

Updated Virophage Taxonomy and Distinction from Polinton-like Viruses

 , , , , and
, , , , and 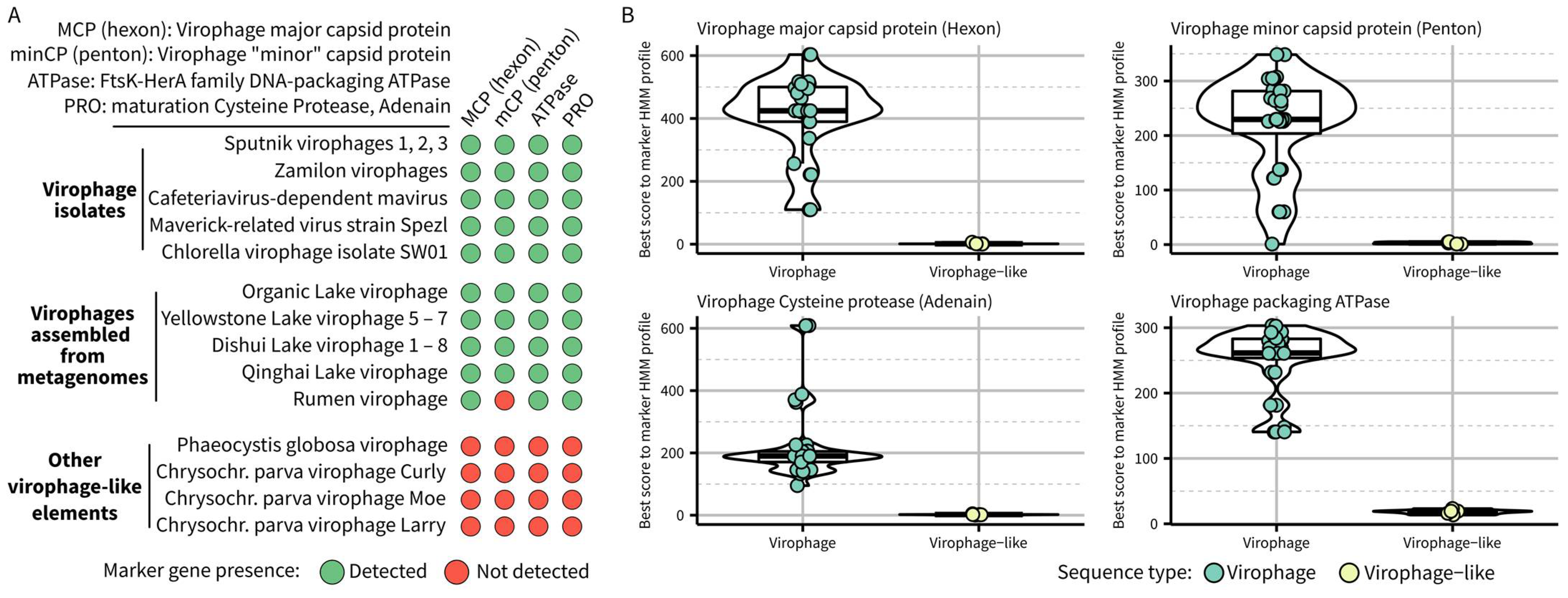{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results & Discussion
2.1. Definition of a Formal Virophage Taxon and Associated Demarcation Criteria
- –
- required features: the complete genome should encode a virophage-like hexon protein, a virophage-like ATPase, and a virophage-like cysteine protease, all of which can be detected based on established HMM profiles for each of these marker genes
- –
- other expected (but not required) features: the genome should consist of dsDNA with a length between 15 kb and 45 kb and encode a virophage-like penton protein detected based on established HMM profile(s).
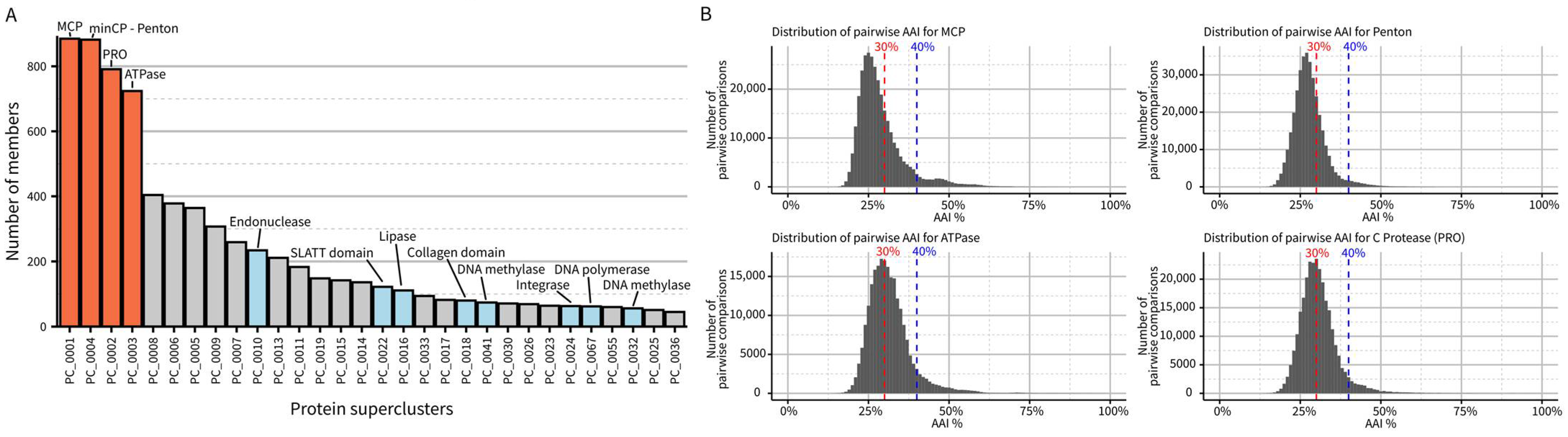
2.2. Common Origin and Genetic Diversity in the Extent Virophage Clade
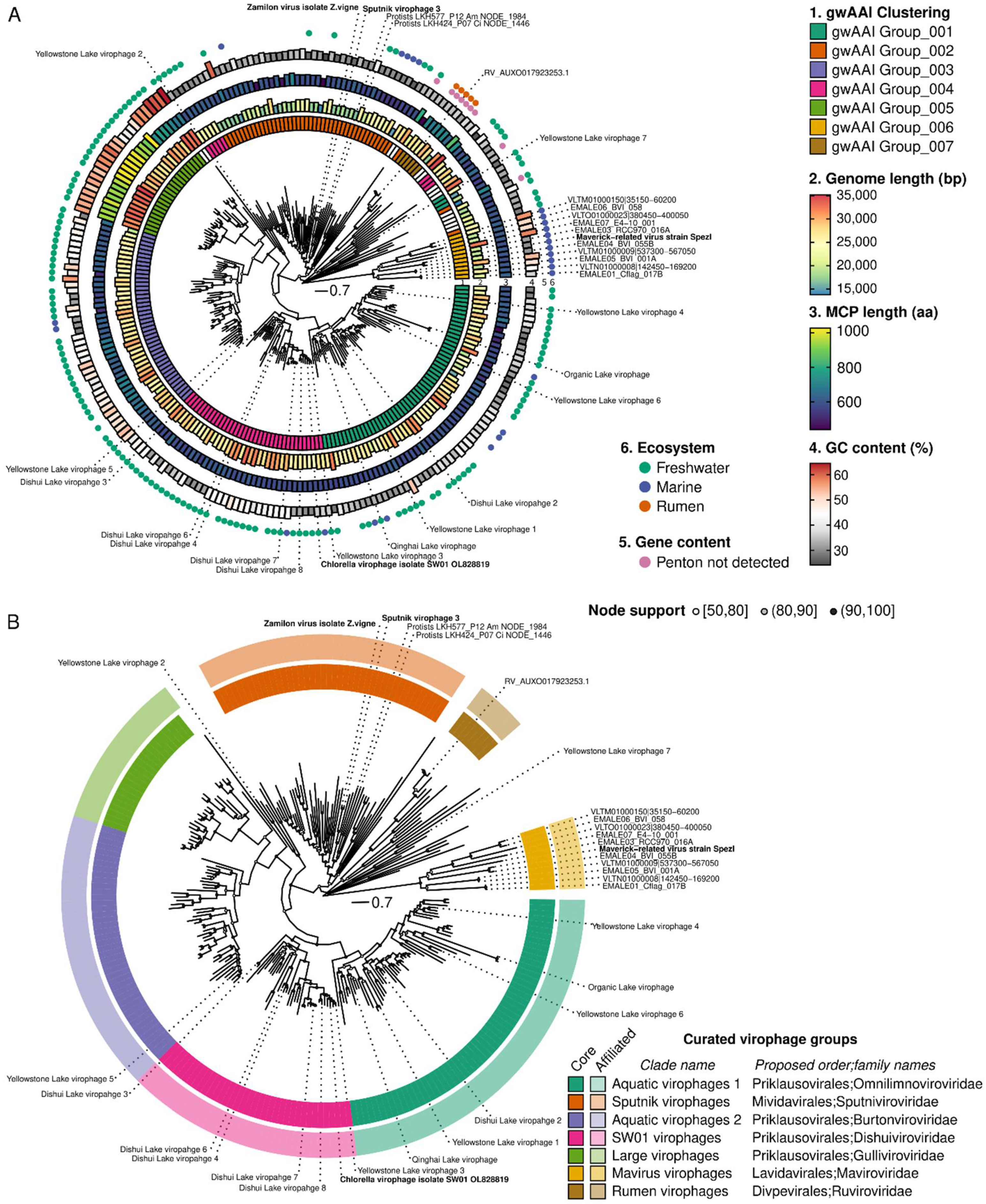
2.3. Challenges and Limitations of Taxonomic Classification within the Virophage Clade
- –
- Rumen virophages as the new Ruviroviridae family in the new Divpevirales order, named for virophages with Divergent penton proteins.
- –
- Mavirus virophages as the new Maviroviridae family after the first isolated member of the taxon, in the new Lavidavirales order, a name adapted from the current Lavidaviridae family named for “Large virus dependent or associated.”
- –
- Sputnik virophages as the new Sputniviroviridae family after the first isolated member of the taxon, in the new Mividavirales order, for “Mimivirus dependent or associated”
- –
- SW01 virophages as the new Dishuiviroviridae family after the lake from which the first member of the taxon (SW01) was isolated, in the Priklausovirales order, which is currently the only order established in the Maveriviricetes class.
- –
- Aquatic virophages 1 as the new Omnilimnoviroviridae family using the prefixes “omni” (“all,” “everywhere”) and “limno” denoting a link to freshwater environments, since members of this clade were detected across a broad geographic range of freshwater lakes, also in the same Priklausovirales order.
- –
- Large virophages as the new Gulliviroviridae family named after Lemuel Gulliver, the main character of “Gulliver’s travel,” since these virophages can be considered as “giant” compared to other virophages but are still relatively small compared to their associated giant viruses, also in the same Priklausovirales order.
- –
- Aquatic virophages 2 as the new Burtonviroviridae family named after Mary Burton, the wife of Lemuel Gulliver in “Gulliver’s travel,” as this clade is the most closely related to the large virophages, also in the same Priklausovirales order.
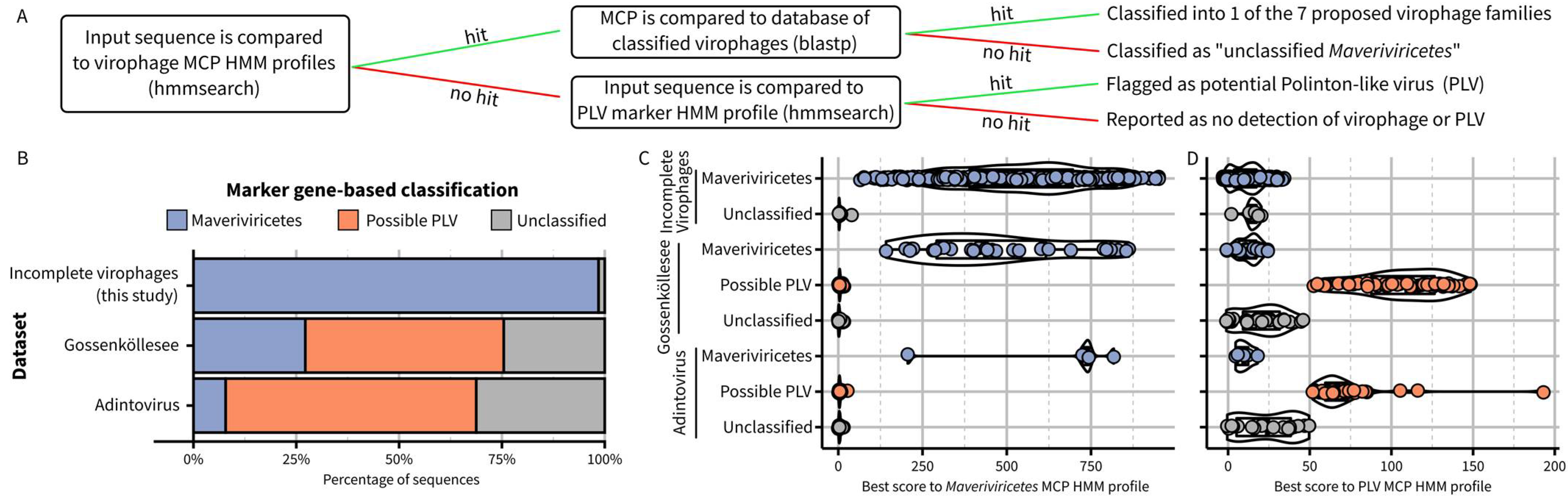
2.4. Examples of Detection and Taxonomic Assignment of Virophages in Mixed Datasets
2.5. Beyond Genome-Based Taxonomy: Virophage Host Range and Interactions
3. Conclusions
4. Methods
4.1. Collection of Virophage and Virophage-Like Sequences
4.2. External Datasets Used to Illustrate Virophage Identification and Classification Approach
4.3. Virophage Genome Clustering, Quality Control, and Trimming
4.4. Genome Annotation and De Novo Protein Clustering
4.5. Rooted Phylogenetic Trees for ATPase and PRO Genes
4.6. Identification and Analysis of Complete and Near-Complete Virophage Genomes
4.7. Identification of New Virophage Taxa, Delineation Criteria, and Classification Approach
4.8. Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Claverie, J.-M.; Abergel, C. Mimivirus and Its Virophage. Annu. Rev. Genet. 2009, 43, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Mougari, S.; Sahmi-Bounsiar, D.; Levasseur, A.; Colson, P.; La Scola, B. Virophages of Giant Viruses: An Update at Eleven. Viruses 2019, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Duponchel, S.; Fischer, M.G. Viva Lavidaviruses! Five Features of Virophages That Parasitize Giant DNA Viruses. PLOS Pathog. 2019, 15, e1007592. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; Raoult, D.; et al. The Virophage as a Unique Parasite of the Giant Mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef]
- Fischer, M.G.; Suttle, C.A. A Virophage at the Origin of Large DNA Transposons. Science 2011, 332, 231–234. [Google Scholar] [CrossRef]
- Sheng, Y.; Wu, Z.; Xu, S.; Wang, Y. Isolation and Identification of a Large Green Alga Virus (Chlorella Virus XW01) of Mimiviridae and Its Virophage (Chlorella Virus Virophage SW01) by Using Unicellular Green Algal Cultures. J. Virol. 2022, 96, e02114-21. [Google Scholar] [CrossRef]
- Gaia, M.; Benamar, S.; Boughalmi, M.; Pagnier, I.; Croce, O.; Colson, P.; Raoult, D.; Scola, B.L. Zamilon, a Novel Virophage with Mimiviridae Host Specificity. PLoS ONE 2014, 9, e94923. [Google Scholar] [CrossRef]
- Fischer, M.G. The Virophage Family Lavidaviridae. Curr. Issues Mol. Biol. 2021, 40, 1–24. [Google Scholar] [CrossRef]
- Krupovic, M.; Kuhn, J.H.; Fischer, M.G. A Classification System for Virophages and Satellite Viruses. Arch. Virol. 2016, 161, 233–247. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The New Scope of Virus Taxonomy: Partitioning the Virosphere into 15 Hierarchical Ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Yutin, N.; Kapitonov, V.V.; Koonin, E.V. A New Family of Hybrid Virophages from an Animal Gut Metagenome. Biol. Direct 2015, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Zhou, J.; Roux, S.; Nayfach, S.; Pavlopoulos, G.A.; Schulz, F.; McMahon, K.D.; Walsh, D.; Woyke, T.; Ivanova, N.N.; et al. Diversity, Evolution, and Classification of Virophages Uncovered through Global Metagenomics. Microbiome 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage Control of Antarctic Algal Host-Virus Dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, W.; Yan, S.; Xiao, J.; Zhang, Y.; Li, B.; Pan, Y.; Wang, Y. Diversity of Virophages in Metagenomic Data Sets. J. Virol. 2013, 87, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, D.; Childers, A.; McDermott, T.R.; Wang, Y.; Liles, M.R. Three Novel Virophage Genomes Discovered from Yellowstone Lake Metagenomes. J. Virol. 2015, 89, 1278–1285. [Google Scholar] [CrossRef]
- Roux, S.; Chan, L.K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of Virophages and Their Giant Virus Hosts Assessed through Time Series Metagenomics. Nat. Commun. 2017, 8, 858. [Google Scholar] [CrossRef]
- Gong, C.; Zhang, W.; Zhou, X.; Wang, H.; Sun, G.; Xiao, J.; Pan, Y.; Yan, S.; Wang, Y. Novel Virophages Discovered in a Freshwater Lake in China. Front. Microbiol. 2016, 7, 5. [Google Scholar] [CrossRef]
- Oh, S.; Yoo, D.; Liu, W.-T.T. Metagenomics Reveals a Novel Virophage Population in a Tibetan Mountain Lake. Microbes Environ. 2016, 31, 173–177. [Google Scholar] [CrossRef]
- Bellas, C.M.; Sommaruga, R. Polinton-like Viruses Are Abundant in Aquatic Ecosystems. Microbiome 2021, 9, 13. [Google Scholar] [CrossRef]
- Yutin, N.; Raoult, D.; Koonin, E.V. Virophages, Polintons, and Transpovirons: A Complex Evolutionary Network of Diverse Selfish Genetic Elements with Different Reproduction Strategies. Virol. J. 2013, 10, 158. [Google Scholar] [CrossRef]
- Yutin, N.; Shevchenko, S.; Kapitonov, V.; Krupovic, M.; Koonin, E.V. A Novel Group of Diverse Polinton-like Viruses Discovered by Metagenome Analysis. BMC Biol. 2015, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E.V. Polintons: A Hotbed of Eukaryotic Virus, Transposon and Plasmid Evolution. Nat. Rev. Microbiol. 2015, 13, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Tisza, M.J.; Welch, N.L.; Belford, A.K.; Peretti, A.; Pastrana, D.V.; Buck, C.B. Adintoviruses: A Proposed Animal-Tropic Family of Midsize Eukaryotic Linear DsDNA (MELD) Viruses. Virus Evol. 2021, 7, veaa055. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V. Genome of Phaeocystis Globosa Virus PgV-16T Highlights the Common Ancestry of the Largest Known DNA Viruses Infecting Eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]
- Roitman, S.; Rozenberg, A.; Lavy, T.; Brussaard, C.P.D.; Kleifeld, O.; Béjà, O. Infection Cycle and Phylogeny of the Polinton-like Virus Phaeocystis Globosa Virus Virophage-14T. bioRxiv 2022. [Google Scholar] [CrossRef]
- Krupovic, M.; Bamford, D.H.; Koonin, E.V. Conservation of Major and Minor Jelly-Roll Capsid Proteins in Polinton (Maverick) Transposons Suggests That They Are Bona Fide Viruses. Biol. Direct 2014, 9, 6. [Google Scholar] [CrossRef]
- Fischer, M.G.; Hackl, T. Host Genome Integration and Giant Virus-Induced Reactivation of the Virophage Mavirus. Nature 2016, 540, 288–291. [Google Scholar] [CrossRef]
- Hackl, T.; Duponchel, S.; Barenhoff, K.; Weinmann, A.; Fischer, M.G. Virophages and Retrotransposons Colonize the Genomes of a Heterotrophic Flagellate. eLife 2021, 10, e72674. [Google Scholar] [CrossRef]
- Stough, J.M.A.; Yutin, N.; Chaban, Y.V.; Moniruzzaman, M.; Gann, E.R.; Pound, H.L.; Steffen, M.M.; Black, J.N.; Koonin, E.V.; Wilhelm, S.W.; et al. Genome and Environmental Activity of a Chrysochromulina Parva Virus and Its Virophages. Front. Microbiol. 2019, 10, 703. [Google Scholar] [CrossRef]
- Roux, S.; Páez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An Integrated Ecological and Evolutionary Framework for Interrogating Genomes of Uncultivated Viruses. Nucleic Acids Res. 2020, 49, D764–D775. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUVIG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Buck, C.B. A Catalog of Tens of Thousands of Viruses from Human Metagenomes Reveals Hidden Associations with Chronic Diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.A.; Coffman, K.A.; Gilbert, C.; Ravindran, S.; Albery, G.F.; Abbott, J.; Argyridou, E.; Bellosta, P.; Betancourt, A.J.; Colinet, H.; et al. The Discovery, Distribution, and Diversity of DNA Viruses Associated with Drosophila Melanogaster in Europe. Virus Evol. 2021, 7, veab031. [Google Scholar] [CrossRef] [PubMed]
- Castillo, Y.M.; Forn, I.; Yau, S.; Morán, X.A.G.; Alonso-Sáez, L.; Arandia-Gorostidi, N.; Vaqué, D.; Sebastián, M. Seasonal Dynamics of Natural Ostreococcus Viral Infection at the Single Cell Level Using VirusFISH. Environ. Microbiol. 2021, 23, 3009–3019. [Google Scholar] [CrossRef]
- del Arco, A.; Fischer, M.; Becks, L. Simultaneous Giant Virus and Virophage Quantification Using Droplet Digital PCR. Viruses 2022, 14, 1056. [Google Scholar] [CrossRef]
- Levasseur, A.; Bekliz, M.; Chabrière, E.; Pontarotti, P.; Scola, B.L.; Raoult, D. MIMIVIRE Is a Defence System in Mimivirus That Confers Resistance to Virophage. Nature 2016, 531, 249–252. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Sundaramurthi, J.C.; Lee, J.; Kandimalla, M.; Chen, I.-M.A.; Kyrpides, N.C.; Reddy, T.B.K. Genomes OnLine Database (GOLD) v.8: Overview and Updates. Nucleic Acids Res. 2021, 49, D723–D733. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to Identify Similar Regions in Large Sequence Sets. Curr. Protoc. Bioinform. 2003, 10, 10.3. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Chandonia, J.-M.; Fox, N.K.; Brenner, S.E. SCOPe: Manual Curation and Artifact Removal in the Structural Classification of Proteins – Extended Database. J. Mol. Biol. 2017, 429, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-Fast Iterative Protein Sequence Searching by HMM-HMM Alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-Suite3 for Fast Remote Homology Detection and Deep Protein Annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Edler, D.; Bohlin, L.; Rosvall, M. Mapping Higher-Order Network Flows in Memory and Multilayer Networks with Infomap. Algorithms 2017, 10, 112. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- van Dongen, S.; Abreu-Goodger, C. Using MCL to Extract Clusters from Networks. In Bacterial Molecular Networks: Methods and Protocols; van Helden, J., Toussaint, A., Thieffry, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2012; pp. 281–295. ISBN 978-1-61779-361-5. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Iii, T.J.B.; Li, Y.; Shen, X.-X.; Rokas, A. ClipKIT: A Multiple Sequence Alignment Trimming Software for Accurate Phylogenomic Inference. PLOS Biol. 2020, 18, e3001007. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Páez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic Compendium of 189,680 DNA Viruses from the Human Gut Microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation, 2022.
- Wickham, H.; Girlich, M. Tidyr: Tidy Messy Data, 2022.
- Yu, G. Using Ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.-Y.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Yu, G. Data Integration, Manipulation and Visualization of Phylogenetic Trees, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2022. [Google Scholar] [CrossRef]
- Xu, S.; Dai, Z.; Guo, P.; Fu, X.; Liu, S.; Zhou, L.; Tang, W.; Feng, T.; Chen, M.; Zhan, L.; et al. GgtreeExtra: Compact Visualization of Richly Annotated Phylogenetic Data. Mol. Biol. Evol. 2021, 38, 4039–4042. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Rokas, A. Ggpubfigs: Colorblind-Friendly Color Palettes and Ggplot2 Graphic System Extensions for Publication-Quality Scientific Figures. Microbiol. Resour. Announc. 2021, 10, e00871-21. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Lavy, A.; McGrath, D.G.; Matheus Carnevali, P.B.; Wan, J.; Dong, W.; Tokunaga, T.K.; Thomas, B.C.; Williams, K.H.; Hubbard, S.S.; Banfield, J.F. Microbial Communities across a Hillslope-riparian Transect Shaped by Proximity to the Stream, Groundwater Table, and Weathered Bedrock. Ecol. Evol. 2019, 9, 6869–6900. [Google Scholar] [CrossRef] [PubMed]
- Linz, A.M.; Aylward, F.O.; Bertilsson, S.; McMahon, K.D. Time-series Metatranscriptomes Reveal Conserved Patterns between Phototrophic and Heterotrophic Microbes in Diverse Freshwater Systems. Limnol. Oceanogr. 2020, 65, S101–S112. [Google Scholar] [CrossRef]
- Waldo, N.B.; Chistoserdova, L.; Hu, D.; Gough, H.L.; Neumann, R.B. Impacts of The Wetland Sedge Carex Aquatilis on Microbial Community and Methane Metabolisms. Plant Soil 2022, 471, 491–506. [Google Scholar] [CrossRef]
- Borges, I.A.; de Assis, F.L.; Silva, L.K.; dos Santos Silva, L.K.; Abrahão, J. Rio Negro Virophage: Sequencing of the near Complete Genome and Transmission Electron Microscopy of Viral Factories and Particles. Braz. J. Microbiol. 2018, 49, 260–261. [Google Scholar] [CrossRef]
- Rozmarynowycz, M.J.; Beall, B.F.N.; Bullerjahn, G.S.; Small, G.E.; Sterner, R.W.; Brovold, S.S.; D’souza, N.A.; Watson, S.B.; McKay, R.M.L. Transitions in Microbial Communities along a 1600 Km Freshwater Trophic Gradient. J. Gt. Lakes Res. 2019, 45, 263–276. [Google Scholar] [CrossRef]
- Kelly, C.N.; Schwaner, G.W.; Cumming, J.R.; Driscoll, T.P. Metagenomic Reconstruction of Nitrogen and Carbon Cycling Pathways in Forest Soil: Influence of Different Hardwood Tree Species. Soil Biol. Biochem. 2021, 156, 108226. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, R.; Liu, L.; Li, B.; Zhang, T. Selective Enrichment of Comammox from Activated Sludge Using Antibiotics. Water Res. 2021, 197, 117087. [Google Scholar] [CrossRef]
- Kantor, R.S.; Huddy, R.J.; Iyer, R.; Thomas, B.C.; Brown, C.T.; Anantharaman, K.; Tringe, S.; Hettich, R.L.; Harrison, S.T.L.; Banfield, J.F. Genome-Resolved Meta-Omics Ties Microbial Dynamics to Process Performance in Biotechnology for Thiocyanate Degradation. Environ. Sci. Technol. 2017, 51, 2944–2953. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Caffrey, S.M.; Soh, J.; Agrawal, A.; Brown, D.; Budwill, K.; Dong, X.; Dunfield, P.F.; Foght, J.; Gieg, L.M.; et al. Metagenomics of Hydrocarbon Resource Environments Indicates Aerobic Taxa and Genes to Be Unexpectedly Common. Environ. Sci. Technol. 2013, 47, 10708–10717. [Google Scholar] [CrossRef] [PubMed]
- Sabuda, M.C.; Putman, L.I.; Hoehler, T.M.; Kubo, M.D.; Brazelton, W.J.; Cardace, D.; Schrenk, M.O. Biogeochemical Gradients in a Serpentinization-Influenced Aquifer: Implications for Gas Exchange Between the Subsurface and Atmosphere. J. Geophys. Res. Biogeosci. 2021, 126, e2020JG006209. [Google Scholar] [CrossRef]
- Bendall, M.L.; Stevens, S.L.; Chan, L.-K.; Malfatti, S.; Schwientek, P.; Tremblay, J.; Schackwitz, W.; Martin, J.; Pati, A.; Bushnell, B.; et al. Genome-Wide Selective Sweeps and Gene-Specific Sweeps in Natural Bacterial Populations. ISME J. 2016, 10, 1589–1601. [Google Scholar] [CrossRef]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex Archaea That Bridge the Gap between Prokaryotes and Eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and Potential Biogeochemical Impacts of Uncultivated Globally Abundant Ocean Viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef]
- MetaHIT Consortium; Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; et al. Identification and Assembly of Genomes and Genetic Elements in Complex Metagenomic Samples without Using Reference Genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Levy-Booth, D.J.; Giesbrecht, I.J.W.; Kellogg, C.T.E.; Heger, T.J.; D’Amore, D.V.; Keeling, P.J.; Hallam, S.J.; Mohn, W.W. Seasonal and Ecohydrological Regulation of Active Microbial Populations Involved in DOC, CO2, and CH4 Fluxes in Temperate Rainforest Soil. ISME J. 2019, 13, 950–963. [Google Scholar] [CrossRef]
- Krüger, K.; Chafee, M.; Ben Francis, T.; Glavina del Rio, T.; Becher, D.; Schweder, T.; Amann, R.I.; Teeling, H. In Marine Bacteroidetes the Bulk of Glycan Degradation during Algae Blooms Is Mediated by Few Clades Using a Restricted Set of Genes. ISME J. 2019, 13, 2800–2816. [Google Scholar] [CrossRef]
- Jeudy, S.; Bertaux, L.; Alempic, J.-M.; Lartigue, A.; Legendre, M.; Belmudes, L.; Santini, S.; Philippe, N.; Beucher, L.; Biondi, E.G.; et al. Exploration of the Propagation of Transpovirons within Mimiviridae Reveals a Unique Example of Commensalism in the Viral World. ISME J. 2020, 14, 727–739. [Google Scholar] [CrossRef]
- St. James, A.R.; Yavitt, J.B.; Zinder, S.H.; Richardson, R.E. Linking Microbial Sphagnum Degradation and Acetate Mineralization in Acidic Peat Bogs: From Global Insights to a Genome-Centric Case Study. ISME J. 2021, 15, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Gazitúa, M.C.; Vik, D.R.; Roux, S.; Gregory, A.C.; Bolduc, B.; Widner, B.; Mulholland, M.R.; Hallam, S.J.; Ulloa, O.; Sullivan, M.B. Potential Virus-Mediated Nitrogen Cycling in Oxygen-Depleted Oceanic Waters. ISME J. 2021, 15, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Goudeau, D.; Olmsted, C.; McMahon, K.D.; Yitbarek, S.; Thweatt, J.L.; Bryant, D.A.; Eloe-Fadrosh, E.A.; Malmstrom, R.R.; Roux, S. Host Population Diversity as a Driver of Viral Infection Cycle in Wild Populations of Green Sulfur Bacteria with Long Standing Virus-Host Interactions. ISME J. 2021, 15, 1569–1584. [Google Scholar] [CrossRef]
- Tsuji, J.M.; Tran, N.; Schiff, S.L.; Venkiteswaran, J.J.; Molot, L.A.; Tank, M.; Hanada, S.; Neufeld, J.D. Anoxygenic Photosynthesis and Iron–Sulfur Metabolic Potential of Chlorobia Populations from Seasonally Anoxic Boreal Shield Lakes. ISME J. 2020, 14, 2732–2747. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.Q.; Bachand, S.C.; McIntyre, P.B.; Kraemer, B.M.; Vadeboncoeur, Y.; Kimirei, I.A.; Tamatamah, R.; McMahon, K.D.; Anantharaman, K. Depth-Discrete Metagenomics Reveals the Roles of Microbes in Biogeochemical Cycling in the Tropical Freshwater Lake Tanganyika. ISME J. 2021, 15, 1971–1986. [Google Scholar] [CrossRef] [PubMed]
- Francis, T.B.; Bartosik, D.; Sura, T.; Sichert, A.; Hehemann, J.-H.; Markert, S.; Schweder, T.; Fuchs, B.M.; Teeling, H.; Amann, R.I.; et al. Changing Expression Patterns of TonB-Dependent Transporters Suggest Shifts in Polysaccharide Consumption over the Course of a Spring Phytoplankton Bloom. ISME J. 2021, 15, 2336–2350. [Google Scholar] [CrossRef]
- Zhou, J.; Theroux, S.M.; Bueno de Mesquita, C.P.; Hartman, W.H.; Tian, Y.; Tringe, S.G. Microbial Drivers of Methane Emissions from Unrestored Industrial Salt Ponds. ISME J. 2022, 16, 284–295. [Google Scholar] [CrossRef]
- Jurgensen, S.K.; Roux, S.; Schwenck, S.M.; Stewart, F.J.; Sullivan, M.B.; Brum, J.R. Viral Community Analysis in a Marine Oxygen Minimum Zone Indicates Increased Potential for Viral Manipulation of Microbial Physiological State. ISME J. 2022, 16, 972–982. [Google Scholar] [CrossRef]
- Angle, J.C.; Morin, T.H.; Solden, L.M.; Narrowe, A.B.; Smith, G.J.; Borton, M.A.; Rey-Sanchez, C.; Daly, R.A.; Mirfenderesgi, G.; Hoyt, D.W.; et al. Methanogenesis in Oxygenated Soils Is a Substantial Fraction of Wetland Methane Emissions. Nat. Commun. 2017, 8, 1567. [Google Scholar] [CrossRef]
- Dombrowski, N.; Teske, A.P.; Baker, B.J. Expansive Microbial Metabolic Versatility and Biodiversity in Dynamic Guaymas Basin Hydrothermal Sediments. Nat. Commun. 2018, 9, 4999. [Google Scholar] [CrossRef]
- Sorensen, J.W.; Dunivin, T.K.; Tobin, T.C.; Shade, A. Ecological Selection for Small Microbial Genomes along a Temperate-to-Thermal Soil Gradient. Nat. Microbiol. 2019, 4, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Daly, R.A.; Roux, S.; Borton, M.A.; Morgan, D.M.; Johnston, M.D.; Booker, A.E.; Hoyt, D.W.; Meulia, T.; Wolfe, R.A.; Hanson, A.J.; et al. Viruses Control Dominant Bacteria Colonizing the Terrestrial Deep Biosphere after Hydraulic Fracturing. Nat. Microbiol. 2019, 4, 352–361. [Google Scholar] [CrossRef]
- Woodcroft, B.J.; Singleton, C.M.; Boyd, J.A.; Evans, P.N.; Emerson, J.B.; Zayed, A.A.F.; Hoelzle, R.D.; Lamberton, T.O.; McCalley, C.K.; Hodgkins, S.B.; et al. Genome-Centric View of Carbon Processing in Thawing Permafrost. Nature 2018, 560, 49–54. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef]
- Hackl, T.; Martin, R.; Barenhoff, K.; Duponchel, S.; Heider, D.; Fischer, M.G. Four High-Quality Draft Genome Assemblies of the Marine Heterotrophic Nanoflagellate Cafeteria Roenbergensis. Sci. Data 2020, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Colatriano, D.; Tran, P.Q.; Guéguen, C.; Williams, W.J.; Lovejoy, C.; Walsh, D.A. Genomic Evidence for the Degradation of Terrestrial Organic Matter by Pelagic Arctic Ocean Chloroflexi Bacteria. Commun. Biol. 2018, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Desnues, C.; Scola, B.L.; Yutin, N.; Fournous, G.; Robert, C.; Azza, S.; Jardot, P.; Monteil, S.; Campocasso, A.; Koonin, E.V.; et al. Provirophages and Transpovirons as the Diverse Mobilome of Giant Viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 18078–18083. [Google Scholar] [CrossRef]
- Tallada, S.; Hall, G.; Barich, D.; Morgan-Kiss, R.M.; Slonczewski, J.L. Antibiotic Resistance Genes and Taxa Analysis from Mat and Planktonic Microbiomes of Antarctic Perennial Ice-Covered Lake Fryxell and Lake Bonney. Antarct. Sci. 2021, 34, 408–422. [Google Scholar] [CrossRef]
- Kantor, R.S.; van Zyl, A.W.; van Hille, R.P.; Thomas, B.C.; Harrison, S.T.L.; Banfield, J.F. Bioreactor Microbial Ecosystems for Thiocyanate and Cyanide Degradation Unravelled with Genome-Resolved Metagenomics: Metagenomics of Thiocyanate/Cyanide Biodegradation. Environ. Microbiol. 2015, 17, 4929–4941. [Google Scholar] [CrossRef]
- Tran, P.; Ramachandran, A.; Khawasik, O.; Beisner, B.E.; Rautio, M.; Huot, Y.; Walsh, D.A. Microbial Life under Ice: Metagenome Diversity and in Situ Activity of Verrucomicrobia in Seasonally Ice-covered Lakes. Environ. Microbiol. 2018, 20, 2568–2584. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Tsementzi, D.; Luo, C.; Konstantinidis, K.T. Iterative Subtractive Binning of Freshwater Chronoseries Metagenomes Identifies over 400 Novel Species and Their Ecologic Preferences. Environ. Microbiol. 2020, 22, 3394–3412. [Google Scholar] [CrossRef] [PubMed]
- Michaud, C.; Hervé, V.; Dupont, S.; Dubreuil, G.; Bézier, A.M.; Meunier, J.; Brune, A.; Dedeine, F. Efficient but Occasionally Imperfect Vertical Transmission of Gut Mutualistic Protists in a Wood-Feeding Termite. Mol. Ecol. 2020, 29, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.-W.; Chokhawala, H.; Schroth, G.; Luo, S.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic Discovery of Biomass-Degrading Genes and Genomes from Cow Rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant Viruses with an Expanded Complement of Translation System Components. Science 2017, 85, 82–85. [Google Scholar] [CrossRef]
- Maresca, J.A.; Miller, K.J.; Keffer, J.L.; Sabanayagam, C.R.; Campbell, B.J. Distribution and Diversity of Rhodopsin-Producing Microbes in the Chesapeake Bay. Appl. Environ. Microbiol. 2018, 84, e00137-18. [Google Scholar] [CrossRef] [PubMed]
- Props, R.; Denef, V.J. Temperature and Nutrient Levels Correspond with Lineage-Specific Microdiversification in the Ubiquitous and Abundant Freshwater Genus Limnohabitans. Appl. Environ. Microbiol. 2020, 86, e00140-20. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, L.; Liang, X.; Zhou, Y.; Chen, H.; Yan, S.; Wang, Y.; Science, M.; Biology, M.C. Novel Cell-Virus-Virophage Tripartite Infection Systems Discovered in the Freshwater Lake Dishui Lake in Shanghai, China. J. Virol. 2020, 94, e00149-20. [Google Scholar] [CrossRef]
- He, S.; Malfatti, S.A.; McFarland, J.W.; Anderson, F.E.; Pati, A.; Huntemann, M.; Tremblay, J.; Glavina del Rio, T.; Waldrop, M.P.; Windham-Myers, L.; et al. Patterns in Wetland Microbial Community Composition and Functional Gene Repertoire Associated with Methane Emissions. mBio 2015, 6, e00066-15. [Google Scholar] [CrossRef]
- Podowski, J.C.; Paver, S.F.; Newton, R.J.; Coleman, M.L. Genome Streamlining, Proteorhodopsin, and Organic Nitrogen Metabolism in Freshwater Nitrifiers. mBio 2022, 13, e02379-21. [Google Scholar] [CrossRef]
- Kolton, M.; Weston, D.J.; Mayali, X.; Weber, P.K.; McFarlane, K.J.; Pett-Ridge, J.; Somoza, M.M.; Lietard, J.; Glass, J.B.; Lilleskov, E.A.; et al. Defining the Sphagnum Core Microbiome across the North American Continent Reveals a Central Role for Diazotrophic Methanotrophs in the Nitrogen and Carbon Cycles of Boreal Peatland Ecosystems. mBio 2022, 13, e03714-21. [Google Scholar] [CrossRef]
- Garcia, S.L.; Buck, M.; Hamilton, J.J.; Wurzbacher, C.; Grossart, H.-P.; McMahon, K.D.; Eiler, A. Model Communities Hint at Promiscuous Metabolic Linkages between Ubiquitous Free-Living Freshwater Bacteria. mSphere 2018, 3, e00202-18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, Y.; Xu, W.; Pan, J.; Luo, Z.-H.; Li, M. Genome- and Community-Level Interaction Insights into Carbon Utilization and Element Cycling Functions of Hydrothermarchaeota in Hydrothermal Sediment. mSystems 2020, 5, e00795-19. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhan, Y.; Marsan, D.; Páez-Espino, D.; Cai, L.; Chen, F. Uncultivated Viral Populations Dominate Estuarine Viromes on the Spatiotemporal Scale. mSystems 2021, 6, e01020-20. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Lazar, C.S.; Teske, A.P.; Dick, G.J. Genomic Resolution of Linkages in Carbon, Nitrogen, and Sulfur Cycling among Widespread Estuary Sediment Bacteria. Microbiome 2015, 3, 14. [Google Scholar] [CrossRef]
- Tschitschko, B.; Erdmann, S.; DeMaere, M.Z.; Roux, S.; Panwar, P.; Allen, M.A.; Williams, T.J.; Brazendale, S.; Hancock, A.M.; Eloe-Fadrosh, E.A.; et al. Genomic Variation and Biogeography of Antarctic Haloarchaea. Microbiome 2018, 6, 113. [Google Scholar] [CrossRef]
- Martins, P.D.; Danczak, R.E.; Roux, S.; Frank, J.; Borton, M.A.; Wolfe, R.A.; Burris, M.N.; Wilkins, M.J. Viral and Metabolic Controls on High Rates of Microbial Sulfur and Carbon Cycling in Wetland Ecosystems. Microbiome 2018, 6, 138. [Google Scholar] [CrossRef]
- Panwar, P.; Allen, M.A.; Williams, T.J.; Hancock, A.M.; Brazendale, S.; Bevington, J.; Roux, S.; Páez-Espino, D.; Nayfach, S.; Berg, M.; et al. Influence of the Polar Light Cycle on Seasonal Dynamics of an Antarctic Lake Microbial Community. Microbiome 2020, 8, 116. [Google Scholar] [CrossRef]
- ter Horst, A.M.; Santos-Medellín, C.; Sorensen, J.W.; Zinke, L.A.; Wilson, R.M.; Johnston, E.R.; Trubl, G.G.; Pett-Ridge, J.; Blazewicz, S.J.; Hanson, P.J.; et al. Minnesota Peat Viromes Reveal Terrestrial and Aquatic Niche Partitioning for Local and Global Viral Populations. Microbiome 2021, 9, 233. [Google Scholar] [CrossRef]
- Amundson, K.K.; Borton, M.A.; Daly, R.A.; Hoyt, D.W.; Wong, A.; Eder, E.; Moore, J.; Wunch, K.; Wrighton, K.C.; Wilkins, M.J. Microbial Colonization and Persistence in Deep Fractured Shales Is Guided by Metabolic Exchanges and Viral Predation. Microbiome 2022, 10, 5. [Google Scholar] [CrossRef]
- Duncan, A.; Barry, K.; Daum, C.; Eloe-Fadrosh, E.; Roux, S.; Schmidt, K.; Tringe, S.G.; Valentin, K.U.; Varghese, N.; Salamov, A.; et al. Metagenome-Assembled Genomes of Phytoplankton Microbiomes from the Arctic and Atlantic Oceans. Microbiome 2022, 10, 67. [Google Scholar] [CrossRef]
- Abraham, B.S.; Caglayan, D.; Carrillo, N.V.; Chapman, M.C.; Hagan, C.T.; Hansen, S.T.; Jeanty, R.O.; Klimczak, A.A.; Klingler, M.J.; Kutcher, T.P.; et al. Shotgun Metagenomic Analysis of Microbial Communities from the Loxahatchee Nature Preserve in the Florida Everglades. Environ. Microbiome 2020, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Gaia, M.; Pagnier, I.; Campocasso, A.; Fournous, G.; Raoult, D.; La Scola, B. Broad Spectrum of Mimiviridae Virophage Allows Its Isolation Using a Mimivirus Reporter. PLoS ONE 2013, 8, e61912. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.S.; Williams, K.H.; Agarwal, D.; Banfield, J.; Beller, H.; Bouskill, N.; Brodie, E.; Carroll, R.; Dafflon, B.; Dwivedi, D.; et al. The East River, Colorado, Watershed: A Mountainous Community Testbed for Improving Predictive Understanding of Multiscale Hydrological–Biogeochemical Dynamics. Vadose Zone J. 2018, 17, 1–25. [Google Scholar] [CrossRef]
- Kelly, C. Quantifying Ecosystem Changes Following American Chestnut Restoration: From Microbes to Ecosystem Function; DOE Joint Genome Institute: Berkeley, CA, USA, 2017. [Google Scholar] [CrossRef]
- Davenport, E.J.; Neudeck, M.J.; Matson, P.G.; Bullerjahn, G.S.; Davis, T.W.; Wilhelm, S.W.; Denney, M.K.; Krausfeldt, L.E.; Stough, J.M.A.; Meyer, K.A.; et al. Metatranscriptomic Analyses of Diel Metabolic Functions During a Microcystis Bloom in Western Lake Erie (United States). Front. Microbiol. 2019, 10, 2081. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.O.; Templer, P.H.; Sorensen, P.O.; Sanders-DeMott, R.; Groffman, P.M.; Bhatnagar, J.M. Soil Microbes Trade-Off Biogeochemical Cycling for Stress Tolerance Traits in Response to Year-Round Climate Change. Front. Microbiol. 2020, 11, 616. [Google Scholar] [CrossRef]
- Figueroa-Gonzalez, P.A.; Bornemann, T.L.V.; Adam, P.S.; Plewka, J.; Révész, F.; von Hagen, C.A.; Táncsics, A.; Probst, A.J. Saccharibacteria as Organic Carbon Sinks in Hydrocarbon-Fueled Communities. Front. Microbiol. 2020, 11, 587782. [Google Scholar] [CrossRef]
- Williams, T.J.; Allen, M.A.; Ivanova, N.; Huntemann, M.; Haque, S.; Hancock, A.M.; Brazendale, S.; Cavicchioli, R. Genome Analysis of a Verrucomicrobial Endosymbiont With a Tiny Genome Discovered in an Antarctic Lake. Front. Microbiol. 2021, 12, 674758. [Google Scholar] [CrossRef]
- Linz, A.M.; He, S.; Stevens, S.L.R.; Anantharaman, K.; Rohwer, R.R.; Malmstrom, R.R.; Bertilsson, S.; McMahon, K.D. Freshwater Carbon and Nutrient Cycles Revealed through Reconstructed Population Genomes. PeerJ 2018, 6, e6075. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roux, S.; Fischer, M.G.; Hackl, T.; Katz, L.A.; Schulz, F.; Yutin, N. Updated Virophage Taxonomy and Distinction from Polinton-like Viruses. Biomolecules 2023, 13, 204. https://doi.org/10.3390/biom13020204
Roux S, Fischer MG, Hackl T, Katz LA, Schulz F, Yutin N. Updated Virophage Taxonomy and Distinction from Polinton-like Viruses. Biomolecules. 2023; 13(2):204. https://doi.org/10.3390/biom13020204
Chicago/Turabian StyleRoux, Simon, Matthias G. Fischer, Thomas Hackl, Laura A. Katz, Frederik Schulz, and Natalya Yutin. 2023. "Updated Virophage Taxonomy and Distinction from Polinton-like Viruses" Biomolecules 13, no. 2: 204. https://doi.org/10.3390/biom13020204
APA StyleRoux, S., Fischer, M. G., Hackl, T., Katz, L. A., Schulz, F., & Yutin, N. (2023). Updated Virophage Taxonomy and Distinction from Polinton-like Viruses. Biomolecules, 13(2), 204. https://doi.org/10.3390/biom13020204