_Kwok.png)
Engineering of a Bispecific Nanofitin with Immune Checkpoint Inhibitory Activity Conditioned by the Cross-Arm Binding to EGFR and PDL1


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construct, Expression and Purification of Proteins
2.2. Quality Control by Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis
2.3. Biolayer Interferometry Analyses
2.4. Enzyme-Linked Immunosorbent Assays
2.5. Cell Culture
2.6. Cell-Surface Binding by Flow Cytometry
2.7. Western Blot Analysis
2.8. PD1/PDL1 Blockade Bioassay
2.9. Agilent X-Celligence Real-Time Cell Analysis (RTCA)
3. Results
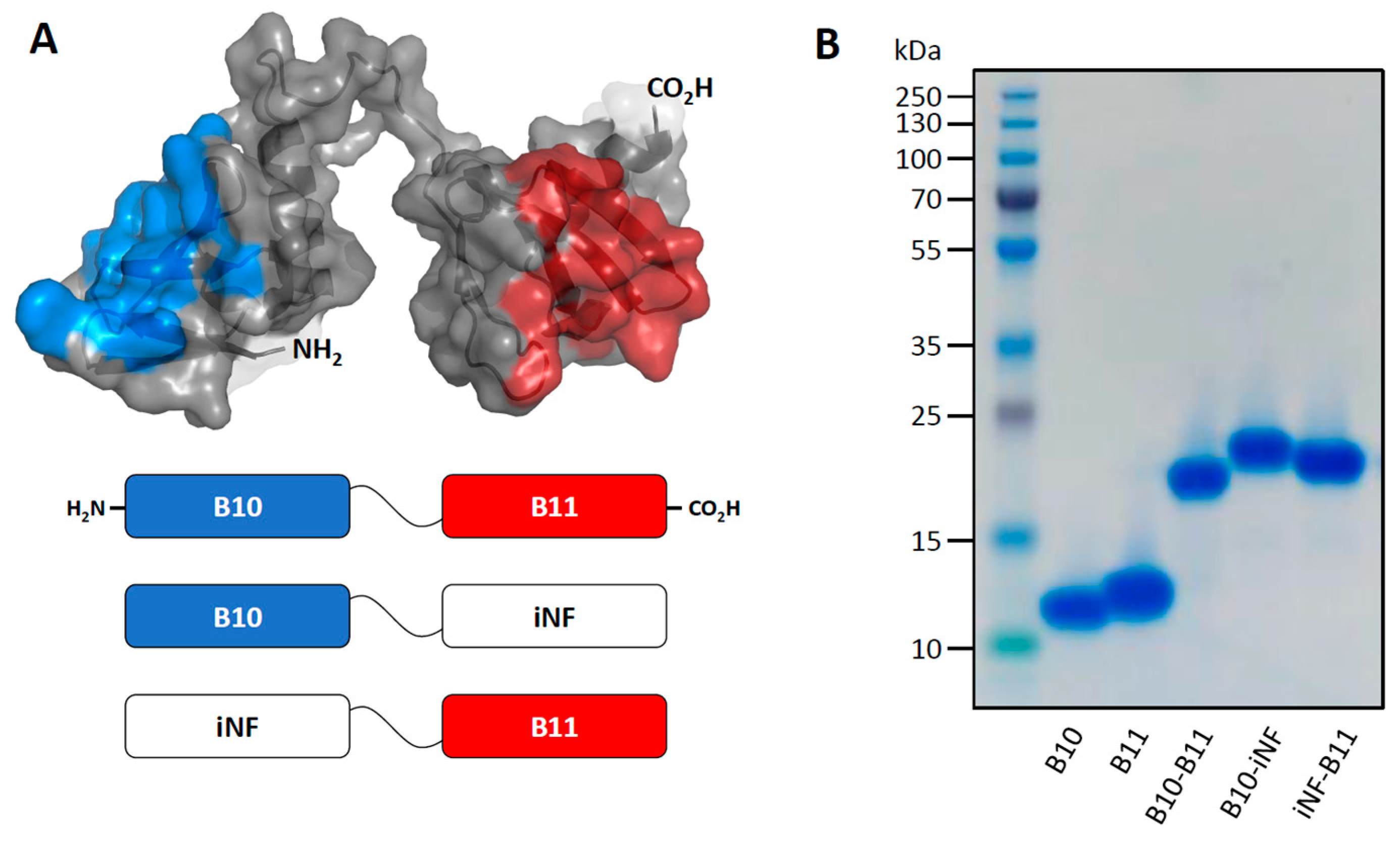
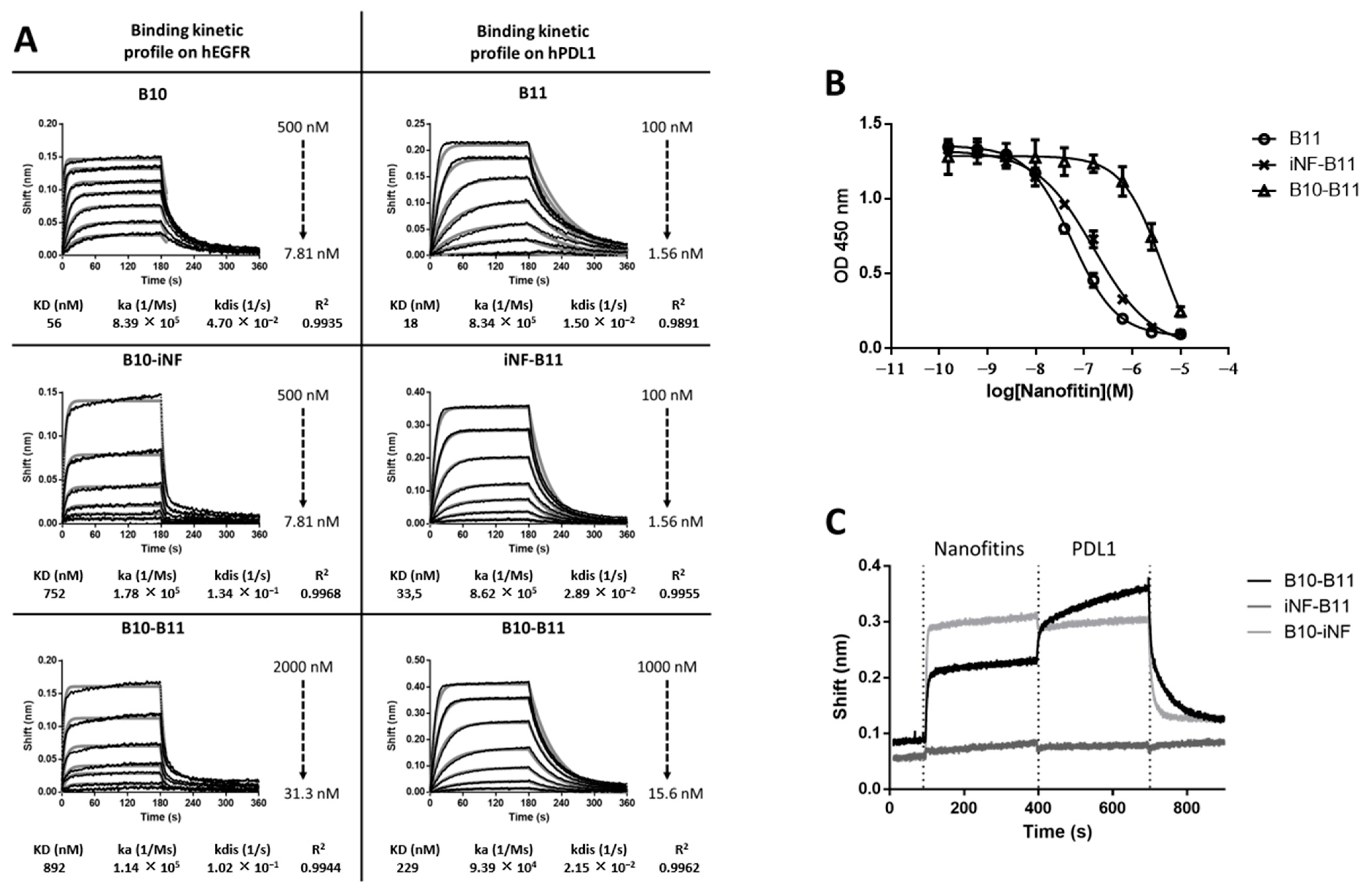
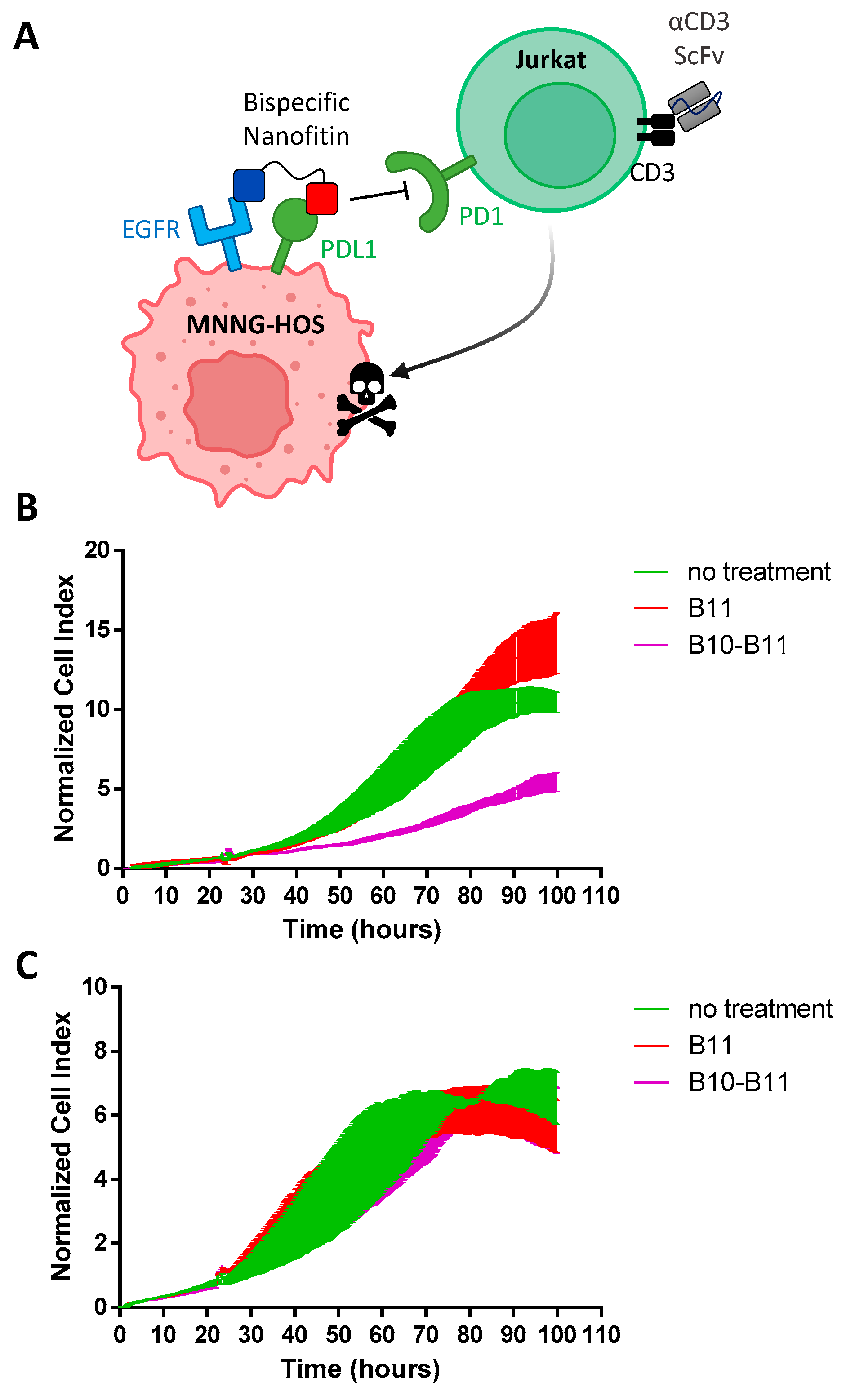
3.1. The Bispecific Nanofitin B10-B11 Can Engage Simultaneously EGFR and PDL1
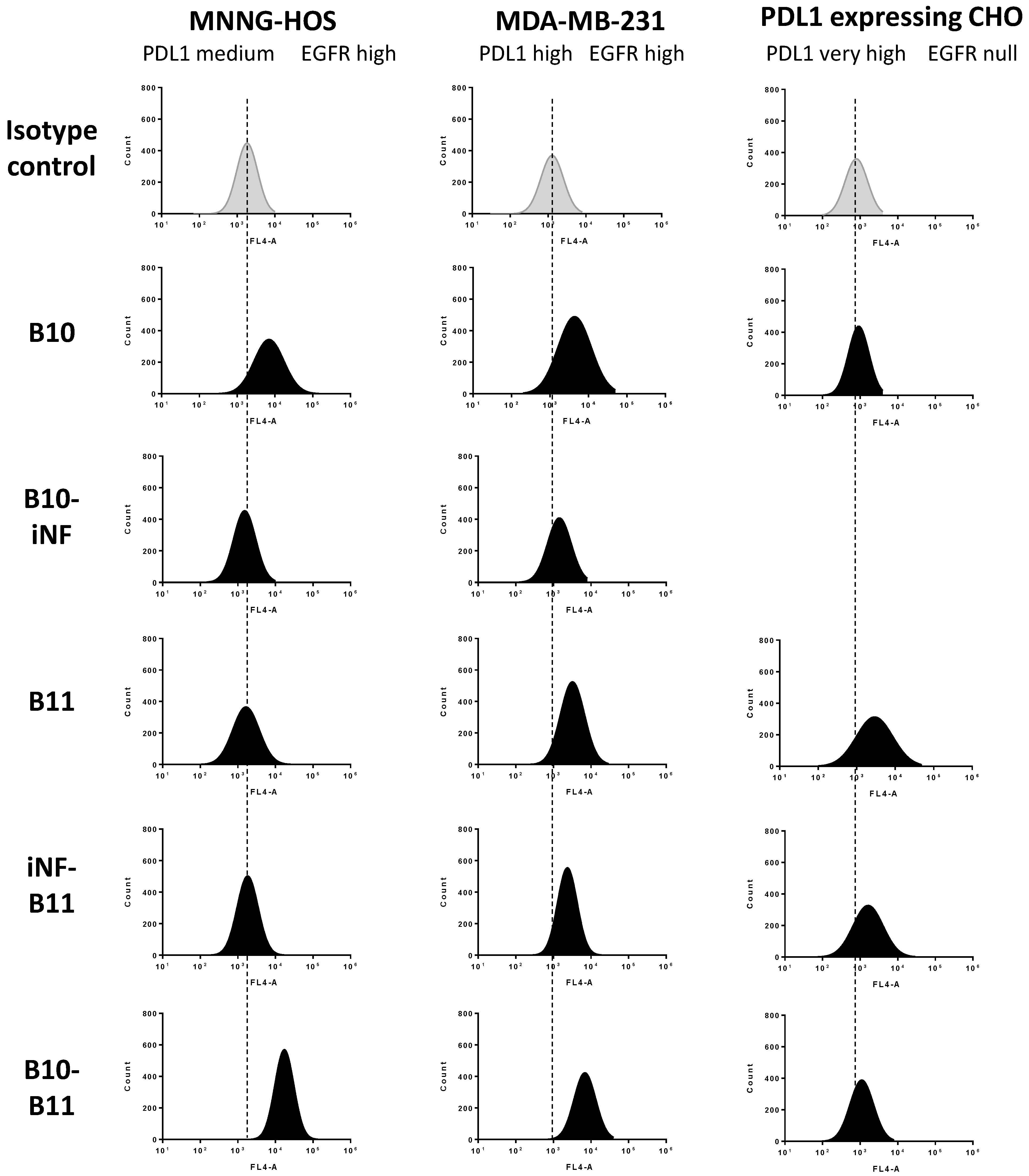
3.2. Cell Binding of the Bispecific Nanofitin B10–B11 Is Conditioned by the Cross-Arm Binding of EGFR and PDL1
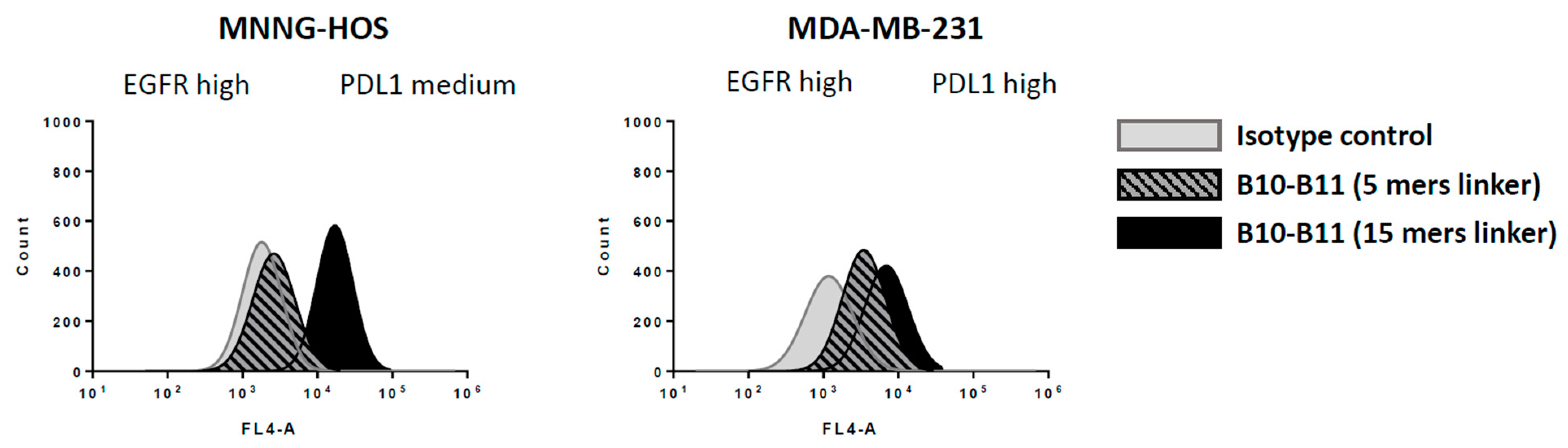
3.3. Decreasing the Linker Size Shows Opposing Effect on Affinity and Cell Binding Efficiency
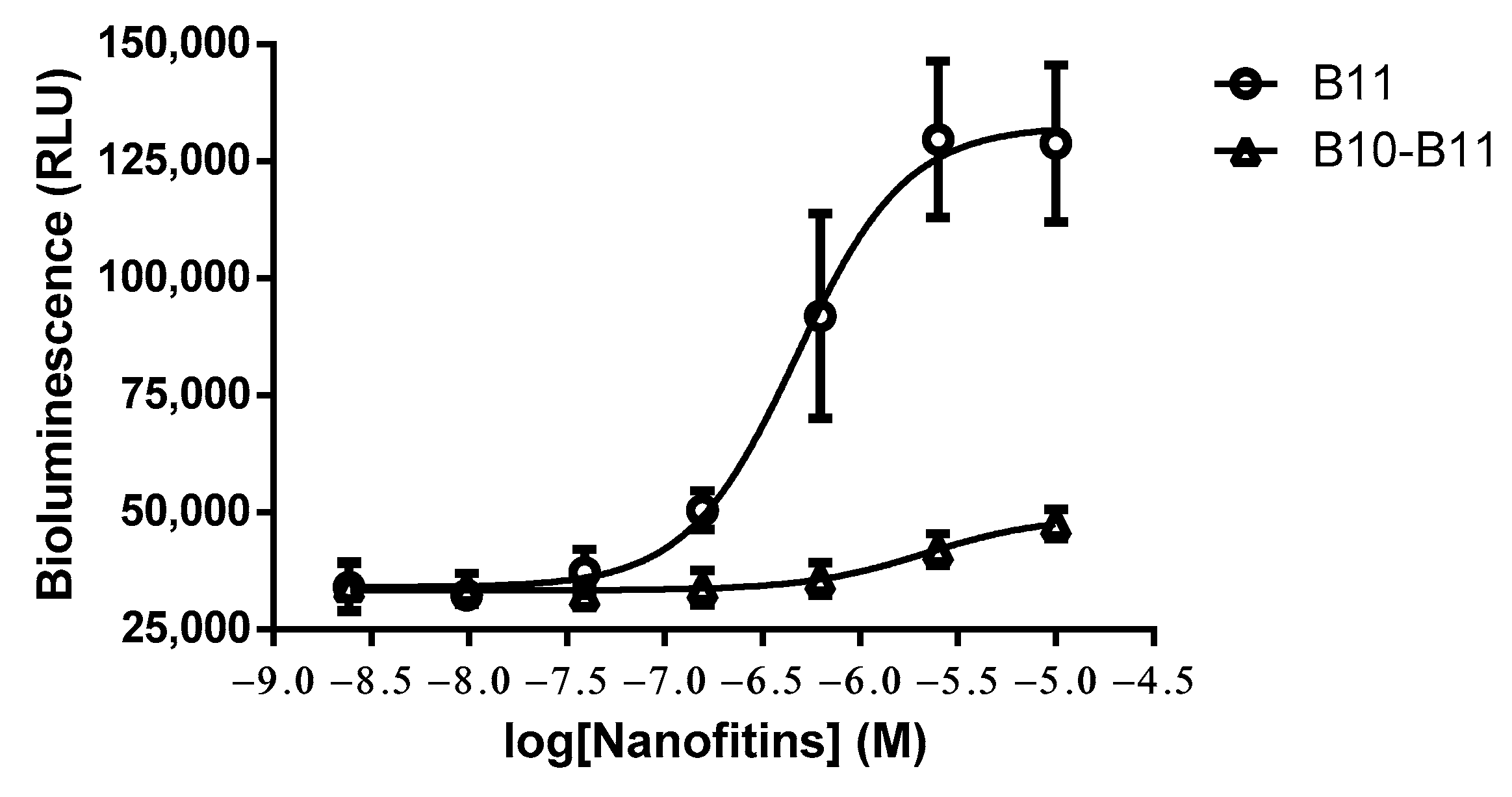
3.4. The Bispecific Nanofitin B10-B11 Displays Immune Checkpoint Inhibitory Activity in an EGFR-Dependent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Huang, S.; van Duijnhoven, S.M.J.; Sijts, A.J.A.M.; van Elsas, A. Bispecific Antibodies Targeting Dual Tumor-Associated Antigens in Cancer Therapy. J. Cancer Res. Clin. Oncol. 2020, 146, 3111–3122. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative Molecular Formats and Therapeutic Applications for Bispecific Antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Batyuk, A.; Honegger, A.; Brandl, F.; Mittl, P.R.E.; Plückthun, A. Rigidly Connected Multispecific Artificial Binders with Adjustable Geometries. Sci. Rep. 2017, 7, 11217. [Google Scholar] [CrossRef] [PubMed]
- Malm, M.; Bass, T.; Gudmundsdotter, L.; Lord, M.; Frejd, F.Y.; Ståhl, S.; Löfblom, J. Engineering of a Bispecific Affibody Molecule towards HER2 and HER3 by Addition of an Albumin-Binding Domain Allows for Affinity Purification and in Vivo Half-Life Extension. Biotechnol. J. 2014, 9, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Peper-Gabriel, J.K.; Pavlidou, M.; Pattarini, L.; Morales-Kastresana, A.; Jaquin, T.J.; Gallou, C.; Hansbauer, E.-M.; Richter, M.; Lelievre, H.; Scholer-Dahirel, A.; et al. The PD-L1/4-1BB Bispecific Antibody–Anticalin Fusion Protein PRS-344/S095012 Elicits Strong T-Cell Stimulation in a Tumor-Localized Manner. Clin. Cancer Res. 2022, 28, 3387–3399. [Google Scholar] [CrossRef]
- Kahl, M.; Settele, F.; Knick, P.; Haupts, U.; Bosse-Doenecke, E. Mabfilin and Fabfilin—New Antibody-Scaffold Fusion Formats for Multispecific Targeting Concepts. Protein Expr. Purif. 2018, 149, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Stüber, J.C.; Richter, C.P.; Bellón, J.S.; Schwill, M.; König, I.; Schuler, B.; Piehler, J.; Plückthun, A. Apoptosis-Inducing Anti-HER2 Agents Operate through Oligomerization-Induced Receptor Immobilization. Commun. Biol. 2021, 4, 762. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, S.L.; Engle, L.J.; Chao, G.; Zhu, R.-R.; Cao, C.; Lin, Z.; Yamniuk, A.P.; Hosbach, J.; Brown, J.; Fitzpatrick, E.; et al. A Fibronectin Scaffold Approach to Bispecific Inhibitors of Epidermal Growth Factor Receptor and Insulin-like Growth Factor-I Receptor. mAbs 2011, 3, 38–48. [Google Scholar] [CrossRef]
- Mouratou, B.; Schaeffer, F.; Guilvout, I.; Tello-Manigne, D.; Pugsley, A.P.; Alzari, P.M.; Pecorari, F. Remodeling a DNA-Binding Protein as a Specific in Vivo Inhibitor of Bacterial Secretin PulD. Proc. Natl. Acad. Sci. USA 2007, 104, 17983–17988. [Google Scholar] [CrossRef] [PubMed]
- Garlich, J.; Cinier, M.; Chevrel, A.; Perrocheau, A.; Eyerman, D.J.; Orme, M.; Kitten, O.; Scheibler, L. Discovery of APL-1030, a Novel, High-Affinity Nanofitin Inhibitor of C3-Mediated Complement Activation. Biomolecules 2022, 12, 432. [Google Scholar] [CrossRef]
- Marcion, G.; Hermetet, F.; Neiers, F.; Uyanik, B.; Dondaine, L.; Dias, A.M.M.; Da Costa, L.; Moreau, M.; Bellaye, P.; Collin, B.; et al. Nanofitins Targeting Heat Shock Protein 110: An Innovative Immunotherapeutic Modality in Cancer. Int. J. Cancer 2021, 148, 3019–3031. [Google Scholar] [CrossRef] [PubMed]
- Goux, M.; Becker, G.; Gorré, H.; Dammicco, S.; Desselle, A.; Egrise, D.; Leroi, N.; Lallemand, F.; Bahri, M.A.; Doumont, G.; et al. Nanofitin as a New Molecular-Imaging Agent for the Diagnosis of Epidermal Growth Factor Receptor Over-Expressing Tumors. Bioconjugate Chem. 2017, 28, 2361–2371. [Google Scholar] [CrossRef]
- Kalichuk, V.; Renodon-Cornière, A.; Béhar, G.; Carrión, F.; Obal, G.; Maillasson, M.; Mouratou, B.; Préat, V.; Pecorari, F. A Novel, Smaller Scaffold for Affitins: Showcase with Binders Specific for EpCAM. Biotechnol. Bioeng. 2018, 115, 290–299. [Google Scholar] [CrossRef]
- Michot, N.; Guyochin, A.; Cinier, M.; Savignard, C.; Kitten, O.; Pascual, M.-H.; Pouzieux, S.; Ozoux, M.-L.; Verdier, P.; Vicat, P.; et al. Albumin Binding Nanofitins, a New Scaffold to Extend Half-Life of Biologics—A Case Study with Exenatide Peptide. Peptides 2022, 152, 170760. [Google Scholar] [CrossRef]
- Behar, G.; Bellinzoni, M.; Maillasson, M.; Paillard-Laurance, L.; Alzari, P.M.; He, X.; Mouratou, B.; Pecorari, F. Tolerance of the Archaeal Sac7d Scaffold Protein to Alternative Library Designs: Characterization of Anti-Immunoglobulin G Affitins. Protein Eng. Des. Sel. 2013, 26, 267–275. [Google Scholar] [CrossRef]
- Huet, S.; Gorre, H.; Perrocheau, A.; Picot, J.; Cinier, M. Use of the Nanofitin Alternative Scaffold as a GFP-Ready Fusion Tag. PLoS ONE 2015, 10, e0142304. [Google Scholar] [CrossRef]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; van Ginkel, R.J.; Heskamp, S.; Wierstra, P.J.; Bremer, E.; Helfrich, W. A Novel Bispecific Antibody for EGFR-Directed Blockade of the PD-1/PD-L1 Immune Checkpoint. OncoImmunology 2018, 7, e1466016. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Cui, Y.; Liu, X.; Liu, G.; Dong, X.; Tang, L.; Hung, Y.; Wang, C.; Feng, M.-Q. A Bispecific Antibody Targeting HER2 and PD-L1 Inhibits Tumor Growth with Superior Efficacy. J. Biol. Chem. 2021, 297, 101420. [Google Scholar] [CrossRef] [PubMed]
- Kotanides, H.; Li, Y.; Malabunga, M.; Carpenito, C.; Eastman, S.W.; Shen, Y.; Wang, G.; Inigo, I.; Surguladze, D.; Pennello, A.L.; et al. Bispecific Targeting of PD-1 and PD-L1 Enhances T-Cell Activation and Antitumor Immunity. Cancer Immunol. Res. 2020, 8, 1300–1310. [Google Scholar] [CrossRef]
- Bossi, P.; Resteghini, C.; Paielli, N.; Licitra, L.; Pilotti, S.; Perrone, F. Prognostic and Predictive Value of EGFR in Head and Neck Squamous Cell Carcinoma. Oncotarget 2016, 7, 74362–74379. [Google Scholar] [CrossRef]
- Troiani, T.; Martinelli, E.; Capasso, A.; Morgillo, F.; Orditura, M.; De Vita, F.; Ciardiello, F. Targeting EGFR in Pancreatic Cancer Treatment. Curr. Drug Targets 2012, 13, 802–810. [Google Scholar] [CrossRef]
- Selvaggi, G.; Novello, S.; Torri, V.; Leonardo, E.; De Giuli, P.; Borasio, P.; Mossetti, C.; Ardissone, F.; Lausi, P.; Scagliotti, G.V. Epidermal Growth Factor Receptor Overexpression Correlates with a Poor Prognosis in Completely Resected Non-Small-Cell Lung Cancer. Ann. Oncol. 2004, 15, 28–32. [Google Scholar] [CrossRef]
- Rokita, M.; Stec, R.; Bodnar, L.; Charkiewicz, R.; Korniluk, J.; Smoter, M.; Cichowicz, M.; Chyczewski, L.; Nikliński, J.; Kozłowski, W.; et al. Overexpression of Epidermal Growth Factor Receptor as a Prognostic Factor in Colorectal Cancer on the Basis of the Allred Scoring System. OncoTargets Ther. 2013, 6, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, N.; Ooi, A. EGFR Protein Overexpression and Gene Amplification in Squamous Cell Carcinomas of the Esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of Epidermal Growth Factor Receptor in Breast Cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Michielin, O.; Lalani, A.-K.; Robert, C.; Sharma, P.; Peters, S. Defining Unique Clinical Hallmarks for Immune Checkpoint Inhibitor-Based Therapies. J. Immunother. Cancer 2022, 10, e003024. [Google Scholar] [CrossRef]
- Song, P.; Zhang, D.; Cui, X.; Zhang, L. Meta-analysis of Immune-related Adverse Events of Immune Checkpoint Inhibitor Therapy in Cancer Patients. Thorac. Cancer 2020, 11, 2406–2430. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving Target Cell Specificity Using a Novel Monovalent Bispecific IgG Design. mAbs 2015, 7, 377–389. [Google Scholar] [CrossRef]
- Robinson, M.K.; Hodge, K.M.; Horak, E.; Sundberg, Å.L.; Russeva, M.; Shaller, C.C.; von Mehren, M.; Shchaveleva, I.; Simmons, H.H.; Marks, J.D.; et al. Targeting ErbB2 and ErbB3 with a Bispecific Single-Chain Fv Enhances Targeting Selectivity and Induces a Therapeutic Effect in Vitro. Br. J. Cancer 2008, 99, 1415–1425. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1+ Activated T Cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the Molecular Basis of a Bispecific Antibody’s Target Selectivity. mAbs 2015, 7, 461–469. [Google Scholar] [CrossRef]
- Harms, B.D.; Kearns, J.D.; Iadevaia, S.; Lugovskoy, A.A. Understanding the Role of Cross-Arm Binding Efficiency in the Activity of Monoclonal and Multispecific Therapeutic Antibodies. Methods 2014, 65, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Razumienko, E.J.; Chen, J.C.; Cai, Z.; Chan, C.; Reilly, R.M. Dual-Receptor–Targeted Radioimmunotherapy of Human Breast Cancer Xenografts in Athymic Mice Coexpressing HER2 and EGFR Using 177 Lu- or 111 In-Labeled Bispecific Radioimmunoconjugates. J. Nucl. Med. 2016, 57, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Goenaga, A.-L.; Harms, B.D.; Zou, H.; Lou, J.; Conrad, F.; Adams, G.P.; Schoeberl, B.; Nielsen, U.B.; Marks, J.D. Impact of Intrinsic Affinity on Functional Binding and Biological Activity of EGFR Antibodies. Mol. Cancer Ther. 2012, 11, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Rhoden, J.J.; Dyas, G.L.; Wroblewski, V.J. A Modeling and Experimental Investigation of the Effects of Antigen Density, Binding Affinity, and Antigen Expression Ratio on Bispecific Antibody Binding to Cell Surface Targets. J. Biol. Chem. 2016, 291, 11337–11347. [Google Scholar] [CrossRef] [PubMed]
- Ainavarapu, S.R.K.; Brujić, J.; Huang, H.H.; Wiita, A.P.; Lu, H.; Li, L.; Walther, K.A.; Carrion-Vazquez, M.; Li, H.; Fernandez, J.M. Contour Length and Refolding Rate of a Small Protein Controlled by Engineered Disulfide Bonds. Biophys. J. 2007, 92, 225–233. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquot, P.; Muñoz-Garcia, J.; Fleury, M.; Cochonneau, D.; Gaussin, R.; Enouf, E.; Roze, C.; Ollivier, E.; Cinier, M.; Heymann, D. Engineering of a Bispecific Nanofitin with Immune Checkpoint Inhibitory Activity Conditioned by the Cross-Arm Binding to EGFR and PDL1. Biomolecules 2023, 13, 636. https://doi.org/10.3390/biom13040636
Jacquot P, Muñoz-Garcia J, Fleury M, Cochonneau D, Gaussin R, Enouf E, Roze C, Ollivier E, Cinier M, Heymann D. Engineering of a Bispecific Nanofitin with Immune Checkpoint Inhibitory Activity Conditioned by the Cross-Arm Binding to EGFR and PDL1. Biomolecules. 2023; 13(4):636. https://doi.org/10.3390/biom13040636
Chicago/Turabian StyleJacquot, Perrine, Javier Muñoz-Garcia, Maurine Fleury, Denis Cochonneau, Rémi Gaussin, Elise Enouf, Caroline Roze, Emilie Ollivier, Mathieu Cinier, and Dominique Heymann. 2023. "Engineering of a Bispecific Nanofitin with Immune Checkpoint Inhibitory Activity Conditioned by the Cross-Arm Binding to EGFR and PDL1" Biomolecules 13, no. 4: 636. https://doi.org/10.3390/biom13040636
APA StyleJacquot, P., Muñoz-Garcia, J., Fleury, M., Cochonneau, D., Gaussin, R., Enouf, E., Roze, C., Ollivier, E., Cinier, M., & Heymann, D. (2023). Engineering of a Bispecific Nanofitin with Immune Checkpoint Inhibitory Activity Conditioned by the Cross-Arm Binding to EGFR and PDL1. Biomolecules, 13(4), 636. https://doi.org/10.3390/biom13040636