
Novel Inhibitory Role of Fenofibric Acid by Targeting Cryptic Site on the RBD of SARS-CoV-2


Abstract
1. Introduction
2. Systems and Methods
2.1. Cavity Search
2.2. MD Simulation System and Setup
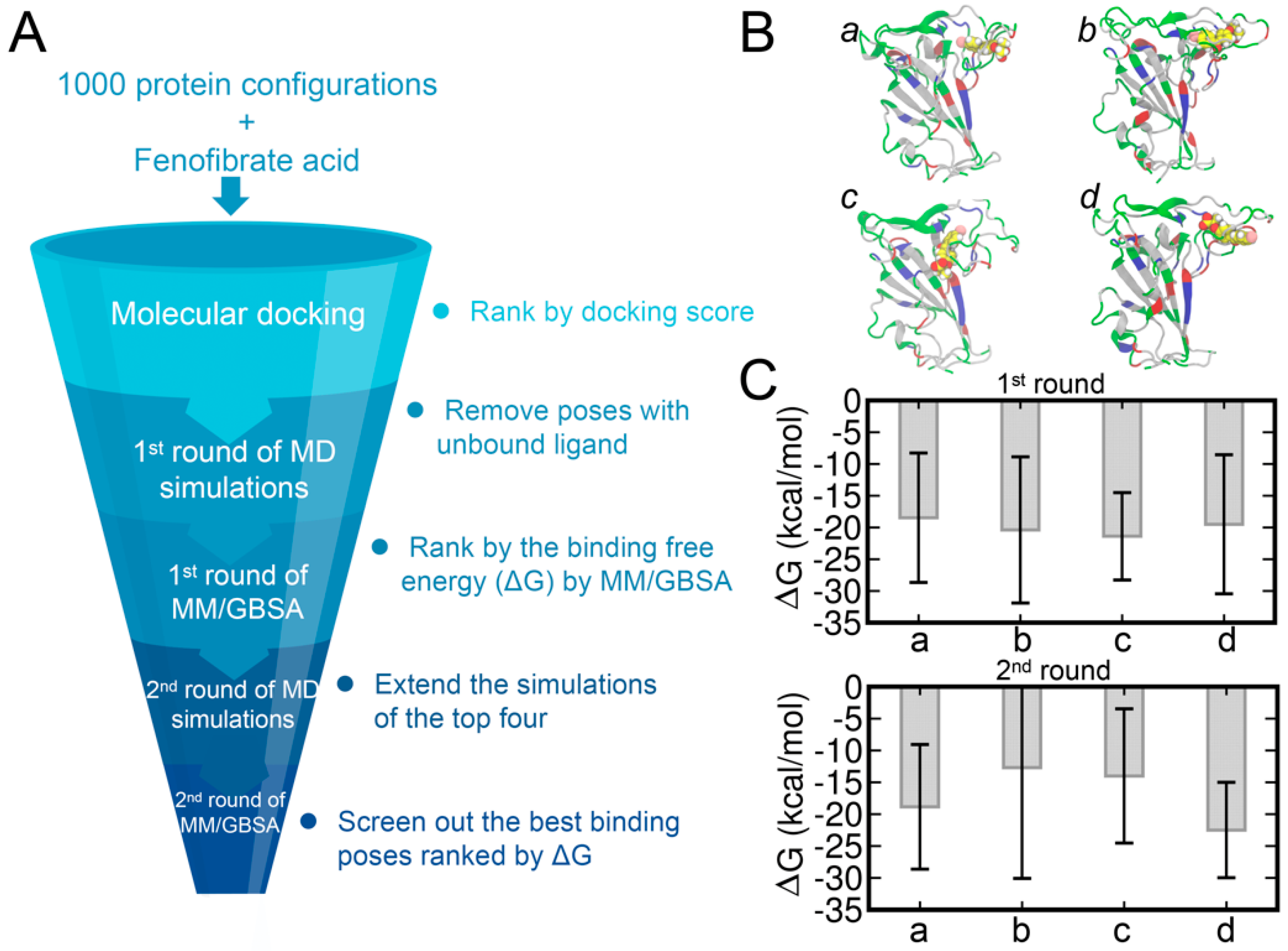
2.3. Molecular Docking and MM/GBSA Calculation
3. Results and Discussion
3.1. Identifying Binding Sites on the RBD of SARS-CoV-2 Spike Protein
3.2. Principal Component Analysis (PCA) of MD Simulation Trajectory and FTMap Analysis
3.3. Molecular Docking of FA to RBD and Binding Affinities Calculated by the MM/GBSA Method
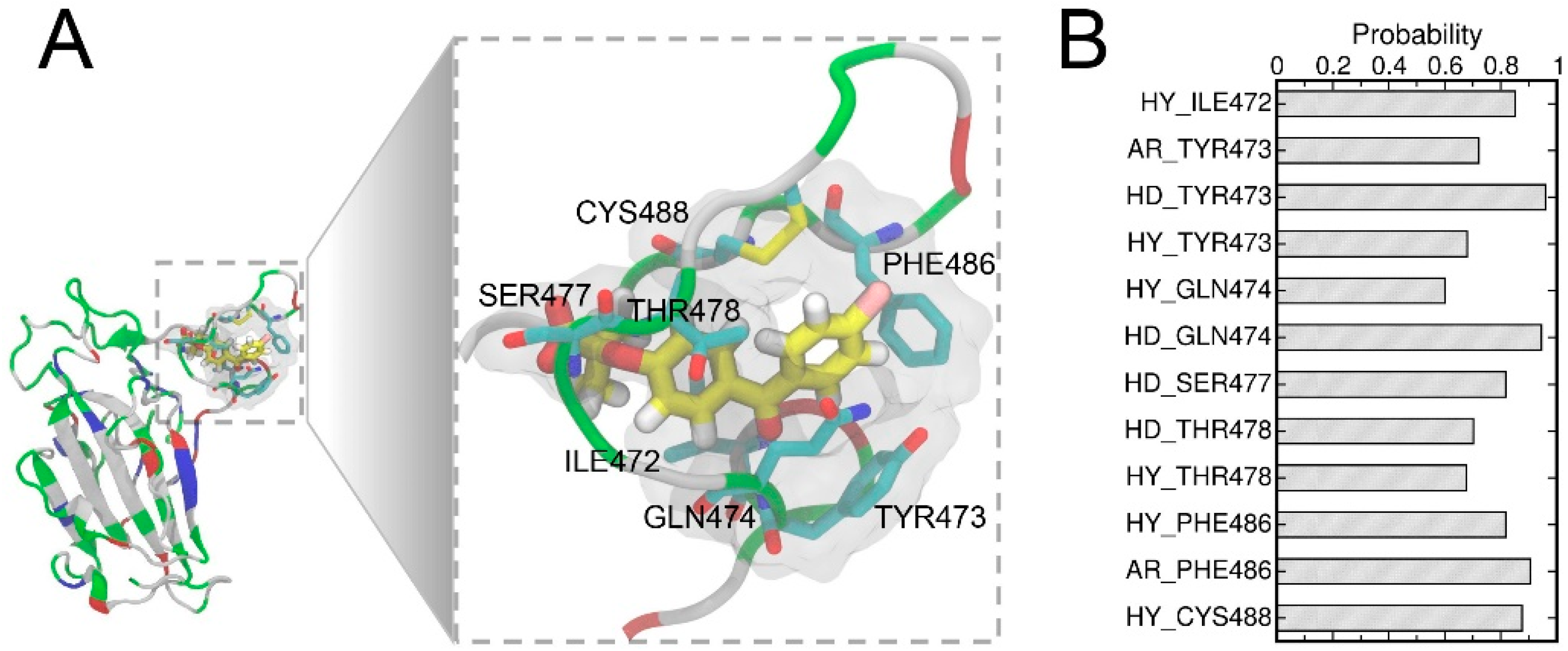
3.4. Structural Analysis of the Complex with the Highest Binding Affinity
3.5. Potential Mechanism of FA Reducing the Complexation of RBD and Human ACE2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Dai, T.; Wang, B.; Zhang, L.; Zeng, L.-H.; Huang, J.; Yan, H.; Zhang, L.; Zhou, F. The way of SARS-CoV-2 vaccine development: Success and challenges. Signal Transduct. Target. Ther. 2021, 6, 387. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Berman, G.; Zhou, H.; Deng, W.; Faughnan, V.; Coronado-Voges, M.; Ding, B.; Dooley, J.; Girard, B.; Hillebrand, W.; et al. Evaluation of mRNA-1273 SARS-CoV-2 Vaccine in Adolescents. N. Engl. J. Med. 2021, 385, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- El Sahly, H.M.; Baden, L.R.; Essink, B.; Doblecki-Lewis, S.; Martin, J.M.; Anderson, E.J.; Campbell, T.B.; Clark, J.; Jackson, L.A.; Fichtenbaum, C.J.; et al. Efficacy of the mRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N. Engl. J. Med. 2021, 385, 1774–1785. [Google Scholar] [CrossRef]
- Golob, J.L.; Lugogo, N.; Lauring, A.S.; Lok, A.S. SARS-CoV-2 vaccines: A triumph of science and collaboration. JCI Insight 2021, 6, e149187. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Akbarzadeh-Khiavi, M.; Torabi, M.; Rahbarnia, L.; Safary, A. Baricitinib combination therapy: A narrative review of repurposed Janus kinase inhibitor against severe SARS-CoV-2 infection. Infection 2022, 50, 295–308. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharm. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef]
- Dotolo, S.; Marabotti, A.; Facchiano, A.; Tagliaferri, R. A review on drug repurposing applicable to COVID-19. Brief. Bioinform. 2021, 22, 726–741. [Google Scholar] [CrossRef]
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, Z.; Li, F.; Xiao, Y.; Wang, Z.; Xu, J.; Dong, F.; Zheng, H.; Yu, R. The study of antiviral drugs targeting SARS-CoV-2 nucleocapsid and spike proteins through large-scale compound repurposing. Heliyon 2021, 7, e06387. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Mycroft-West, C.J.; Pagani, I.; Hill, H.J.; Chen, Y.-H.; Karlsson, R.; Bagdonaite, I.; Guimond, S.E.; Stamataki, Z.; De Lima, M.A.; et al. The Hyperlipidaemic Drug Fenofibrate Significantly Reduces Infection by SARS-CoV-2 in Cell Culture Models. Front. Pharmacol. 2021, 12, 660490. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef]
- Fernandes, M.S.; da Silva, F.S.; Freitas, A.C.S.G.; de Melo, E.B.; Trossini, G.H.G.; Paula, F.R. Insights on 3D Structures of Potential Drug-targeting Proteins of SARS-CoV-2: Application of Cavity Search and Molecular Docking. Mol. Inform. 2021, 40, 2000096. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. Binding Site Detection and Druggability Prediction of Protein Targets for Structure- Based Drug Design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Chen, J.; Lai, L. Pocket v.2: Further Developments on Receptor-Based Pharmacophore Modeling. J. Chem. Inf. Model. 2006, 46, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Meng, H.; Lai, L. Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J. Chem. Inf. Model. 2016, 56, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Statistical Analysis and Prediction of Covalent Ligand Targeted Cysteine Residues. J. Chem. Inf. Model. 2017, 57, 1453–1460. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Prodrugs and Targeted Delivery: Towards Better ADME Properties; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ahmed, R.; Omidian, Z.; Giwa, A.; Cornwell, B.; Majety, N.; Bell, D.R.; Lee, S.; Zhang, H.; Michels, A.; Desiderio, S.; et al. A Public BCR Present in a Unique Dual-Receptor-Expressing Lymphocyte from Type 1 Diabetes Patients Encodes a Potent T Cell Autoantigen. Cell 2019, 177, 1583–1599. [Google Scholar] [CrossRef]
- Luan, B.Q.; Zhou, R.H. Spontaneous ssDNA stretching on graphene and hexagonal boron nitride in plane heterostructures. Nat. Commun. 2019, 10, 4610. [Google Scholar] [CrossRef]
- Luan, B.Q.; Zhou, R.H. Atomic-Scale Fluidic Diodes Based on Triangular Nanopores in Bilayer Hexagonal Boron Nitride. Nano Lett. 2019, 19, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.; Eleftheriou, M.; Zhou, R. Hydration and dewetting near fluorinated superhydrophobic plates. J. Am. Chem. Soc. 2006, 128, 12439–12447. [Google Scholar] [CrossRef]
- Xia, Z.; Clark, P.; Huynh, T.; Loher, P.; Zhao, Y.; Chen, H.W.; Rigoutsos, I.; Zhou, R. Molecular dynamics simulations of Ago silencing complexes reveal a large repertoire of admissible ‘seed-less’ targets. Sci. Rep. 2012, 2, 569. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald—An n.log(n) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Eyal, E.; Lum, G.; Bahar, I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kang, Y.; Duan, M.; Hou, T. Regulation Mechanism for the Binding between the SARS-CoV-2 Spike Protein and Host Angiotensin-Converting Enzyme II. J. Phys. Chem. Lett. 2021, 12, 6252–6261. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Wang, B.; Sam, A.; Hoop, C.L.; Case, D.A.; Baum, J. Molecular dynamics analysis of a flexible loop at the binding interface of the SARS-CoV-2 spike protein receptor-binding domain. Proteins Struct. Funct. Bioinf. 2022, 90, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.R.; Maity, A.; Chakraborty, S.; Chakrabarti, R. Computational design of stapled peptide inhibitor against SARS-CoV-2 receptor binding domain. Pept. Sci. 2022, 114, e24267. [Google Scholar] [CrossRef]
- Beglov, D.; Hall David, R.; Wakefield Amanda, E.; Luo, L.; Allen Karen, N.; Kozakov, D.; Whitty, A.; Vajda, S. Exploring the structural origins of cryptic sites on proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3416–E3425. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Bowman, G.R.; Juarez-Jimenez, J.; Michel, J.; Gervasio, F.L. Investigating Cryptic Binding Sites by Molecular Dynamics Simulations. Acc. Chem. Res. 2020, 53, 654–661. [Google Scholar] [CrossRef]
- Ngan, C.H.; Bohnuud, T.; Mottarella, S.E.; Beglov, D.; Villar, E.A.; Hall, D.R.; Kozakov, D.; Vajda, S. FTMAP: Extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 2012, 40, W271–W275. [Google Scholar] [CrossRef]
- Kimura, S.R.; Hu, H.P.; Ruvinsky, A.M.; Sherman, W.; Favia, A.D. Deciphering Cryptic Binding Sites on Proteins by Mixed-Solvent Molecular Dynamics. J. Chem. Inf. Model. 2017, 57, 1388–1401. [Google Scholar] [CrossRef]
- Kokh, D.B.; Doser, B.; Richter, S.; Ormersbach, F.; Cheng, X.; Wade, R.C. A workflow for exploring ligand dissociation from a macromolecule: Efficient random acceleration molecular dynamics simulation and interaction fingerprint analysis of ligand trajectories. J. Chem. Phys. 2020, 153, 125102. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Taka, E.; Yilmaz, S.Z.; Golcuk, M.; Kilinc, C.; Aktas, U.; Yildiz, A.; Gur, M. Critical Interactions Between the SARS-CoV-2 Spike Glycoprotein and the Human ACE2 Receptor. J. Phys. Chem. B 2021, 125, 5537–5548. [Google Scholar] [CrossRef] [PubMed]
- Gheeraert, A.; Vuillon, L.; Chaloin, L.; Moncorgé, O.; Very, T.; Perez, S.; Leroux, V.; Chauvot de Beauchêne, I.; Mias-Lucquin, D.; Devignes, M.-D.; et al. Singular Interface Dynamics of the SARS-CoV-2 Delta Variant Explained with Contact Perturbation Analysis. J. Chem. Inf. Model. 2022, 62, 3107–3122. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Song, Y.; Xu, Z.; Shang, C.; Zhou, R. SARS-CoV-2 Delta Variant: Interplay between Individual Mutations and Their Allosteric Synergy. Biomolecules 2022, 12, 1742. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zhang, Z.; Gao, J.; Wang, Y. SARS-CoV-2 spike protein N501Y mutation causes differential species transmissibility and antibody sensitivity: A molecular dynamics and alchemical free energy study. Mol. Syst. Des. Eng. 2021, 6, 964–974. [Google Scholar] [CrossRef]
- He, J.; Tao, H.; Yan, Y.; Huang, S.-Y.; Xiao, Y. Molecular Mechanism of Evolution and Human Infection with SARS-CoV-2. Viruses 2020, 12, 428. [Google Scholar] [CrossRef]
- Zuzic, L.; Samsudin, F.; Shivgan, A.T.; Raghuvamsi, P.V.; Marzinek, J.K.; Boags, A.; Pedebos, C.; Tulsian, N.K.; Warwicker, J.; MacAry, P.; et al. Uncovering cryptic pockets in the SARS-CoV-2 spike glycoprotein. Structure 2022, 30, 1062–1074. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔEele | ΔEvdW | ΔGGB | ΔGSA | ΔGeff * |
|---|---|---|---|---|---|
| FA-bound RBD-ACE2 | −535.22 | −65.21 | 577.13 | −9.03 | −33.32 |
| RBD-ACE2 | −551.12 | −89.79 | 607.62 | −13.06 | −47.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Chan, K.C.; Zhou, R. Novel Inhibitory Role of Fenofibric Acid by Targeting Cryptic Site on the RBD of SARS-CoV-2. Biomolecules 2023, 13, 359. https://doi.org/10.3390/biom13020359
Huang J, Chan KC, Zhou R. Novel Inhibitory Role of Fenofibric Acid by Targeting Cryptic Site on the RBD of SARS-CoV-2. Biomolecules. 2023; 13(2):359. https://doi.org/10.3390/biom13020359
Chicago/Turabian StyleHuang, Jianxiang, Kevin C. Chan, and Ruhong Zhou. 2023. "Novel Inhibitory Role of Fenofibric Acid by Targeting Cryptic Site on the RBD of SARS-CoV-2" Biomolecules 13, no. 2: 359. https://doi.org/10.3390/biom13020359
APA StyleHuang, J., Chan, K. C., & Zhou, R. (2023). Novel Inhibitory Role of Fenofibric Acid by Targeting Cryptic Site on the RBD of SARS-CoV-2. Biomolecules, 13(2), 359. https://doi.org/10.3390/biom13020359