association of atp synthase α-chain with neurofibrillary degeneration in alzheimer’s disease. Neuroscience 2003, 117, 293–303. [Google Scholar] [CrossRef]
- Xing, S.-L.; Chen, B.; Shen, D.-Z.; Zhu, C.-Q. β-amyloid Peptide Binds and Regulates Ectopic ATP Synthase α-Chain on Neural Surface. Int. J. Neurosci. 2012, 122, 290–297. [Google Scholar] [CrossRef]
- Chou, J.L.; Shenoy, D.V.; Thomas, N.; Choudhary, P.K.; LaFerla, F.M.; Goodman, S.R.; Breen, G.A. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J. Proteom. 2011, 74, 466–479. [Google Scholar] [CrossRef]
- Delmas, P.D.; Davis, S.R.; Hensen, J.; Adami, S.; van Os, S.; Nijland, E.A. Effects of tibolone and raloxifene on bone mineral density in osteopenic postmenopausal women. Osteoporos. Int. 2008, 19, 1153–1160. [Google Scholar] [CrossRef]
- Landgren, M.; Helmond, F.; Engelen, S. Tibolone relieves climacteric symptoms in highly symptomatic women with at least seven hot flushes and sweats per day. Maturitas 2005, 50, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Albertazzi, P.; Natale, V.; Barbolini, C.; Teglio, L.; Di Micco, R. The effect of tibolone versus continuous combined norethisterone acetate and oestradiol on memory, libido and mood of postmenopausal women: A pilot study. Maturitas 2000, 36, 223–229. [Google Scholar] [CrossRef]
- E de Gooyer, M.; Deckers, G.H.; Schoonen, W.G.; Verheul, H.A.; Kloosterboer, H.J. Receptor profiling and endocrine interactions of tibolone. Steroids 2003, 68, 21–30. [Google Scholar] [CrossRef]
- Hota, K.B.; Hota, S.K.; Srivastava, R.B.; Singh, S.B. Neuroglobin Regulates Hypoxic Response of Neuronal Cells through Hif-1α- and Nrf2-mediated Mechanism. J. Cereb. Blood Flow Metab. 2012, 32, 1046–1060. [Google Scholar] [CrossRef]
- Liu, N.; Yu, Z.; Xiang, S.; Zhao, S.; Tjärnlund-Wolf, A.; Xing, C.; Zhang, J.; Wang, X. Transcriptional regulation mechanisms of hypoxia-induced neuroglobin gene expression. Biochem. J. 2012, 443, 153–164. [Google Scholar] [CrossRef]
- Hansson, L.O.; Friedler, A.; Freund, S.; Rüdiger, S.; Fersht, A.R. Two sequence motifs from HIF-1α bind to the DNA-binding site of p53. Proc. Natl. Acad. Sci. USA 2002, 99, 10305–10309. [Google Scholar] [CrossRef]
- Brunori, M.; Giuffrè, A.; Nienhaus, K.; Nienhaus, G.U.; Scandurra, F.M.; Vallone, B. Neuroglobin, nitric oxide, and oxygen: Functional pathways and conformational changes. Proc. Natl. Acad. Sci. USA 2005, 102, 8483–8488. [Google Scholar] [CrossRef]
- Baez-Jurado, E.; Guio-Vega, G.; Hidalgo-Lanussa, O.; González, J.; Echeverria, V.; Ashraf, G.M.; Sahebkar, A.; Barreto, G.E. Mitochondrial Neuroglobin Is Necessary for Protection Induced by Conditioned Medium from Human Adipose-Derived Mesenchymal Stem Cells in Astrocytic Cells Subjected to Scratch and Metabolic Injury. Mol. Neurobiol. 2019, 56, 5167–5187. [Google Scholar] [CrossRef]
- Baez-Jurado, E.; Vega, G.G.; Aliev, G.; Tarasov, V.V.; Esquinas, P.; Echeverria, V.; Barreto, G.E. Blockade of Neuroglobin Reduces Protection of Conditioned Medium from Human Mesenchymal Stem Cells in Human Astrocyte Model (T98G) Under a Scratch Assay. Mol. Neurobiol. 2018, 55, 2285–2300. [Google Scholar] [CrossRef]
- Baez, E.; Echeverria, V.; Cabezas, R.; Ávila-Rodriguez, M.; Garcia-Segura, L.M.; Barreto, G.E. Protection by Neuroglobin Expression in Brain Pathologies. Front. Neurol. 2016, 7, 146. [Google Scholar] [CrossRef]
- Brittain, T.; Skommer, J.; Raychaudhuri, S.; Birch, N. An Antiapoptotic Neuroprotective Role for Neuroglobin. Int. J. Mol. Sci. 2010, 11, 2306–2321. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Skommer, J.; Henty, K.; Birch, N.; Brittain, T. Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis 2010, 15, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Canali, G.; Garcia, M.; Hivert, B.; Pinatel, D.; Goullancourt, A.; Oguievetskaia, K.; Saint-Martin, M.; Girault, J.-A.; Faivre-Sarrailh, C.; Goutebroze, L. Genetic variants in autism-related CNTNAP2 impair axonal growth of cortical neurons. Hum. Mol. Genet. 2018, 27, 1941–1954. [Google Scholar] [CrossRef]
- Szymanski, M.; Wang, R.; Fallin, M.D.; Bassett, S.S.; Avramopoulos, D. Neuroglobin and Alzheimer’s dementia: Genetic association and gene expression changes. Neurobiol. Aging 2010, 31, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liu, N.; Li, Y.; Xu, J.; Wang, X. Neuroglobin overexpression inhibits oxygen–glucose deprivation-induced mitochondrial permeability transition pore opening in primary cultured mouse cortical neurons. Neurobiol. Dis. 2013, 56, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Duong, T.T.H.; Witting, P.K.; Antao, S.T.; Parry, S.N.; Kennerson, M.; Lai, B.; Vogt, S.; Lay, P.A.; Harris, H.H. Multiple protective activities of neuroglobin in cultured neuronal cells exposed to hypoxia re-oxygenation injury. J. Neurochem. 2009, 108, 1143–1154. [Google Scholar] [CrossRef]
- Lechauve, C.; Augustin, S.; Cwerman-Thibault, H.; Bouaita, A.; Forster, V.; Célier, C.; Rustin, P.; Marden, M.C.; Sahel, J.-A.; Corral-Debrinski, M. Neuroglobin involvement in respiratory chain function and retinal ganglion cell integrity. Biochim. Biophys. Acta 2012, 1823, 2261–2273. [Google Scholar] [CrossRef]
- Long, J.; He, P.; Shen, Y.; Li, R. New Evidence of Mitochondria Dysfunction in the Female Alzheimer’s Disease Brain: Deficiency of Estrogen Receptor-β. J. Alzheimer’s Dis. 2012, 30, 545–558. [Google Scholar] [CrossRef]
- Lambrinoudaki, I.; Karaflou, M.; Kaparos, G.; Alexandrou, A.; Creatsa, M.; Aravantinos, L.; Augoulea, A.; Kouskouni, E. Effect of tibolone and raloxifene on serum markers of apoptosis in postmenopausal women. Climacteric 2013, 16, 258–264. [Google Scholar] [CrossRef]
- Christodoulakos, G.E.; Lambrinoudaki, I.V.; Creatsa, M.G.; Economou, E.V.; Siasou, Z.; Panoulis, C.P.; Kalligerou, I.; Papadias, C. Circulating levels of atherogenesis-associated adipocytokines and apoptotic markers are differentially influenced by hormone therapy, tibolone and raloxifene in healthy postmenopausal women. Climacteric 2008, 11, 155–165. [Google Scholar] [CrossRef]
- Kummer, K.K.; Zeidler, M.; Kalpachidou, T.; Kress, M. Role of IL-6 in the regulation of neuronal development, survival and function. Cytokine 2021, 144, 155582. [Google Scholar] [CrossRef]
- Gonzalez, Y.; Garzón-Benitez, A.V.; Forero, D.; Barreto, G.E. TERTinhibition leads to reduction ofIL-6 expression induced by palmitic acid and interferes with the protective effects of tibolone in an astrocytic cell model. J. Neuroendocr. 2019, 31, e12768. [Google Scholar] [CrossRef]
- McGovern, A.J.; Barreto, G.E. Network pharmacology identifies IL6 as an important hub and target of tibolone for drug repurposing in traumatic brain injury. Biomed. Pharmacother. 2021, 140, 111769. [Google Scholar] [CrossRef]
- Schubert, C.; Raparelli, V.; Westphal, C.; Dworatzek, E.; Petrov, G.; Kararigas, G.; Regitz-Zagrosek, V. Reduction of apoptosis and preservation of mitochondrial integrity under ischemia/reperfusion injury is mediated by estrogen receptor β. Biol. Sex Differ. 2016, 7, 53. [Google Scholar] [CrossRef]
- Vesga-Jiménez, D.J.; Martín-Jiménez, C.A.; Rodríguez, A.G.; Aristizábal-Pachón, A.F.; Pinzón, A.; Barreto, G.E.; Ramírez, D.; González, J. Tibolone Pre-Treatment Ameliorates the Dysregulation of Protein Translation and Transport Generated by Palmitic Acid-Induced Lipotoxicity in Human Astrocytes: A Label-Free MS-Based Proteomics and Network Analysis. Int. J. Mol. Sci. 2022, 23, 6454. [Google Scholar] [CrossRef]
- Frohnert, C.; Hutten, S.; Wälde, S.; Nath, A.; Kehlenbach, R.H. Importin 7 and Nup358 Promote Nuclear Import of the Protein Component of Human Telomerase. PLoS ONE 2014, 9, e88887. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. [Google Scholar] [CrossRef]
- González-Giraldo, Y.; Forero, D.A.; Echeverria, V.; Gonzalez, J.; Ávila-Rodriguez, M.; Garcia-Segura, L.M.; Barreto, G.E. Neuroprotective effects of the catalytic subunit of telomerase: A potential therapeutic target in the central nervous system. Ageing Res. Rev. 2016, 28, 37–45. [Google Scholar] [CrossRef]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.-K.; et al. Telomerase directly regulates NF-κB-dependent transcription. Nature 2012, 14, 1270–1281. [Google Scholar] [CrossRef]
- Dhanoya, A.; Wang, T.; Keshavarz-Moore, E.; Fassati, A.; Chain, B.M. Importin-7 Mediates Nuclear Trafficking of DNA in Mammalian Cells. Traffic 2012, 14, 165–175. [Google Scholar] [CrossRef]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; de Walque, R.; De Vleeschouwer, C.; Lafontaine, D.L.J. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.S.; Kayed, R.; Abate, G.; Uberti, D.; Kinnon, P.; Piccirella, S. Post-translational Modifications of the p53 Protein and the Impact in Alzheimer’s Disease: A Review of the Literature. Front. Aging Neurosci. 2022, 14, 835288. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Tu, K.J.; Wei, A.; Lau, A.J.; Gonzalez-Gil, A.; Cao, T.; Braunstein, K.; Ling, J.P.; Troncoso, J.C.; Wong, P.C.; et al. Amyloid-beta and tau pathologies act synergistically to induce novel disease stage-specific microglia subtypes. Mol. Neurodegener. 2022, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Rabin, J.S.; Yang, H.-S.; Schultz, A.P.; Hanseeuw, B.J.; Hedden, T.; Viswanathan, A.; Gatchel, J.R.; Marshall, G.A.; Bs, E.K.; Klein, H.; et al. Vascular Risk and β -Amyloid Are Synergistically Associated with Cortical Tau. Ann. Neurol. 2018, 85, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Lovestone, S.; Reynolds, C.; Latimer, D.; Davis, D.R.; Anderton, B.H.; Gallo, J.-M.; Hanger, D.; Mulot, S.; Marquardt, B.; Stabel, S.; et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994, 4, 1077–1086. [Google Scholar] [CrossRef]
- Segura-Uribe, J.J.; la Torre, P.G.-D.; Castillo-Mendieta, T.; Bribiesca-Cruz, I.; Orozco-Suárez, S.; Soriano-Ursúa, M.A.; Pinto-Almazán, R.; Fuentes-Venado, C.E.; Guerra-Araiza, C. Tibolone Improves Memory and Decreases the Content of Amyloid-β Peptides and Tau Protein in the Hippocampus of a Murine Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 90, 1437–1447. [Google Scholar] [CrossRef]
- Yang, W.; Xu, Q.-Q.; Yuan, Q.; Xian, Y.-F.; Lin, Z.-X. Sulforaphene, a CDK5 Inhibitor, attenuates cognitive deficits in a transgenic mouse model of Alzheimer’s disease via reducing Aβ Deposition, tau hyperphosphorylation and synaptic dysfunction. Int. Immunopharmacol. 2023, 114, 109504. [Google Scholar] [CrossRef]
- Crespo-Biel, N.; Canudas, A.; Camins, A.; Pallàs, M. Kainate induces AKT, ERK and cdk5/GSK3β pathway deregulation, phosphorylates tau protein in mouse hippocampus. Neurochem. Int. 2007, 50, 435–442. [Google Scholar] [CrossRef]
- Guerra-Araiza, C.; Neri-Gómez, T.; Espinosa-Raya, J.; Díaz-Cintra, S.; Segura-Uribe, J.; Orozco-Suárez, S.; Gallardo, J.M. Tibolone modulates neuronal plasticity through regulating Tau, GSK3β/Akt/PI3K pathway and CDK5 p35/p25 complexes in the hippocampus of aged male mice. Neural Regen. Res. 2017, 12, 588–595. [Google Scholar] [CrossRef]
- Espinosa-Raya, J.; Neri-Gómez, T.; Orozco-Suárez, S.; Campos, M.G.; Guerra-Araiza, C. Chronic administration of tibolone modulates anxiety-like behavior and enhances cognitive performance in ovariectomized rats. Horm. Behav. 2012, 61, 76–83. [Google Scholar] [CrossRef]
- García, E.D.F.; Castillo-Hernández, M.C.; Pinto-Almazán, R.; Rivas-Arancibia, S.; Gallardo, J.M.; Guerra-Araiza, C. Tibolone Prevents Oxidation and Ameliorates Cholinergic Deficit Induced by Ozone Exposure in the Male Rat Hippocampus. Neurochem. Res. 2014, 39, 1776–1786. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.; Davis, J.G.; Constantine, G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cognitive correlates of alterations in acetylcholinesterase in Alzheimer’s disease. Neurosci. Lett. 2005, 380, 127–132. [Google Scholar] [CrossRef]
- Sims, N.R.; Bowen, D.M.; Allen, S.J.; Smith, C.C.T.; Neary, D.; Thomas, D.J.; Davison, A.N. Presynaptic Cholinergic Dysfunction in Patients with Dementia. J. Neurochem. 1983, 40, 503–509. [Google Scholar] [CrossRef]
- Wilcock, G.; Esiri, M.; Bowen, D.; Smith, C. Alzheimer’s disease: Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 1982, 57, 407–417. [Google Scholar] [CrossRef]
- Perry, E.K.; E Tomlinson, B.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. BMJ 1978, 2, 1457–1459. [Google Scholar] [CrossRef]
- Almey, A.; Milner, T.A.; Brake, W.G. Estrogen receptors in the central nervous system and their implication for dopamine-dependent cognition in females. Horm. Behav. 2015, 74, 125–138. [Google Scholar] [CrossRef]
- Pau, C.Y.; Pau, K.-Y.; Spies, H.G. Putative estrogen receptor β and α mRNA expression in male and female rhesus macaques. Mol. Cell. Endocrinol. 1998, 146, 59–68. [Google Scholar] [CrossRef]
- Khan, A.A.; Mao, X.O.; Banwait, S.; Jin, K.; Greenberg, D.A. Neuroglobin attenuates β-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19114–19119. [Google Scholar] [CrossRef]
- Chen, L.-M.; Xiong, Y.-S.; Kong, F.-L.; Qu, M.; Wang, Q.; Chen, X.-Q.; Wang, J.-Z.; Zhu, L.-Q. Neuroglobin attenuates Alzheimer-like tau hyperphosphorylation by activating Akt signaling. J. Neurochem. 2011, 120, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, Y.-B.; Sun, J.-Y.; Xiang, Y.; Yang, J.; Dai, S.-Y.; Zhang, X. Neuroglobin Attenuates Beta Amyloid-Induced Apoptosis Through Inhibiting Caspases Activity by Activating PI3K/Akt Signaling Pathway. J. Mol. Neurosci. 2015, 58, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Mao, X.; Xie, L.; Greenberg, D.A.; Jin, K. Neuroglobin Protein is Upregulated in Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 36, 659–663. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreto, G.E. Repurposing of Tibolone in Alzheimer’s Disease. Biomolecules 2023, 13, 1115. https://doi.org/10.3390/biom13071115
Barreto GE. Repurposing of Tibolone in Alzheimer’s Disease. Biomolecules. 2023; 13(7):1115. https://doi.org/10.3390/biom13071115
Chicago/Turabian StyleBarreto, George E. 2023. "Repurposing of Tibolone in Alzheimer’s Disease" Biomolecules 13, no. 7: 1115. https://doi.org/10.3390/biom13071115
APA StyleBarreto, G. E. (2023). Repurposing of Tibolone in Alzheimer’s Disease. Biomolecules, 13(7), 1115. https://doi.org/10.3390/biom13071115