
Dysregulation of AMPA Receptor Trafficking and Intracellular Vesicular Sorting in the Prefrontal Cortex of Dopamine Transporter Knock-Out Rats

 ,
,  , ,
, ,  , and
, and
Abstract
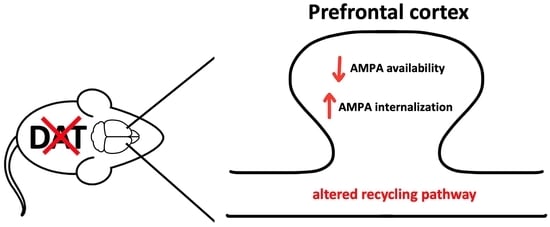
{kind=link}
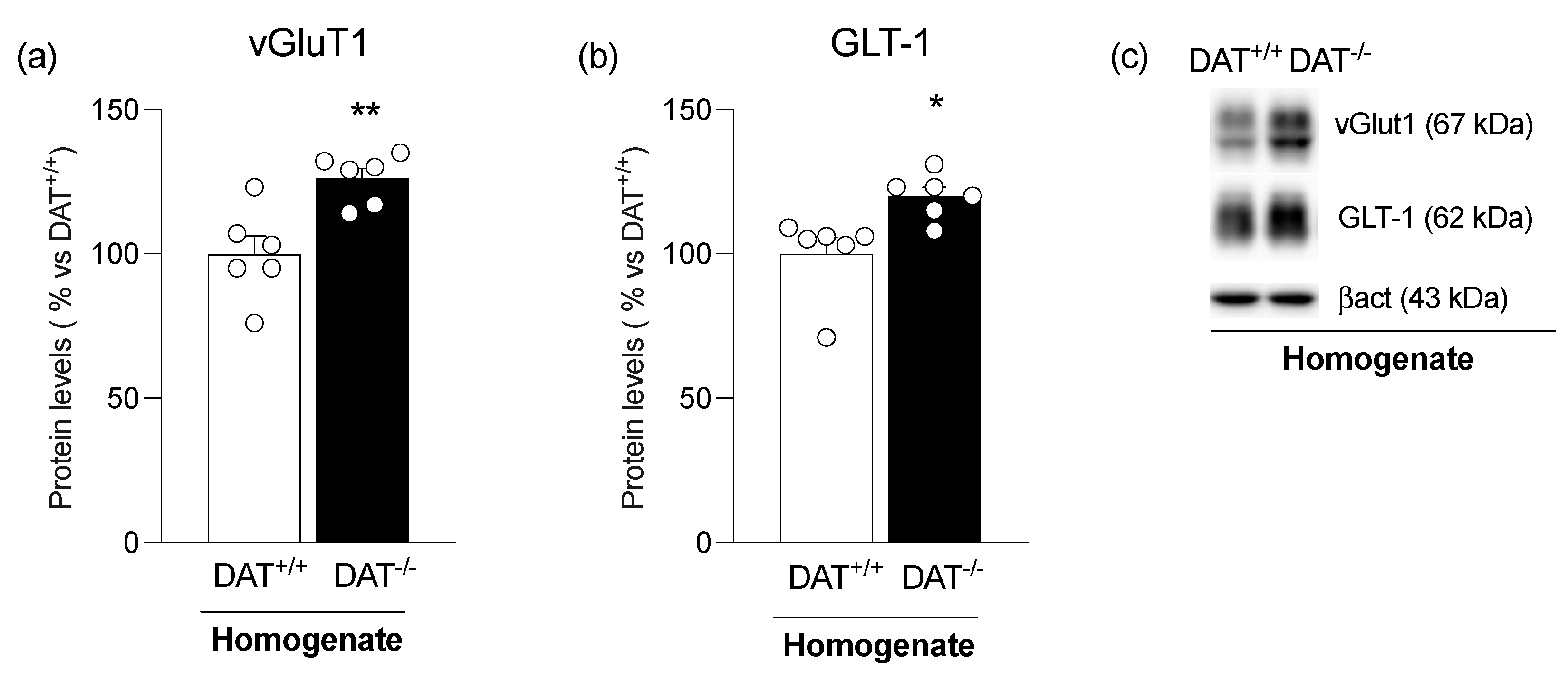{kind=link}
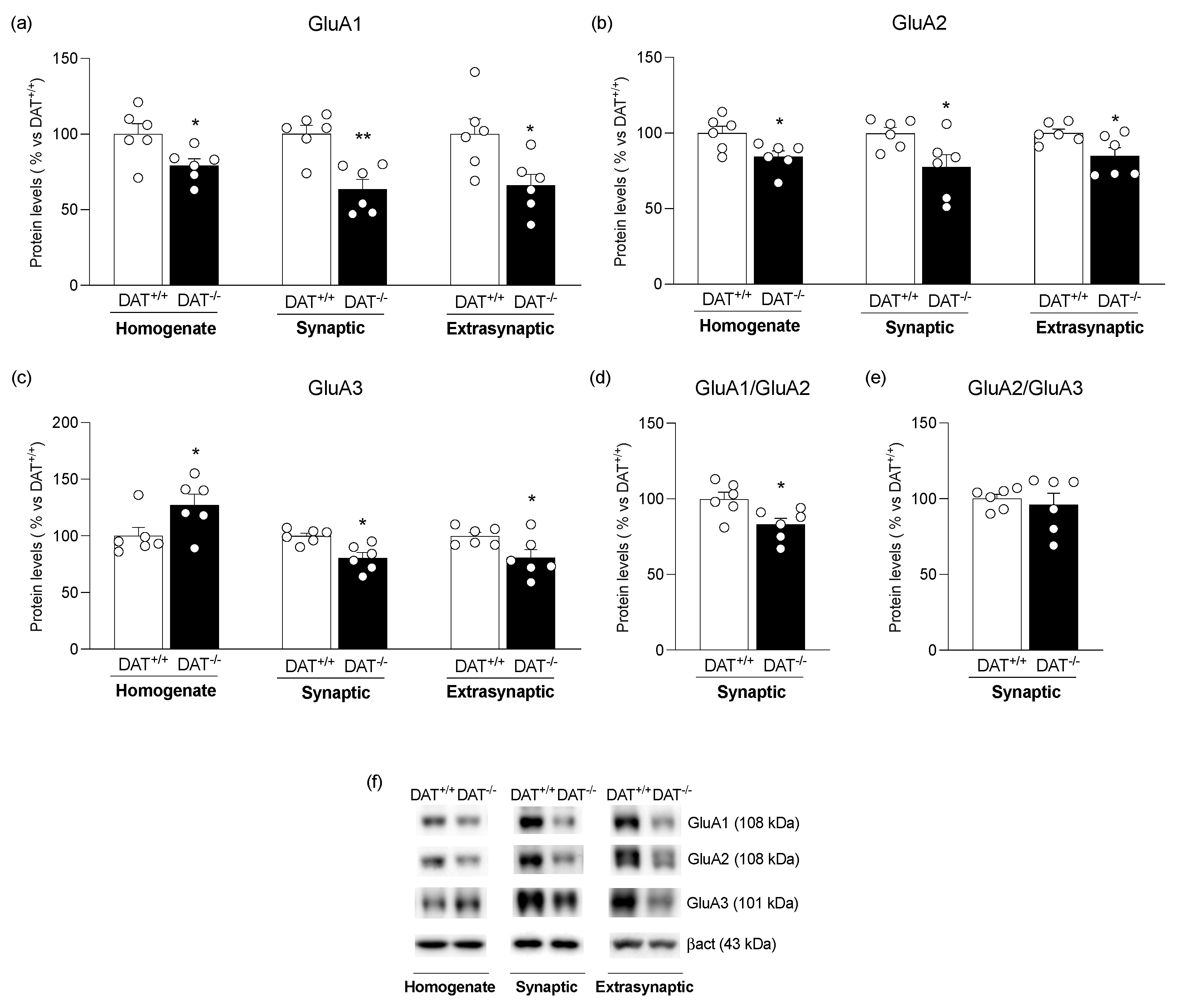{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Housing
2.2. Protein Extract Preparation and Western Blot Analysis
2.3. Statistical Analysis
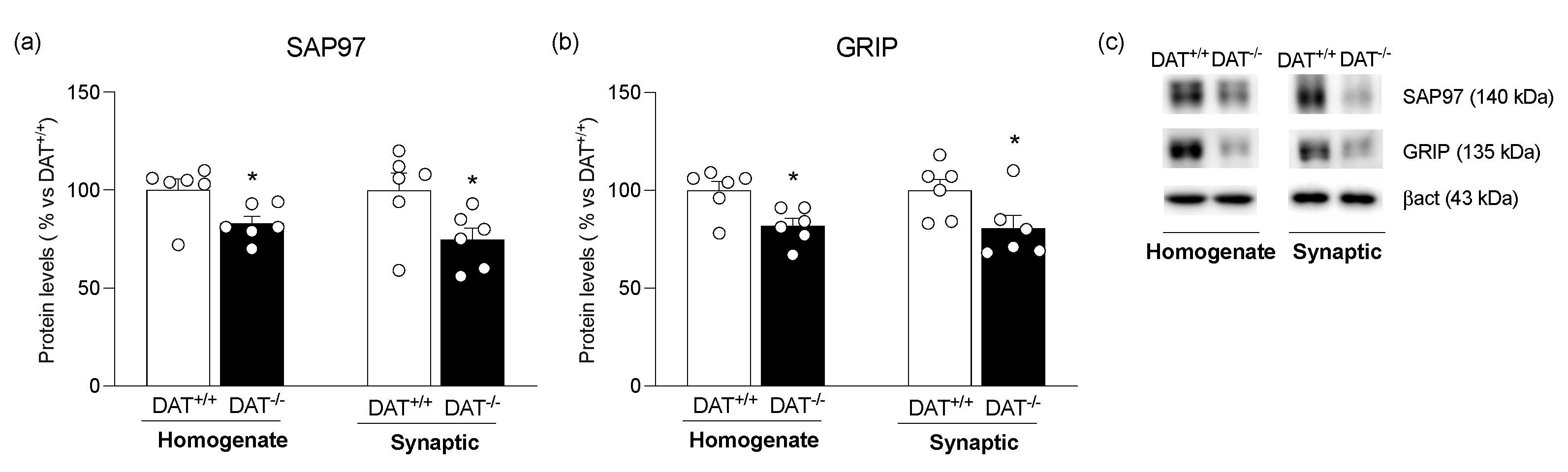
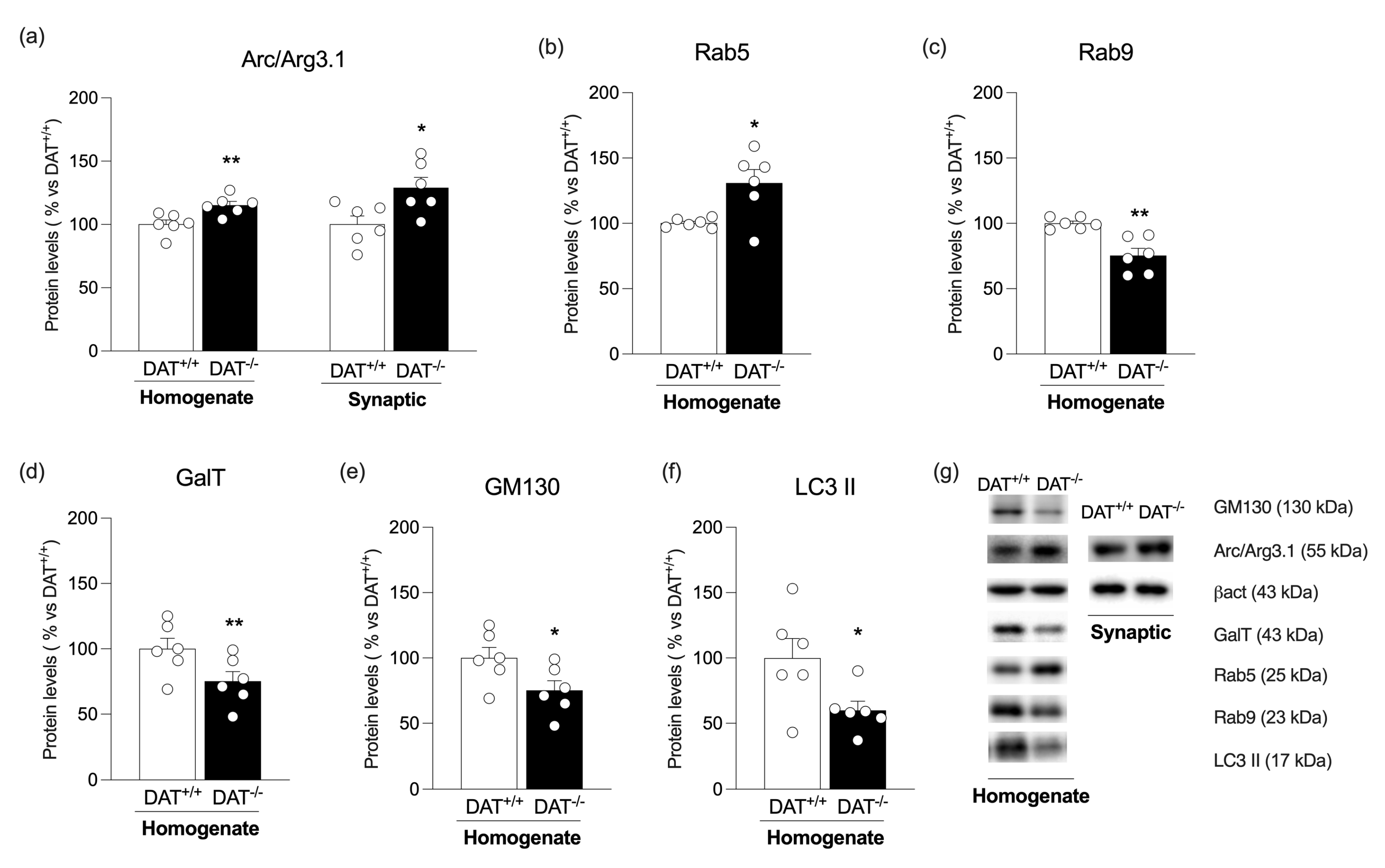
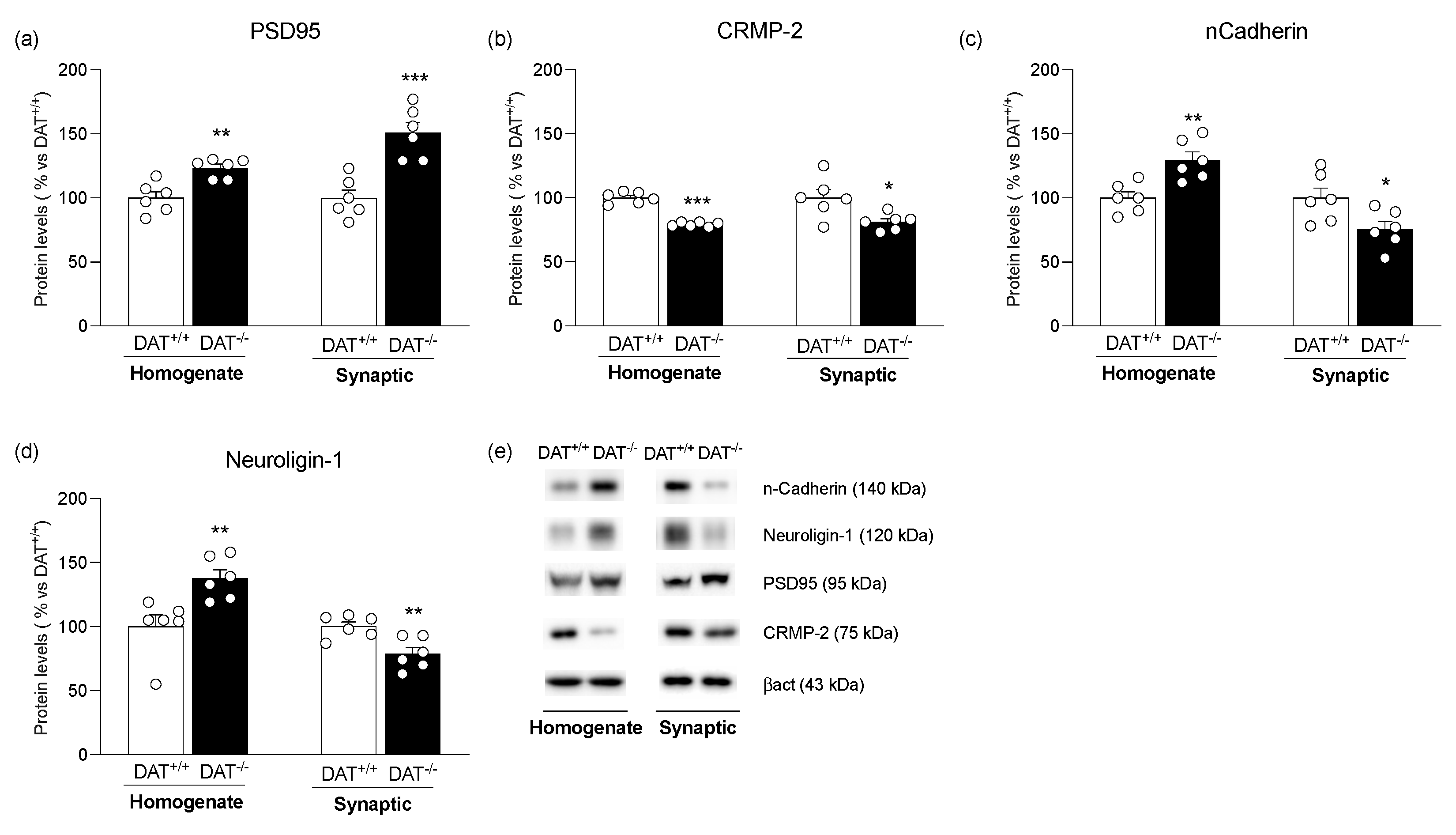
3. Results
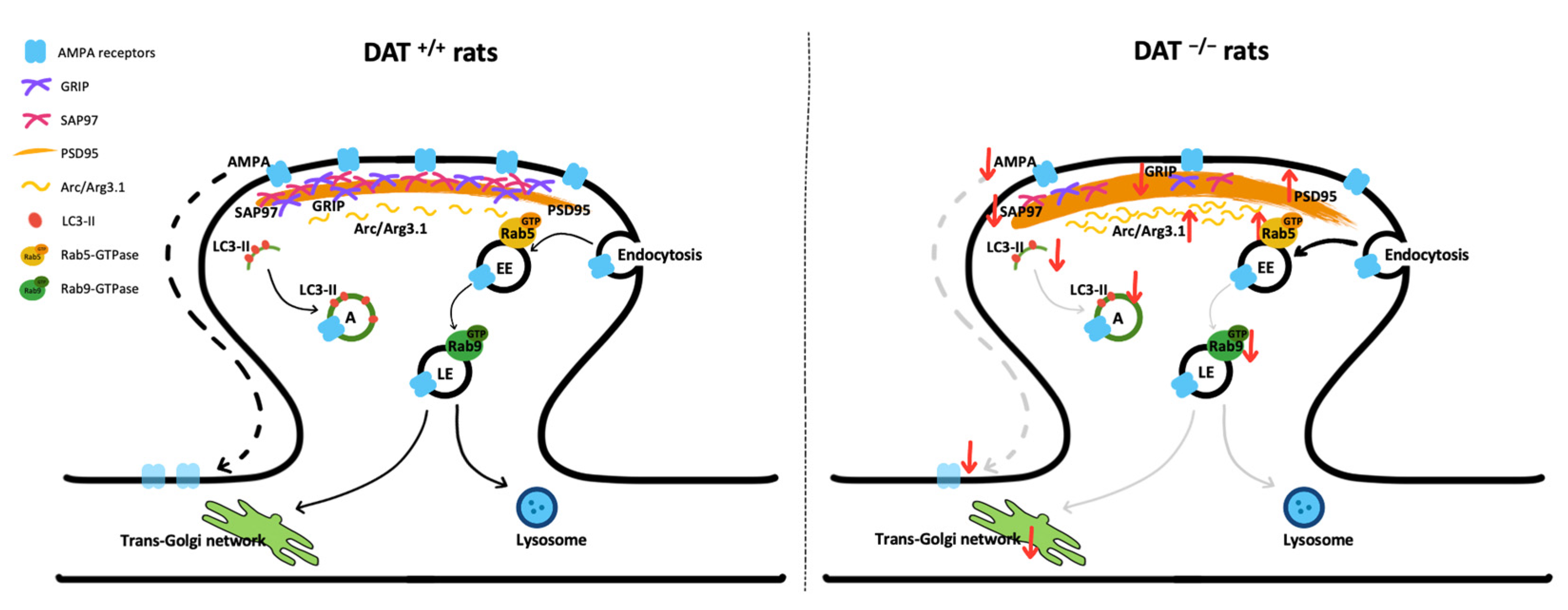
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hui, M.; Beier, K.T. Defining the Interconnectivity of the Medial Prefrontal Cortex and Ventral Midbrain. Front. Mol. Neurosci. 2022, 15, 1–8. [Google Scholar] [CrossRef]
- Bromberg-Martin, E.S.; Matsumoto, M.; Hikosaka, O. Dopamine in Motivational Control: Rewarding, Aversive, and Alerting. Neuron 2010, 68, 815–834. [Google Scholar] [CrossRef] [PubMed]
- Cools, R. Role of Dopamine in the Motivational and Cognitive Control of Behavior. Neuroscientist 2008, 14, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W. Predictive Reward Signal of Dopamine Neurons. J. Neurophysiol. 1998, 80, 1–27. [Google Scholar] [CrossRef]
- Fabrizio, G.; Camilla, B. Modulation of the Glutamatergic Transmission by Dopamine: A Focus on Parkinson, Huntington and Addiction Diseases. Front. Cell Neurosci. 2015, 9, 25. [Google Scholar] [CrossRef]
- Seamans, J.K.; Yang, C.R. The Principal Features and Mechanisms of Dopamine Modulation in the Prefrontal Cortex. Prog. Neurobiol. 2004, 74, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.M. Dopamine: A Potential Substrate for Synaptic Plasticity and Memory Mechanisms. Prog. Neurobiol. 2003, 69, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Pirot, S.; Glowinski, J.; Thierry, A.-M. Excitatory Responses Evoked in Prefrontal Cortex by Mediodorsal Thalamic Nucleus Stimulation: Influence of Anaesthesia. Eur. J. Pharm. 1995, 285, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Heidbreder, C.A.; Groenewegen, H.J. The Medial Prefrontal Cortex in the Rat: Evidence for a Dorso-Ventral Distinction Based upon Functional and Anatomical Characteristics. Neurosci. Biobehav. Rev. 2003, 27, 555–579. [Google Scholar] [CrossRef]
- Xue, B.; Mao, L.M.; Jin, D.Z.; Wang, J.Q. Pharmacological Modulation of AMPA Receptor Phosphorylation by Dopamine and Muscarinic Receptor Agents in the Rat Medial Prefrontal Cortex. Eur. J. Pharm. 2018, 820, 45–52. [Google Scholar] [CrossRef]
- Buck, S.A.; Quincy Erickson-Oberg, M.; Logan, R.W.; Freyberg, Z. Relevance of Interactions between Dopamine and Glutamate Neurotransmission in Schizophrenia. Mol. Psychiatry 2022, 27, 3583–3591. [Google Scholar] [CrossRef]
- Fischer, K.D.; Knackstedt, L.A.; Rosenberg, P.A. Glutamate Homeostasis and Dopamine Signaling: Implications for Psychostimulant Addiction Behavior. Neurochem. Int. 2021, 144, 104896. [Google Scholar] [CrossRef]
- Grace, A.A. Dysregulation of the Dopamine System in the Pathophysiology of Schizophrenia and Depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Pitman, R.K.; Rasmusson, A.M.; Koenen, K.C.; Shin, L.M.; Orr, S.P.; Gilbertson, M.W.; Milad, M.R.; Liberzon, I. Biological Studies of Post-Traumatic Stress Disorder. Nat. Rev. Neurosci. 2012, 13, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Sagvolden, T.; Johansen, E.B.; Aase, H.; Russell, V.A. A Dynamic Developmental Theory of Attention-Deficit/Hyperactivity Disorder (ADHD) Predominantly Hyperactive/Impulsive and Combined Subtypes. Behav. Brain Sci. 2005, 28, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA Receptor Composition in Development, Plasticity and Disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef]
- Chater, T.E.; Goda, Y. The Role of AMPA Receptors in Postsynaptic Mechanisms of Synaptic Plasticity. Front. Cell Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef]
- Fernandes, D.; Carvalho, A.L. Mechanisms of Homeostatic Plasticity in the Excitatory Synapse. J. Neurochem. 2016, 139, 973–996. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA Receptor Trafficking and Synaptic Plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Ehlers, M.D. Reinsertion or Degradation of AMPA Receptors Determined by Activity-Dependent Endocytic Sorting the Accumulation and Half-Life of Postsynaptic AMPARs at Synapses (O’Brien et al. Suggesting Activity-Dependent Regula-Tion of AMPAR Degradation. More Rapid Loss of Synaptic AMPARs Has Been Observed Following Synaptic Stimula. Neuron 2000, 28, 511–525. [Google Scholar]
- van der Sluijs, P.; Hoogenraad, C.C. New Insights in Endosomal Dynamics and AMPA Receptor Trafficking. Semin. Cell Dev. Biol. 2011, 22, 499–505. [Google Scholar] [CrossRef]
- Waung, M.W.; Pfeiffer, B.E.; Nosyreva, E.D.; Ronesi, J.A.; Huber, K.M. Rapid Translation of Arc/Arg3.1 Selectively Mediates MGluR-Dependent LTD through Persistent Increases in AMPAR Endocytosis Rate. Neuron 2008, 59, 84–97. [Google Scholar] [CrossRef]
- Chowdhury, S.; Shepherd, J.D.; Okuno, H.; Lyford, G.; Petralia, R.S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 Interacts with the Endocytic Machinery to Regulate AMPA Receptor Trafficking. Neuron 2006, 52, 445–459. [Google Scholar] [CrossRef]
- Hausser, A.; Schlett, K. Coordination of AMPA Receptor Trafficking by Rab GTPases. Small GTPases 2019, 10, 419–432. [Google Scholar] [CrossRef]
- Shehata, M.; Matsumura, H.; Okubo-Suzuki, R.; Ohkawa, N.; Inokuchi, K. Neuronal Stimulation Induces Autophagy in Hippocampal Neurons That Is Involved in AMPA Receptor Degradation after Chemical Long-Term Depression. J. Neurosci. 2012, 32, 10413–10422. [Google Scholar] [CrossRef]
- Parkinson, G.T.; Hanley, J.G. Mechanisms of AMPA Receptor Endosomal Sorting. Front. Mol. Neurosci. 2018, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.E. Alterations in the Morphology of Dendrites and Dendritic Spines in the Nucleus Accumbens and Prefrontal Cortex Following Repeated Treatment with Amphetamine or Cocaine. Eur. J. Neurosci. 1999, 11, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhao, Y.; Wolf, M.E. Dopamine Receptor Stimulation Modulates AMPA Receptor Synaptic Insertion in Prefrontal Cortex Neurons. J. Neurosci. 2005, 25, 7342–7351. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.T.; Murray, C.H.; Reimers, J.M.; Chauhan, N.M.; Woo, K.K.Y.; Molla, H.M.; Loweth, J.A.; Wolf, M.E. Trafficking of Calcium-Permeable and Calcium-Impermeable AMPA Receptors in Nucleus Accumbens Medium Spiny Neurons Co-Cultured with Prefrontal Cortex Neurons. Neuropharmacology 2017, 116, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Caffino, L.; Mottarlini, F.; van Reijmersdal, B.; Telese, F.; Verheij, M.M.M.; Fumagalli, F.; Homberg, J.R. The Role of the Serotonin Transporter in Prefrontal Cortex Glutamatergic Signaling Following Short- and Long-Access Cocaine Self-Administration. Addict. Biol. 2021, 26, e12896. [Google Scholar] [CrossRef]
- Leo, D.; Sukhanov, I.; Zoratto, F.; Illiano, P.; Caffino, L.; Sanna, F.; Messa, G.; Emanuele, M.; Esposito, A.; Dorofeikova, M.; et al. Pronounced Hyperactivity, Cognitive Dysfunctions, and BDNF Dysregulation in Dopamine Transporter Knock-out Rats. J. Neurosci. 2018, 38, 1959–1972. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Jones, S.R.; Fumagalli, F.; Wightman, R.M.; Caron, M.G. Re-Evaluation of the Role of the Dopamine Transporter in Dopamine System Homeostasis 1. Brain Res. Rev. 1998, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Fumagalli, F.; Jones, S.R.; Caron, M.G. Dopamine Transporter Is Required for in Vivo MPTP Neurotoxicity: Evidence from Mice Lacking the Transporter. J. Neurochem. 1997, 69, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Gainetdinov, R.R.; Valenzano, K.J.; Caron, M.G. Role of Dopamine Transporter in Methamphetamine-Induced Neurotoxicity: Evidence from Mice Lacking the Transporter. J. Neurosci. 1998, 18, 4861–4869. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin Regulation of Neural Circuit Development and Function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Xu, T.X.; Sotnikova, T.D.; Liang, C.; Zhang, J.; Jung, J.U.; Spealman, R.D.; Gainetdinov, R.R.; Yao, W.D. Hyperdopaminergic Tone Erodes Prefrontal Long-Term Potential via a D 2 Receptor-Operated Protein Phosphatase Gate. J. Neurosci. 2009, 29, 14086–14099. [Google Scholar] [CrossRef] [PubMed]
- Geurts, A.M.; Cost, G.J.; Freyvert, Y.; Zeitler, B.; Miller, J.C.; Choi, V.M.; Jenkins, S.S.; Wood, A.; Cui, X.; Meng, X.; et al. Knockout Rats via Embryo Microinjection of Zinc-Finger Nucleases. Science 2009, 325, 433. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]
- Mottarlini, F.; Targa, G.; Bottan, G.; Tarenzi, B.; Fumagalli, F.; Caffino, L. Cortical Reorganization of the Glutamate Synapse in the Activity-Based Anorexia Rat Model: Impact on Cognition. J. Neurochem. 2022, 161, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Caffino, L.; Verheij, M.M.M.; Roversi, K.; Targa, G.; Mottarlini, F.; Popik, P.; Nikiforuk, A.; Golebiowska, J.; Fumagalli, F.; Homberg, J.R. Hypersensitivity to Amphetamine’s Psychomotor and Reinforcing Effects in Serotonin Transporter Knockout Rats: Glutamate in the Nucleus Accumbens. Br. J. Pharm. 2020, 177, 4532–4547. [Google Scholar] [CrossRef]
- Fremeau, R.T.; Voglmaier, S.; Seal, R.P.; Edwards, R.H. VGLUTs Define Subsets of Excitatory Neurons and Suggest Novel Roles for Glutamate. Trends. Neurosci. 2004, 27, 98–103. [Google Scholar] [CrossRef]
- Martineau, M.; Guzman, R.E.; Fahlke, C.; Klingauf, J. VGLUT1 Functions as a Glutamate/Proton Exchanger with Chloride Channel Activity in Hippocampal Glutamatergic Synapses. Nat. Commun. 2017, 8, 2279. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, T.S.; Rosenberg, P.A. GLT-1: The Elusive Presynaptic Glutamate Transporter. Neurochem. Int. 2016, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Zerial, M.; Mcbride, H. Rab Proteins as Membrane Organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A. Unlocking Golgi: Why Does Morphology Matter? Biochemistry 2019, 84, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e8. [Google Scholar] [CrossRef]
- Berry, K.P.; Nedivi, E. Spine Dynamics: Are They All the Same? Neuron 2017, 96, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, A.C.; Ferrario, C.R.; Glucksman, M.J.; Wolf, M.E. Signaling Pathway Adaptations and Novel Protein Kinase A Substrates Related to Behavioral Sensitization to Cocaine. J. Neurochem. 2009, 110, 363–377. [Google Scholar] [CrossRef]
- Inagaki, N.; Chihara, K.; Arimura, N.; Ménager, C.; Kawano, Y.; Matsuo, N.; Nishimura, T.; Amano, M.; Kaibuchi, K. CRMP-2 Induces Axons in Cultured Hippocampal Neurons. Nat. Neurosci. 2001, 4, 781–782. [Google Scholar] [CrossRef]
- Kadowaki, M.; Nakamura, S.; Machon, O.; Krauss, S.; Radice, G.L.; Takeichi, M. N-Cadherin Mediates Cortical Organization in the Mouse Brain. Dev. Biol. 2007, 304, 22–33. [Google Scholar] [CrossRef]
- Song, J.-Y.; Ichtchenko, K.; Su¨dhofsu¨dhof, T.C.; Brose, N. Neuroligin 1 Is a Postsynaptic Cell-Adhesion Molecule of Excitatory Synapses. Proc. Natl. Acad. Sci. USA 1999, 96, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.W.; Arnsten, A.F.T. The Neuropsychopharmacology of Fronto-Executive Function: Monoaminergic Modulation. Annu. Rev. Neurosci. 2009, 32, 267–287. [Google Scholar] [CrossRef] [PubMed]
- Cools, R.; D’Esposito, M. Inverted-U-Shaped Dopamine Actions on Human Working Memory and Cognitive Control. Biol. Psychiatry 2011, 69, e112–e125. [Google Scholar] [CrossRef] [PubMed]
- Corti, E.; Duarte, C.B. The Role of Post-Translational Modifications in Synaptic AMPA Receptor Activity. Biochem. Soc. Trans. 2023, 51, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.E.; Berridge, K.C. The Neuroscience of Natural Rewards: Relevance to Addictive Drugs. J. Neurosci. 2002, 22, 3306–3311. [Google Scholar] [CrossRef]
- Bai, J.; Blot, K.; Tzavara, E.; Nosten-Bertrand, M.; Giros, B.; Otani, S. Inhibition of Dopamine Transporter Activity Impairs Synaptic Depression in Rat Prefrontal Cortex through Over-Stimulation of D1 Receptors. Cereb. Cortex 2014, 24, 945–955. [Google Scholar] [CrossRef]
- Huang, Y.H.; Lin, Y.; Mu, P.; Lee, B.R.; Brown, T.E.; Wayman, G.; Marie, H.; Liu, W.; Yan, Z.; Sorg, B.A.; et al. In Vivo Cocaine Experience Generates Silent Synapses. Neuron 2009, 63, 40–47. [Google Scholar] [CrossRef]
- Koya, E.; Cruz, F.C.; Ator, R.; Golden, S.A.; Hoffman, A.F.; Lupica, C.R.; Hope, B.T. Silent Synapses in Selectively Activated Nucleus Accumbens Neurons Following Cocaine Sensitization. Nat. Neurosci. 2012, 15, 1556–1562. [Google Scholar] [CrossRef]
- Wright, W.J.; Graziane, N.M.; Neumann, P.A.; Hamilton, P.J.; Cates, H.M.; Fuerst, L.; Spenceley, A.; MacKinnon-Booth, N.; Iyer, K.; Huang, Y.H.; et al. Silent Synapses Dictate Cocaine Memory Destabilization and Reconsolidation. Nat. Neurosci. 2020, 23, 32–46. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Nicoll, R.A. Silent Synapses and the Emergence of a Postsynaptic Mechanism for LTP. Nat. Rev. Neurosci. 2008, 9, 813–825. [Google Scholar] [CrossRef]
- Borgdorff, A.J.; Choquet, D. Regulation of AMPA Receptor Lateral Movements. Nature 2002, 417, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Triller, A.; Choquet, D. Surface Trafficking of Receptors between Synaptic and Extrasynaptic Membranes: And yet They Do Move! Trends Neurosci. 2005, 28, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, G. Dynamic Control of Synaptic Adhesion and Organizing Molecules in Synaptic Plasticity. Neural Plast. 2017, 2017, 6526151. [Google Scholar] [CrossRef]
- Abe, H.; Jitsuki, S.; Nakajima, W.; Murata, Y.; Jitsuki-Takahashi, A.; Katsuno, Y.; Tada, H.; Sano, A.; Suyama, K.; Mochizuki, N.; et al. CRMP2-Binding Compound, Edonerpic Maleate, Accelerates Motor Function Recovery from Brain Damage. Science 2018, 360, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Mondin, M.; Labrousse, V.; Hosy, E.; Heine, M.; Tessier, B.; Levet, F.; Poujol, C.; Blanchet, C.; Choquet, D.; Thoumine, O. Neurexin-Neuroligin Adhesions Capture Surface-Diffusing AMPA Receptors through PSD-95 Scaffolds. J. Neurosci. 2011, 31, 13500–13515. [Google Scholar] [CrossRef]
- MacGillavry, H.D.; Song, Y.; Raghavachari, S.; Blanpied, T.A. Nanoscale Scaffolding Domains within the Postsynaptic Density Concentrate Synaptic Ampa Receptors. Neuron 2013, 78, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, I.; Malinow, R. Postsynaptic Density 95 Controls AMPA Receptor Incorporation during Long-Term Potentiation and Experience-Driven Synaptic Plasticity. J. Neurosci. 2004, 24, 916–927. [Google Scholar] [CrossRef]
- Shi, S.-H.; Hayashi, Y.; Esteban, J.A.; Malinow, R. Subunit-Specific Rules Governing AMPA Receptor Trafficking to Synapses in Hippocampal Pyramidal Neurons. Cell 2001, 105, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Huang, Y.H.; Balakrishnan, S.; Liu, L.; Wang, Y.T.; Nestler, E.J.; Schlüter, O.M.; Dong, Y. Ampa and Nmda Receptor Trafficking at Cocaine-Generated Synapses. J. Neurosci. 2021, 41, 1996–2011. [Google Scholar] [CrossRef]
- Petrini, E.M.; Lu, J.; Cognet, L.; Lounis, B.; Ehlers, M.D.; Choquet, D. Endocytic Trafficking and Recycling Maintain a Pool of Mobile Surface AMPA Receptors Required for Synaptic Potentiation. Neuron 2009, 63, 92–105. [Google Scholar] [CrossRef]
- Steward, O.; Worley, P.F. Selective Targeting of Newly Synthesized Arc MRNA to Active Synapses Requires NMDA Receptor Activation. Neuron 2001, 30, 227–240. [Google Scholar] [CrossRef]
- Shepherd, J.D.; Rumbaugh, G.; Wu, J.; Chowdhury, S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 Mediates Homeostatic Synaptic Scaling of AMPA Receptors. Neuron 2006, 52, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Rial Verde, E.M.; Lee-Osbourne, J.; Worley, P.F.F.; Malinow, R.; Cline, H.T.T. Increased Expression of the Immediate-Early Gene Arc/Arg3.1 Reduces AMPA Receptor-Mediated Synaptic Transmission. Neuron 2006, 52, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Giannotti, G.; Caffino, L.; Calabrese, F.; Racagni, G.; Riva, M.A.; Fumagalli, F. Prolonged Abstinence from Developmental Cocaine Exposure Dysregulates BDNF and Its Signaling Network in the Medial Prefrontal Cortex of Adult Rats. Int. J. Neuropsychopharmacol. 2014, 17, 625–634. [Google Scholar] [CrossRef]
- Caffino, L.; Giannotti, G.; Malpighi, C.; Racagni, G.; Filip, M.; Fumagalli, F. Long-Term Abstinence from Developmental Cocaine Exposure Alters Arc/Arg3.1 Modulation in the Rat Medial Prefrontal Cortex. Neurotox Res. 2014, 26, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The Small GTPase Rab5 Functions as a Regulatory Factor in the Early Endocytic Pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef]
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of Endosomal Motility and Degradation by Amyotrophic Lateral Sclerosis 2/Alsin. Mol. Brain 2009, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Soldati, T.; Rancano, C.; Geissler, H.; Pfeffer, S.R. Rab7 and Rab9 Are Recruited onto Late Endosomes by Biochemically Distinguishable Processes. J. Biol. Chem. 1995, 270, 25541–25548. [Google Scholar] [CrossRef] [PubMed]
- Schreij, A.M.A.; Fon, E.A.; McPherson, P.S. Endocytic Membrane Trafficking and Neurodegenerative Disease. Cell. Mol. Life Sci. 2016, 73, 1529–1545. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.; Sheta, R.; Idi, W.; Oueslati, A. Alpha-Synuclein and the Endolysosomal System in Parkinson’s Disease: Guilty by Association. Biomolecules 2021, 11, 1333. [Google Scholar] [CrossRef]
- Morón, J.A.; Brockington, A.; Wise, R.A.; Rocha, B.A.; Hope, B.T. Dopamine Uptake through the Norepinephrine Transporter in Brain Regions with Low Levels of the Dopamine Transporter: Evidence from Knock-Out Mouse Lines. J. Neurosci. 2002, 22, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Carboni, E.; Silvagni, A.; Vacca, C.; di Chiara, G. Cumulative Effect of Norepinephrine and Dopamine Carrier Blockade on Extracellular Dopamine Increase in the Nucleus Accumbens Shell, Bed Nucleus of Stria Terminalis and Prefrontal Cortex. J. Neurochem. 2006, 96, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Carvelli, L.; Blakely, R.D.; Defelice, L.J. Dopamine Transporter/Syntaxin 1A Interactions Regulate Transporter Channel Activity and Dopaminergic Synaptic Transmission. Proc. Natl. Acad. Sci. USA 2008, 105, 14192–14197. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, J.; Jørgensen, T.N.; Gether, U. Regulation of Dopamine Transporter Function by Protein-Protein Interactions: New Discoveries and Methodological Challenges. J. Neurochem. 2010, 113, 27–41. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Targa, G.; Mottarlini, F.; Rizzi, B.; Leo, D.; Caffino, L.; Fumagalli, F. Dysregulation of AMPA Receptor Trafficking and Intracellular Vesicular Sorting in the Prefrontal Cortex of Dopamine Transporter Knock-Out Rats. Biomolecules 2023, 13, 516. https://doi.org/10.3390/biom13030516
Targa G, Mottarlini F, Rizzi B, Leo D, Caffino L, Fumagalli F. Dysregulation of AMPA Receptor Trafficking and Intracellular Vesicular Sorting in the Prefrontal Cortex of Dopamine Transporter Knock-Out Rats. Biomolecules. 2023; 13(3):516. https://doi.org/10.3390/biom13030516
Chicago/Turabian StyleTarga, Giorgia, Francesca Mottarlini, Beatrice Rizzi, Damiana Leo, Lucia Caffino, and Fabio Fumagalli. 2023. "Dysregulation of AMPA Receptor Trafficking and Intracellular Vesicular Sorting in the Prefrontal Cortex of Dopamine Transporter Knock-Out Rats" Biomolecules 13, no. 3: 516. https://doi.org/10.3390/biom13030516
APA StyleTarga, G., Mottarlini, F., Rizzi, B., Leo, D., Caffino, L., & Fumagalli, F. (2023). Dysregulation of AMPA Receptor Trafficking and Intracellular Vesicular Sorting in the Prefrontal Cortex of Dopamine Transporter Knock-Out Rats. Biomolecules, 13(3), 516. https://doi.org/10.3390/biom13030516