Abstract
Mitochondria play a key role in cellular functions, including energy production and oxidative stress regulation. For this reason, maintaining mitochondrial homeostasis and proteostasis (homeostasis of the proteome) is essential for cellular health. Therefore, there are different mitochondrial quality control mechanisms, such as mitochondrial biogenesis, mitochondrial dynamics, mitochondrial-derived vesicles (MDVs), mitophagy, or mitochondrial unfolded protein response (mtUPR). The last item is a stress response that occurs when stress is present within mitochondria and, especially, when the accumulation of unfolded and misfolded proteins in the mitochondrial matrix surpasses the folding capacity of the mitochondrion. In response to this, molecular chaperones and proteases as well as the mitochondrial antioxidant system are activated to restore mitochondrial proteostasis and cellular function. In disease contexts, mtUPR modulation holds therapeutic potential by mitigating mitochondrial dysfunction. In particular, in the case of neurodegenerative diseases, such as primary mitochondrial diseases, Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS), or Friedreich’s Ataxia (FA), there is a wealth of evidence demonstrating that the modulation of mtUPR helps to reduce neurodegeneration and its associated symptoms in various cellular and animal models. These findings underscore mtUPR’s role as a promising therapeutic target in combating these devastating disorders.
1. Introduction
Mitochondria are double-membrane organelles that originated from symbiosis between an α-proteobacteria-like progenitor and a pre-eukaryotic cell [1]. The most common function of mitochondria is their role in energy metabolism. Reducing equivalents are formed during the tricarboxylic acid (TCA) cycle and are subsequently used by the mitochondrial electron transport chain (mtETC) to generate a proton gradient, which is finally used for ATP formation in the process known as oxidative phosphorylation (OXPHOS) [2,3]. Beyond energy production, mitochondria are also involved in other functions such as fatty acid synthesis [4], iron sulfur cluster [5] and heme biogenesis [6], calcium homeostasis [7], apoptosis regulation [8], amino acid and lipid biosynthesis [9,10], etc. All these functions are carried out by mitochondrial proteins. The mitochondrial proteome is composed of about 1500 proteins, although only 13 of them are encoded by mitochondrial DNA (mtDNA). The rest are encoded by nuclear DNA (nDNA), synthesized on cytosolic ribosomes as precursors, and must be transported to the mitochondria [11]. The fact that mitochondrial function depends on both genomes necessitates precise coordination and communication between them.
Mutations in mitochondrial proteins encoded by mtDNA or nDNA, the overproduction of reactive oxygen species (ROS), the deregulation of import processes, or an uncoupled mtETC lead to a disruption in mitochondrial function, which is responsible for primary mitochondrial diseases [12] and, secondarily, for other illnesses and physiological processes related to mitochondrial dysfunction, such as neurodegenerative diseases [13], ageing [14], metabolic diseases [15], cardiac disorders [16], and cancer [17].
To ensure the correct functioning of mitochondria and cells in general, cells employ different protein quality control mechanisms to prevent mitochondrial perturbations. In this review, we will summarize these mechanisms, beginning with mitochondrial biogenesis and the cytosolic quality control of mitochondrial protein import. We will then proceed to mitochondrial dynamics, encompassing fusion and fission processes, followed by mitophagy, with special attention paid to the role of the mitochondrial unfolded protein response (mtUPR) and its therapeutic potential.
2. Mitochondrial Quality Control
Since mitochondria are essential for eukaryotic organisms, these organelles possess several quality control mechanisms for counteracting dysregulated mitochondrial activity. Among them, the cytosolic protein quality control system prevents abnormal polypeptides from entering the mitochondria during mitochondrial biogenesis [18]. Mitophagy removes parts or entire mitochondria in response to mitochondrial damage [19]. Mitochondrial-derived vesicles (MDVs) are generated when mitochondrial membranes become oxidized due to mitochondrial stress, causing them to be loaded into vesicles, which are then transferred to lysosomes or peroxisomes for degradation [20]. The mtUPR induces the activation of mitochondrial chaperones and proteases, as well as antioxidant enzymes, to counteract mitochondrial stress [21]. Eventually, mitochondrial dynamics, including fusion and fission processes, regulate mitochondrial homeostasis [22].
2.1. Mitochondrial Biogenesis
Mitochondrial biogenesis requires special coordination between nuclear and mitochondrial genomes. It is defined as a self-renewal pathway by which new mitochondria are formed from pre-existing ones [23]. Mitochondrial biogenesis involves two different subprocesses: (1) mtDNA transcription and translation and (2) the transcription, translation, import, and assembly of nDNA-encoded mitochondrial proteins [24].
mtDNA transcription is activated by the family of peroxisome proliferator-activated receptors, among which peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) is considered the master regulator [25]. PGC-1α can be activated via phosphorylation through AMP-activated kinase (AMPK) or deacetylation via SIRT1 [26]. PGC-1α can also be activated by cAMP response element-binding protein (CREB), which, in turn, is activated through phosphorylation via protein kinase A (PKA) [27], calcium-calcium/calmodulin-dependent protein kinase (CaMK) [28], or protein kinase B, also known as Akt [29]. The activation of PGC-1α is followed by the stimulation of nuclear respiratory factors 1 and 2 (NRF1 and NRF2), which promote the transcription of multiple mitochondrial genes, including mitochondrial transcription factor A (TFAM), the first well-characterized transcription factor from mammalian mitochondria. This leads to mtDNA replication and transcription [30]. Finally, the translation of the mtDNA-encoded genes into proteins takes place.
On the other hand, nDNA-encoded mitochondrial proteins are synthesized on cytosolic ribosomes as precursors in response to the activation of nuclear transcription factors such as NRFs. These preproteins traverse the outer mitochondrial membrane (OMM) through the translocase of the outer membrane (TOM) complex system, which recognizes internal and cleavage mitochondrial targeting signal (MTS) present in the N-terminal [31]. Then, the precursors cross the inner mitochondrial membrane (IMM) because of the positive charge present in their MTS, which initiates the electrophoretic translocation process. This process involves their entry into the mitochondria through the translocase of the inner membrane (TIM) complex, relying on the electric potential across the IMM [32]. Subsequently, mitochondrial precursors are transported to the mitochondrial matrix with the assistance of mitochondrial chaperones, which facilitate import and folding [33]. Finally, proteins undergo proteolytic removal of their MTS sequence, an important step in their maturation, and attain their final conformation through the action of mitochondrial chaperones [34]. There are other specific import pathways depending on the location of the mitochondrial proteins. For more information, various articles are available for reference [35,36,37,38].
Therefore, it is necessary to synchronize the transcription and translation of two different and physically separated genomes for mitochondrial biogenesis, something that occurs due to the action of PGC-1α and NRF transcription factors (see Figure 1).
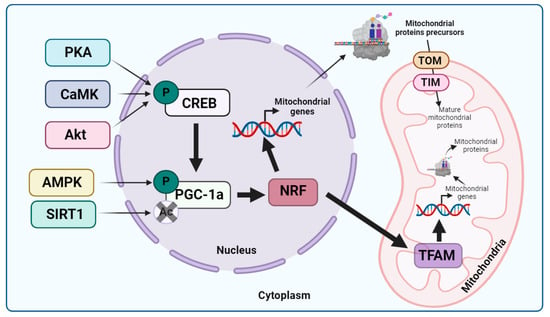
Figure 1.
Mitochondrial biogenesis. Considering that PGC-1α is the main regulator of mitochondrial biogenesis, it can be activated through phosphorylation via CREB, which, in turn, is phosphorylated by PKA, CaMK, or Akt, or via AMPK. PGC-1α can also be activated via deacetylation through SIRT1. Once this transcription factor is activated, it stimulates NRF transcription factors that activate the transcription of nDNA-encoded mitochondrial genes and TFAM. Subsequently, TFAM promotes mtDNA replication and transcription, while mitochondrial proteins encoded by nDNA are synthesized on cytosolic ribosomes as precursors to allow them to cross the OMM and IMM through the TOM and TIM systems, respectively. Once in the mitochondrial matrix, the mitochondrial precursors undergo proteolytic processing necessary for their maturation, reaching their final conformation due to the action of mitochondrial chaperones. Arrow: activation or induction of the subsequent process.
Mitochondrial biogenesis can be activated by various stimuli, such as exercise, diet, hormones, and stressors. In the last case, mitochondrial biogenesis might be upregulated to enhance mitochondrial function or to compensate for the increased rates of mitophagy [39].
Cytosolic Quality Control of Mitochondrial Protein Import
During mitochondrial biogenesis, mitochondrial proteins precursors synthesized on cytosolic ribosomes can exceed the capacity of the mitochondrial translocation machinery, leading to an accumulation of non-imported mitochondrial proteins precursors in the cytoplasm and, consequently, proteotoxic stress [40]. This accumulation, named mitochondrial precursor overaccumulation stress (mPOS), can be alleviated through several mechanisms [41]. The immediate response triggered is known as the unfolded protein response activated by the mistargeting of proteins (UPRam), which involves the assembly of the proteasome through the factors Irc24 and Poc4 [42]. This leads to an increase in proteasomal activity that degrades mitochondrial precursors in the cytosol, contributing to a response known as the mitoprotein-induced stress response. The latter is dependent on heat shock transcription factor 1 (HSF1), which, under stress conditions, is released from chaperone complexes [18].
On the other hand, the mitoprotein-induced stress response leads to a decrease in translation activity through the phosphorylation of the eukaryotic translation initiation factor 2α (eif2α) [43]. Thus, the increased degradation activity and decreased translation promote the clearance of mitochondrial protein precursors, reducing proteotoxic stress. Moreover, the cytosolic quality control of mitochondrial protein import is strongly associated with the mtUPR, since ATFS-1 in C. elegans and ATF5 in mammalian cells are both MTSs and nuclear localization signals (NLSs). When mitochondrial import fails, these factors are transported to the nucleus, where they promote the transcription of molecular chaperones and proteases, leading to mtUPR activation [42].
2.2. Mitochondrial Dynamics
Mitochondrial dynamics are defined as a balance between fission and fusion processes [22] that change mitochondrial shape and size under pathological and physiological conditions. These processes are responsible for mtDNA stability, mitochondrial morphology and homeostasis maintenance, mitochondrial respiratory capacity, and response to cellular stress [44]. Therefore, mitochondrial fusion and fission are part of the quality control system, and the disruption of these processes is the cause of several human diseases.
Mitochondrial fusion is defined as the merging of two mitochondria into one [45]. For this process to transpire, three enzymes of the dynamin-like family of GTPases are required: Mitofusins 1 and 2 (Mfn1, Mfn2), located in the OMM, and Optic Atrophy 1 (Opa1), anchored in the IMM [46]. Mitochondrial fusion begins with the interaction of the mitofusins present in the two fusing mitochondria, which form homo- or hetero-oligomers [47]. After that, Opa1 mediates the fusion of the IMMs of both mitochondria. However, unlike in the case of mitofusins, Opa1 must only be present in one of the two fusing mitochondria [48]. Finally, the contents of both mitochondria mix, giving rise to a new larger mitochondrion with the mitochondrial matrix components of the two parent mitochondria diffused throughout it, including mtDNA [49]. Therefore, mitochondrial fusion can aid in the rescue of damaged mitochondria by diluting aberrant proteins or allowing wild-type mtDNA to compensate for flaws [50].
Mitochondrial fusion can be triggered in response to several factors, such as exposure to ultraviolet C (UV-C) irradiation, hydrogen peroxide, cycloheximide, actinomycin D, or azinomycin or starvation, among others. It leads to hyperfusion, a process centered on maintaining ATP levels within mitochondria and responding to stress in a pro-survival process known as stress-induced mitochondrial hyperfusion (SIMH) [51]. In this process, stromatin-like protein 2 (SLP-2) is required, as when SLP-2-depleted cells are exposed to mitochondrial stress, mitochondria do not become hyperfused but fragmented. This is due to the requirement of SLP-2 to prevent the proteolytic cleavage of Opa1, indicating that long isoforms of Opa1 are necessary for the SIMH process [52]. SIMH can also be stimulated by the unfolded protein response in the endoplasmic reticulum (UPRER) in a manner dependent on the phosphorylation of eif2α, one of the key factors in the mtUPR, as we will discuss throughout this review [53].
Additionally, when exposed to a virus, mitochondrial fusion may be activated, and it supports the innate immune response [54].
On the other hand, mitochondrial fission, the opposing process of mitochondrial fusion, is defined as the separation of one mitochondrion into two or more daughter mitochondria [45]. Several proteins are necessary in this process, with dynamin-related protein 1 (Drp1) playing a central role. Drp1 is usually located in the cytosol, from which it translocates to the OMM. Then, Drp1 is anchored to the mitochondrial surface with the help of mitochondrial fission 1 protein (FIS1), mitochondrial dynamics proteins 49 (MID49) and 51 (MID51), or mitochondrial fission factor (MFF) [55]. Once Drp1 is on the OMM, it forms large homomultimeric complexes to facilitate constriction and scission, processes that are dependent on GTP hydrolysis [56]. To ensure the correct segregation of mtDNA, it must be replicated synchronously with fission.
Furthermore, two types of mitochondrial fission have recently been described, namely, ‘peripheral fission’ (less than 25% from an extreme) and ‘midzone fission’ (inside the middle 50%), depending on the Drp1 adaptors involved. In the case of peripheral divisions, FIS1 is the protein involved, not serving as a Drp1 adaptor to the OMM but acting as a recruiter of lysosomes for subsequent mitochondrial degradation through mitophagy. Nevertheless, in the case of midzone fissions, the Drp1 adaptor implicated is MFF. However, the protein involved is not the sole distinction between both types of mitochondrial fission. It has been reported that daughter mitochondria from peripheral divisions are targeted for degradation through mitophagy, while those originating from midzone divisions contribute to mitochondrial biogenesis during cell proliferation [57].
Mitochondrial fission can be activated after shorts periods of exercise due to increased ROS levels. However, after the recovery period, mitochondria become increasingly networked, implying that the activation of mitochondrial fission acts as a process of acclimation to an exercise state [58].
In general terms, mitochondrial fusion is activated in response to low-level or chronic exposure to stressors, while mitochondrial fission is stimulated when there is acute high-level exposure [59]. Nevertheless, if mitochondrial damage cannot be repaired via mitochondrial dynamics, the mitochondria are eliminated via mitophagy, the selective autophagy of mitochondria.
2.3. Mitophagy
Mitophagy is defined as the specific autophagic elimination of mitochondria [60]. This process was first identified in mammalian cells through electron microscopy studies, which revealed enhanced mitochondrial sequestration in lysosomes after glucagon-stimulated hepatocyte catabolism [61]. When mitochondria become damaged, the IMM experiences prolonged depolarization, leading to the stabilization of the PTEN-induced kinase 1 (PINK1) protein at the OMM. PINK1 is a protein with an MTS, a transmembrane domain, and a kinase domain. When PINK1 is transported to healthy mitochondria, the MTS is processed by mitochondrial protease-like (MPP) protein in the mitochondrial matrix, and the transmembrane segment is processed by the presenilin-associated rhomboid-like (PARL) protein in the IMM. Consequently, PINK1 is degraded [62]. Nevertheless, when PINK1 is transported to damaged mitochondria, its processing does not occur, leading to the accumulation of its long isoform on the OMM, forming a supercomplex of 850 kDa that executes autophosphorylation [63]. Once PINK1 is activated, it phosphorylates the E3 ubiquitin ligase Parkin. However, this phosphorylation alone is not sufficient to recruit Parkin to the mitochondria and activate it. On the contrary, it has been described that PINK1 phosphorylates ubiquitin, which binds to Parkin, facilitating its recruitment to the OMM and activating its ubiquitin ligase activity [64]. Once Parkin is activated, it ubiquitinates multiple proteins on the mitochondrial surface, which are subsequently detected by ubiquitin-binding proteins such as optineurin (OPTN), p62, NDP52, Tax1-binding protein 1 (TAX1BP1), and NRB1. These proteins interact with the microtubule-associated protein 1A/1B light chain 3 (LC3) protein to form autophagosomes [65], which transport the enclosed materials to the lysosome for extensive degradation via lysosomal hydrolases, a fusion stage mediated by Pleckstrin homology domain-containing family M member 1 (PLEKHM1) and HOPS LC3-binding proteins, and the lysosome membrane-associated protein Rab7 [66]. In addition to this classical mitophagy pathway (PINK1/Parkin-related mitophagy), several other mitophagy receptors have been identified in PINK1/Parkin-independent mitophagy, including Fun14 domain-containing protein 1 (FUNDC1), autophagy and beclin 1 regulator 1 (AMBRA1), and Nix/BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3). AMBRA1 has been proposed to aid in the removal of damaged mitochondria by guiding them to autophagosomes through its interaction with LC3 [67]. Nix/BNIP3, on the other hand, is critical for the elimination of mitochondria during erythroid maturation [68]. Finally, FUNDC1, physically interacts with LC3, increasing mitophagic flux [69] (see Figure 2).
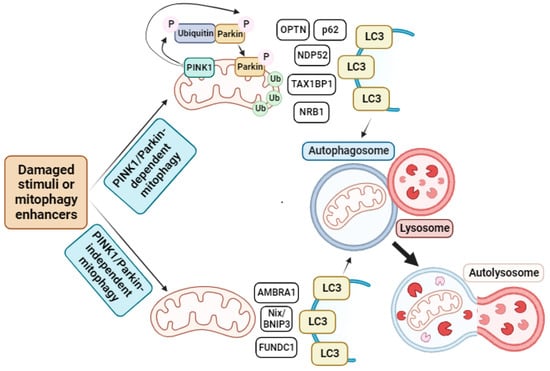
Figure 2.
Mitophagy pathway. In response to damaged stimuli or through mitophagy enhancer supplementation, cells activate mitophagy. This process can be divided into two different subprocesses depending on the participation of PINK1 and Parkin proteins. In the PINK1/Parkin-dependent cascade, PINK1 at the OMM is activated and phosphorylates Parkin. However, this phosphorylation is not sufficient to recruit Parkin to the OMM. On the contrary, PINK1 phosphorylates ubiquitin, which binds to Parkin, facilitating its recruitment to the mitochondria. Then, Parkin ubiquitinates several proteins on the mitochondrial surface, which are recognized by different receptors such as OPTN, p62, NDP52, TAX1BP1, or NRB1 that, in turn, interact with LC3 present in the autophagosome membrane and stimulate the formation of autophagosome. These autophagosome fuse with lysosomes, leading to the formation of autolysosomes, where mitochondrial degradation occurs. On the other hand, in PINK1/Parkin-independent mitophagy, damaged mitochondria are recognized by other receptors such as AMBRA1, Nix/BNIP3, or FUNDC1 that, as in the previous case, interact with LC3 to form the autophagosome, which again fuses with a lysosome to form the autolysosome, where complete degradation of the mitochondrion take place. Arrow: activation or induction of the subsequent process.
Several mitophagy stimulators have been described, such as hypoxia [70], starvation, caloric restriction, and exercise [71], as well as compounds like urolithin A [72], actinonin [73], NAD+-boosters [74], trehalose [75], resveratrol [76], bexarotene [77], β-asarone [78], melatonin [79], spermidine [73], UMI-77 [80], kaempferol and rhapontigenin [81], and metformin [82].
In general, when there is a dissipation or uncoupling of mitochondrial membrane potential, mitochondrial protein import fails, a phenomenon used by cells as a sensor of mitochondrial dysfunction in order to activate their compensatory mechanisms [83]. If the stress is severe, cells first activate mitochondrial fission, and if this is not able to negate the stress, they activate mitophagy [19]. The activation of mitophagy promotes the maintenance of mitochondrial function as it serves as a sensor allowing the cells to induce mitochondrial biogenesis with the aim of maintaining mitochondrial content [84]. However, in situations of moderate stress, cells activate mitochondrial fusion and another mitochondrial quality control process, the mtUPR, leading to the promotion of genes that stimulate mitochondrial function recovery as well as innate immunity and metabolic adaptation, among others [21] (see Figure 3).
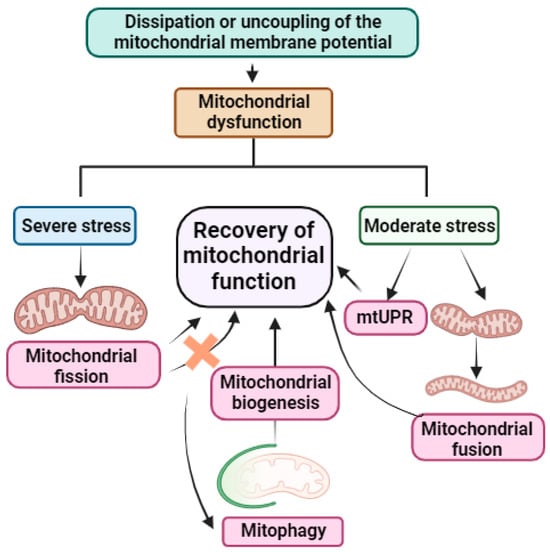
Figure 3.
Summary of mitochondrial stress response mechanisms. When mitochondrial dysfunction occurs because of dissipation or uncoupling of mitochondrial membrane potential, mitochondrial protein import fails. This fact is used by cells as a sensor to activate their mitochondrial quality control mechanisms. If the stress is severe, the cells first activate mitochondrial fission. If this does not resolve the stress, cells activate mitophagy, promoting the elimination of damaged mitochondria to preserve those free of damage and inducing mitochondrial biogenesis for the recovery of mitochondrial function. However, if the stress is moderate, cells activate mitochondrial fusion or mtUPR, leading to the recovery of mitochondrial function. Quality control mechanisms of mitochondria are highlighted in pink. Arrow: activation or induction of the subsequent process.
2.4. Mitochondrial Unfolded Protein Response (mtUPR)
The mtUPR is a vital pathway that maintains mitochondrial homeostasis and proteostasis via stimulating the transcription of nuclear-encoded genes that protect and support mitochondria. This pathway was initially described in rat hepatoma cells by Martinus et al. [85], who discovered that inducing mitochondrial stress through the depletion of mtDNA led to the transcription of mitochondrial molecular chaperones but not those found in other cellular compartments.
Subsequently, Zhao et al. [86] proposed that the activation of this stress response in mammalian cells, induced by factors such as an ornithine transcarbamylase mutant, not only activates chaperones but also mitochondrial proteases such as ClpP or Lon protease-like 1 (LonP1). Additionally, these researchers identified C/EBP Homologous Protein (CHOP) as a critical transcription factor for mtUPR.
However, the mtUPR has been predominantly studied in C. elegans. In this model organism, the process is initiated when there is an accumulation of misfolded or unfolded proteins within the mitochondrial matrix that exceeds the chaperones’ capacity to handle them as well as through other mitochondrial stress-producing factors such as toxins produced by pathogens or mtETC malfunctions, among others [87]. When aberrant proteins accumulate, a complex known as caseinolytic protease P (CLPP-1) hydrolyzes them into short peptides consisting of 8–20 amino acids [88]. Subsequently, the inner-membrane-spanning ATP-binding cassette transporter HAF-1 transports these small peptides from the mitochondrial matrix to the intermembrane space (IMS), reducing the import of the activating transcription factor associated with stress-1 (ATFS-1) into the mitochondria [89]. The activation of response genes and the transfer of mtUPR signals from the mitochondria to the nucleus are both orchestrated by ATFS-1, which contains an NLS and an MTS. Under physiological conditions, ATFS-1 enters the mitochondria using its MTS, where it is degraded by the LonP1 protease. However, during stressful situations, the efficiency of mitochondrial import is compromised, resulting in reduced degradation of ATFS-1 within the mitochondria and its accumulation in the cytosol. This accumulation leads to its transport into the nucleus, where it activates the transcription of mitochondrial molecular chaperones and proteases [90] (see Figure 4).
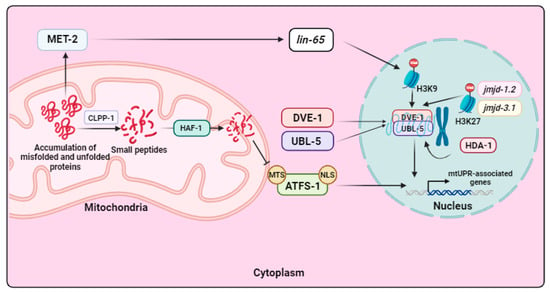
Figure 4.
mtUPR signaling in C. elegans. When an accumulation of misfolded and unfolded proteins occurs in mitochondria, the CLPP-1 protease is activated and hydrolyzes these proteins into smaller peptides, which are then transported to the IMS by HAF-1. This decreases the transport of the transcription factor ATFS-1 into the mitochondria, causing it to accumulate in the cytoplasm and translocate to the nucleus. In this organelle, ATFS-1 activates the transcription of mtUPR-related genes. Moreover, mtUPR involves two transcription factors, DVE-1 and UBL-5, that, in response to mitochondrial stress, are transported from the cytoplasm to the nucleus, where they form a complex that promotes the transcription of mtUPR-associated genes. In addition, mtUPR is dependent on several mechanisms of chromatin remodeling. Firstly, this stress response pathway is subject to methylation regulation by the histone methylase MET-2, which is activated in response to mitochondrial damage. This activation allows its cofactor LIN-65 to enter the nucleus, where it carries out H3K9 methylation. While most of the chromatin condenses as a result, some regions remain open, and it is in these regions that the complex formed by DVE-1 and UBL-5 binds to promote transcription of mtUPR-related genes. Furthermore, the histone deacetylase HDA-1 also contributes to chromatin decondensation, as do the histone demethylases JMJD-1.2 and JMJD-3.1, allowing for the binding of the DVE-1/UBL-5 complex and triggering the transcription of molecular chaperones and proteases to restore mitochondrial function. Arrow: activation or induction of the subsequent process.
Additionally, under mitochondrial stress conditions, the homeodomain-containing transcription factor “defective proventriculus” (DVE-1) and ubiquitin-like protein 5 (UBL-5) are translocated into the nucleus, where they form a complex that activates the transcription of mitochondrial chaperones [91]. Finally, chaperones and proteases are transported to the damaged mitochondria to fold or degrade, respectively, unfolded, misfolded, and aberrant proteins [21] (see Figure 4).
Furthermore, it has been described that chromatin remodeling is necessary for the activation of the mtUPR in C. elegans. Two histone lysine demethylases are involved in this stress response, namely, JMJD-1.2 and JMJD-3.1. These enzymes lead to the demethylation of H3K27, consequently promoting the transcription of mtUPR-related genes [92]. In addition, the mtUPR is also subject to acetylation regulation. The histone deacetylase HDA-1 collaborates with DVE-1 to activate the transcription of genes linked to mtUPR [93]. Moreover, chromatin remodeling depends on the activity of the histone methyltransferase MET-2 and its nuclear cofactor LIN-65. Generally, methylation globally silences chromatin but leaves some regions accessible, allowing transcription factors to attach themselves. This occurs under mitochondrial stress when MET-2 is activated, promoting LIN-65’s entrance into the nucleus and, as a result, H3K9 methylation. The portions of the chromatin that have not been repressed remain open, and it is in these portions where the UBL-5/DVE-1 complex interacts to enhance the transcription of genes related to the mtUPR [94] (see Figure 4).
It has been described that in C. elegans, the mtUPR not only upregulates mitochondrial chaperones and proteases but also promotes the transcription of genes involved in OXPHOS and glycolysis, establishing coordinated regulation of mitochondrial proteostasis and metabolic state [90]. In fact, while mitochondrial damage is being inflicted, ATFS-1 reduces the transcription of OXPHOS and TCA cycle genes while simultaneously promoting the assembly of OXPHOS complexes, leading to an improvement in mitochondrial proteome health [90].
Moreover, Durieux et al. [95] and Berendzen et al. [96] demonstrated that the mtUPR can be activated cell-non-autonomously via mitokines. The presence of mitochondrial stressors within neurons in worms leads to the communication of stress from neuronal mitochondria to peripheral tissues such as the intestine. This process requires serotonin as a mitokine and a functional mtUPR. Consequently, this cell-non-autonomous mode of mtUPR can provide an organism with superior protection against local mitochondrial stressors.
In mammals, mtUPR regulation is similar to that in nematodes but is far more complex and still not fully understood. Several stressors have been described that can activate the mtUPR in mammalian cells beyond the accumulation of misfolded and unfolded proteins, such as disruptions in OXPHOS, the overproduction of ROS, several compounds, and mitochondrial translation inhibition, among others [97]. In these organisms, a transcription factor ortholog of ATFS-1, known as Activating Transcription Factor 5 (ATF5), has been identified as the main regulator of the mtUPR [98].
Additionally, other transcription factors, including Activating Transcription Factor 4 (ATF4) and CHOP, also play essential roles in regulating the mtUPR in mammals, as they promote the transcriptional expression of ATF5 [99,100,101]. Nevertheless, the communication among these factors is not yet understood. In these organisms, the mtUPR is initiated with the phosphorylation of eif2α, which leads to increased transcription of CHOP, ATF4, and ATF5. These transcription factors are then translocated to the nucleus, where they activate the transcription of mitochondrial chaperones such as Hsp70, Hsp60, and Hsp10, as well as mitochondrial proteases such as ClpP and LonP1 [21,102]. The dependence of the mtUPR on the transcription factors CHOP, ATF4, and ATF5 integrates it with the Integrated Stress Response (ISR), as the primary transcriptional regulator of the latter is ATF4. Moreover, these transcription factors are also shared with UPRER. Both pathways, UPRER and mtUPR, are responses to stress caused by the accumulation of unfolded or misfolded proteins [103]. However, these processes occur in different cellular compartments, with UPRER taking place in the endoplasmic reticulum (ER) and mtUPR specifically transpiring in the mitochondria [104]. In fact, one is independent of the other, as the treatment of cells with mitochondrial-specific stressors induced mitochondrial chaperones and proteases without affecting chaperones in the ER. In addition, it has been reported that during mitochondrial stress, ATF5 acts upstream of ATF4, the reverse of what occurs during ER stress [105].
Furthermore, it has recently been described that the proteolytic release of the factor DELE1 by the mitochondrial protease OMA1 carries the signaling of stress from the mitochondria to the cytosol [106], thereby adding a new factor to mtUPR signaling.
Other factors are also implicated in mtUPR regulation in mammalian cells. In situations of mitochondrial stress, the mitochondrial single-stranded DNA binding protein 1 (SSBP1), which is typically found in the mitochondrial matrix, is transported to the cytoplasm via the voltage/dependent anion channel 1 (VDAC1) protein. There, it forms a complex with HSF1, ultimately promoting the transcription of molecular chaperones [107].
It has also been observed that in conditions of mtUPR activation, the amount of MRPP3 protein is reduced, as it is degraded by LonP1. MRPP3 is part of the RNA-free mitochondrial RNase P complex, which also includes MRPP1 and MRPP2, serving as the catalytic subunit of the complex. The reduction in MRPP3 coincides with a decrease in pre-RNA processing following mtUPR induction, leading to a reduction in mtDNA translation, thereby protecting mitochondria from deleterious proteins [108].
Later, in 2018, Münch reviewed other axes of mtUPR [109]. The first is the transcriptional canonical mtUPR axis, which includes the signaling cascade described above, involving the transcription factors CHOP, ATF4, and ATF5, and increased expression of mitochondrial chaperones and proteases [110]. The second is the translational mtUPR axis mentioned earlier, where, in mtUPR induction situations, MRPP3 transcript and protein levels decrease, leading to a reduction in pre-RNA processing and, consequently, a lower level of mtDNA translation [108]. The translational mtUPR axis complements the transcriptional canonical mtUPR axis in combating mitochondrial protein misfolding.
Thirdly, a sirtuin mtUPR axis has been reported. Sirtuins are lysine deacetylases that control several cellular processes. In mammals, there are seven sirtuins: SIRT1, 6, and 7, primarily localized in the nucleus; SIRT2, which predominantly localizes in the cytosol; and SIRT3, 4, and 5, primarily present in mitochondria [111,112,113]. The roles of all sirtuins in the mtUPR have been described [114]. However, SIRT1 and SIRT3 have been reported to have predominant functions. These sirtuins exert an antioxidant effect by deacetylating FOXO3A, which then translocates to the nucleus and promotes the transcription of antioxidant enzymes such as catalase and mitochondrial superoxide dismutase 2 (SOD2) [115,116]. As NAD+ levels decline with mitochondrial dysfunction and reduced NAD+/NADH ratios are implicated in mitochondrial disorders, NAD+-boosting agents (niacine, nicotinamide riboside, etc.) may induce SIRT activation, which promotes the antioxidant response [117]. Moreover, it has been proposed that different mitochondrial stressors, including the accumulation of misfolded and unfolded protein within the mitochondrial matrix, trigger the activation of SIRT3, resulting in a sirtuin mtUPR axis complementary to the transcriptional and translational canonical mtUPR axes [118].
Finally, another mtUPR axis has been reported; it is activated when mitochondrial stress is localized in the IMS. This axis is mediated by estrogen receptor α (ERα), which is phosphorylated by Akt or increased ROS. Once activated, ERα stimulates NRF1, a critical factor in mitochondrial biogenesis, and OMI, an IMS-specific protease that degrades aberrant proteins, leading to activation of the IMS–mtUPR axis [119].
In contrast to the other mtUPR axes, which inhibit mitochondrial complex synthesis and promote mitochondrial assembly, the activation of the sirtuin or IMS mtUPR axes also induces mitochondrial biogenesis via SIRT1 and NRF1, respectively, thus increasing mitochondrial complex synthesis. In this way, two mitochondrial quality control mechanisms, mitochondrial biogenesis and mtUPR, act simultaneously to reinforce the restoration of mitochondrial function [97].
A representative figure of these four axes of the mtUPR can be found in the review by Cilleros-Holgado et al. [97]. In Table 1, we summarize the markers of the different mtUPR axes as well as the location where mitochondrial stress occurs in each of them.

Table 1.
Summary of the different mtUPR axes with the locations where mitochondrial stress takes place and markers of each of these pathways.
Finally, it is important to consider that, in contrast to the mtUPR, mitophagy removes damaged mitochondria, reducing the mitochondrial population and consequently leading to shortages in ATP and cell death. Therefore, the mtUPR appears to be a more effective mechanism for preserving mitochondrial function [124]. Nevertheless, it is worth noting that the prolonged activation of the mtUPR can have detrimental effects due to the propagation of damaged mitochondria. To address this issue, mitochondria have a damage threshold at which they cease mtUPR activation and initiate mitophagy. This necessitates the precise regulation of both compensatory mechanisms to ensure proper cellular function [125].
3. mtUPR as a Therapeutic Target
Numerous studies support the therapeutic potential of mtUPR modulation for the treatment of a wide range of diseases, including primary and secondary mitochondrial diseases [87,97,126,127,128,129,130].
3.1. Primary Mitochondrial Diseases
In the case of primary mitochondrial diseases, those caused by mutations in nDNA or mtDNA genes that code for mitochondrial proteins, the activation of mtUPR leads to increased ATP production, the assembly of OXPHOS-related subunits, improved mitochondrial antioxidant capacity, and overall recovery of mitochondrial function [131]. For example, Perry et al. demonstrated that mtUPR activation using tetracyclines, especially doxycycline, improved the survival and proliferation of MELAS cybrids and benefitted Rieske disease (mitochondrial complex III knockout) mouse fibroblasts and ND1 mutant cybrids under low-glucose conditions. They also tested the effect of other mtUPR activators such as pentamidine, known to inhibit mitochondrial translation, and retapamulin, which inhibits prokaryotic translation, and found that cell survival increased in all cellular models of mitochondrial diseases under nutrient stress conditions. Furthermore, the cited authors tested the effect of tetracyclines in a mouse model of Leigh syndrome lacking the mitochondrial complex I protein NDUFS4, resulting in improvements in body weight loss, longevity, and the amelioration of neurological decline [132]. Subsequently, Suárez-Rivero et al. confirmed the positive effect of tetracyclines and the consequent activation of mtUPR in fibroblasts derived from patients with mutations in the GFM1 gene [133]. Suárez-Rivero et al. also described that the activation of mtUPR via pterostilbene and mitochondrial cofactors improved mitochondrial function in different cellular models of primary mitochondrial diseases, enhancing the sirtuin mtUPR and transcriptional canonical mtUPR axes [134].
Nevertheless, it is important to consider that the use of antibiotics and, especially, tetracyclines, for the treatment of mitochondrial diseases is controversial [135].
On the other hand, it has been reported that mtUPR activation in urine-derived stem cells obtained from patients with the m.3243A>G mutation characteristic of MELAS syndrome induced mitochondrial dysfunction, while its inhibition through silencing ATF5 resulted in an improvement in cellular pathophysiology via increasing mitochondrial membrane potential and reducing ROS levels [136]. This difference in outcomes could be due to varying levels of heteroplasmy that occur when mitochondrial diseases are caused by mtDNA mutations. For this reason, further studies are needed to confirm the effect of mtUPR activation on primary mitochondrial diseases.
3.2. Cardiac and Metabolic Disorders
In cardiac disorders, the mtUPR plays a protective role [137]. Wang et al. observed that treatment with oligomycin or doxycycline in mice was beneficial with respect to cardiac ischemia–reperfusion injury, reducing infarct size. However, for this treatment to be effective, ATF5 and therefore mtUPR activation was required [138]. In addition, Smyrnias et al. proposed that mtUPR induction through six different methods, namely, treatment with paraquat, treatment with isoproterenol (a β-adrenoreceptor agonist), treatment with G-TPP (a chaperone inhibitor), mutation of the ornithine transcarbamylase protein, treatment with Olaparib (a PARP inhibitor), or treatment with nicotinamide, improved mitochondrial respiration in stressed cardiomyocytes in mice and attenuated contractile failure and hypertrophy [139]. Similarly, Xu et al. demonstrated that choline treatment in a rat model of heart disease activated the sirtuin mtUPR axis and inhibited both myocardial metabolic dysfunction and myocyte hypertrophy [140]. Likewise, Wang et al. showed that mtUPR induction via oligomycin had a protective role in septic cardiomyopathy induced by lipopolysaccharide, reducing cardiac dysfunction and mitochondrial damage [124]. Furthermore, Zhang et al. reported that the activation of mtUPR via treatment with tetrahydrocurcumin in mice subjected to transverse aortic constriction surgery reduced cardiac hypertrophy and surgery-induced oxidative stress [141]. Nevertheless, as Smyrnias et al. described, the activation of the mtUPR is not only beneficial when induced by external agents, as cells themselves activate this compensatory pathway to reduce damage. Myocardial cells derived from patients with aortic stenosis exhibited high levels of mtUPR markers such as ATF5, Hsp60, or LonP1. Those with even higher levels showed reduced rates of cardiomyocyte death, decreased levels of abnormal fibrosis, and fewer markers of cardiac damage in plasma [139].
On the other hand, metabolic diseases are related to alterations in OXPHOS gene expression and general mitochondrial dysfunction [142]. The most widely studied metabolic disease is diabetes, characterized by high levels of glucose in the blood and insulin resistance [143]. Elevated blood sugar levels over time inhibit the function of chaperones and proteases, leading to the accumulation of aberrant proteins and protein aggregates [144]. For this reason, the activation of the mtUPR could be beneficial in the treatment of diabetes. Wardelmann et al. observed that treatment with intranasal insulin activated mtUPR in insulin-deficient mice, improving mitochondrial function, inhibiting autophagy, and preventing weight gain under high-fat diet conditions [145]. In addition, Lee et al. demonstrated that LonP1 protease deficiency promoted liver gluconeogenesis and insulin resistance, while its overexpression mitigated liver insulin resistance induced by treatment with cholesterol and palmitate in liver SK-HEP-1 cells derived from humans [146]. Moreover, Wu et al. suggested that high-fat or high-glucose diets increased ClpP protease expression in pancreatic Min6 β-cells, leading to a decrease in cell apoptosis and ROS production, as well as the stabilization of insulin signaling [147]. Also, Kleinridders et al. reported that patients and mice with type 2 diabetes had lower Hsp60 expression levels in the brain, and this phenomenon was associated with insulin resistance [148].
mtUPR modulation also provides protective effects in the metabolic liver disease setting. These pathologies are associated with alterations in OXPHOS and elevated ROS production. Gariani et al. described that treatment with nicotinamide adenine dinucleotide in mouse models of fatty liver disease reverted non-alcoholic fatty liver disease by activating SIRT1 and SIRT3, ultimately increasing hepatic β-oxidation and OXPHOS activity [149]. In addition, the induction of the sirtuin mtUPR axes via nicotinamide precursor supplementation attenuated body weight gain and fat mass among mice fed a high-fat diet [150].
3.3. Cancer
Finally, mtUPR modulation has also been proposed as a therapeutic target for the treatment of different types of cancer. However, in this case, the therapies are not based on the activation of this stress response but on its inhibition, as it has been shown that mtUPR markers such as ATF5, Hsp60, Hsp70, ClpP, or LonP1 are upregulated in a wide variety of cancers. There are many studies on this subject, which can be consulted in the following references: [151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184].
In conclusion, a substantial body of literature supports the modulation of mtUPR as a therapeutic target in the treatment of various diseases, all of which have mitochondrial dysfunction as a primary underlying cause. We will emphasize the utilization of this approach in the context of ageing and age-related diseases, particularly neurodegenerative disorders. These conditions have a significant societal impact, with a growing number of affected individuals and a notable lack of curative treatments.
3.4. Ageing and Neurodegenerative Diseases
Neurodegenerative diseases encompass a diverse group of disorders characterized by the progressive and selective loss of anatomically or physiologically connected neuronal systems [185].
A major risk factor for neurodegenerative disorders is the process of ageing. It has been hypothesized that mitochondria play a central role in this context by producing ROS and accumulating mutations in mtDNA, both of which are believed to accelerate the physiological ageing process [14]. It is well established that during ageing, mtDNA mutations accumulate, contributing to the decline in mitochondrial function [186]. mtDNA is more vulnerable to damage due to the absence of histones and other chromatin proteins as well as its proximity to sites of ROS production [187]. Furthermore, ROS imbalance has a significant impact on cognitive loss associated with ageing [188]. The brain is particularly susceptible to oxidative damage due to its high oxygen consumption rate, high concentration of extremely peroxidizable unsaturated fatty acids, and relative lack of antioxidant enzymes compared to other organs [189]. Additionally, ageing is accompanied by a reduction in mitophagy, which leads to the accumulation of damaged mitochondria, increased oxidative stress, and the induction of apoptosis [190].
Given the pivotal role of mitochondrial dysfunction in ageing, it is not surprising that mitochondrial quality control mechanisms play a crucial role in this process. Increasing mitochondrial biogenesis by activating SIRT1 has been shown to delay ageing and extend lifespan in animal models [152,191]. In line with this, numerous studies suggest that the induction of the mtUPR can extend lifespan. For instance, the lifespan of a complex IV mtETC mutant C. elegans specimen exceeded that of control worms, and this extension was attributed to the activation of mtUPR, as evidenced by elevated markers of this stress response, such as Hsp6 and Hsp10 [95]. Moreover, mtUPR activation through mitochondrial translation inhibition, resveratrol, or PARP inhibitors promoted longevity among worms [192]. Moreover, supplementation with NAD+ precursors or the inhibition of NAD+-consuming enzymes also prolonged lifespan in C. elegans [193]. Importantly, C. elegans is not the sole model organism in which the activation of the mtUPR is beneficial for ageing. In mice, damage to mitochondrial ribosomes and the subsequent mtUPR induction also delayed ageing [194]. Furthermore, in D. melanogaster, mtUPR stimulation via the overexpression of the TRAP1 chaperone promoted both longevity and healthspan [195]. This beneficial effect was also observed in the fungal ageing model P. anserina, where the overexpression of the mitochondrial protease LonP1, a marker of mtUPR, extended lifespan [196].
In summary, there is sufficient evidence to suggest that the activation of the mtUPR can be helpful in delaying ageing and prolonging lifespan and that it may also play a beneficial role in the treatment of neurodegenerative diseases, as discussed below.
3.4.1. Alzheimer’s Disease
Alzheimer’s Disease (AD) is the most prevalent form of neurodegeneration, affecting more than 47.5 million people worldwide. AD is characterized by two neuropathological hallmarks: (1) neurofibrillary tangles formed by neurofilaments and hyperphosphorylated tau protein and (2) extracellular senile plaques formed via the accumulation of amyloid β (Aβ) peptide. Together, these hallmarks result in an irreversible loss of neurons, especially in the cortex and hippocampus, leading to memory and cognition impairments. The majority of AD cases occur sporadically among individuals over 65 years of age [197].
Numerous studies support the role of mitochondrial dysfunction in the pathogenesis of AD. For instance, the treatment of pig neurons with hydrogen peroxide increased intracellular Aβ levels [198], and treatment with CCCP caused intracellular Aβ accumulation in cultured astrocytes [199]. It has been suggested that oxidative damage precedes Aβ deposition in animal models [200]. Additionally, post-mortem analysis of AD brains has revealed impaired activities of three essential mitochondrial enzymes: the pyruvate dehydrogenase complex, the α-ketoglutarate dehydrogenase complex, and isocitrate dehydrogenase [201,202,203]. Moreover, defects in complexes of the mtETC have been reported in AD cases [204]. These mtETC abnormalities reduced ATP production and increased ROS [205]. Moreover, mitochondrial dysfunction, along with the resulting ROS overproduction, may alter amyloid precursor protein (APP) processing, leading to the intracellular accumulation of Aβ, a characteristic feature of AD [198]. Aβ aggregation can block the import of nuclear-encoded mitochondrial proteins, causing oxidative damage to mitochondrial and cellular proteins, nucleic acids, and lipids. This sets off a vicious cycle, ultimately resulting in apoptosis and cell death [206,207]. Moreover, mitochondrial size and shape are altered in AD patients, with mitochondria exhibiting an elongated, interconnected appearance referred to as “mitochondria on a string” [208]. This is linked to disruptions in mitochondrial dynamics, particularly reduced mitochondrial fission and mitophagy, as noted by Zhang et al. [209] (see Figure 5).
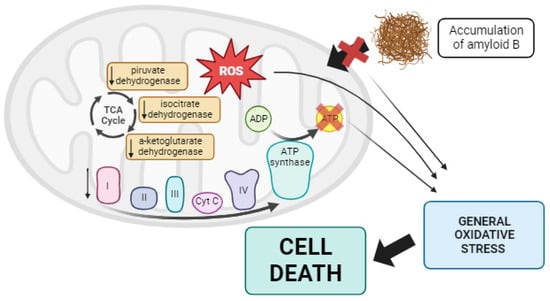
Figure 5.
Summary of mitochondrial dysfunction in AD. In Alzheimer’s disease, lower activity of some TCA enzymes such as pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase has been observed, along with alterations in the mtETC. These changes collectively result in reduced ATP production and the overproduction of ROS. Additionally, this promotes the aberrant processing of APP, leading to the accumulation of Aβ, which disrupts the import of mitochondrial proteins into the mitochondria. All of these factors contribute to generalized oxidative stress in the cell, ultimately causing cell death via apoptosis. Arrow: activation or induction of the subsequent process.
Considering the pivotal role of mitochondria in AD, the mtUPR could be a potential therapeutic target for this disease. Indeed, Beck et al. demonstrated an increase in the expression levels of mtUPR-associated proteins in the frontal cortex of sporadic AD patients [210]. This finding has also been reported by other authors, such as Sorrentino et al., who observed an upregulation of mtUPR-associated genes in AD patients, mouse models, and C. elegans as a protective response during disease progression [211]. Moreover, in an AD C. elegans model, ATFS-1 depletion via RNAi led to severe cognitive impairment, reduced mitochondrial respiration, and exacerbated Aβ aggregation, highlighting the protective role of the mtUPR in this disease [211]. Treatment with doxycycline or nicotinamide precursors to induce the mtUPR in the cited worm model and in mice improved overall health and lifespan by enhancing both animal motility and the clearance of Aβ peptides [211]. These authors also tested the positive effect of doxycycline on SHSY5Y cells with an APP mutation, observing a reduction in Aβ deposits and an improvement in AD pathophysiology [211]. Using the CL2006 C. elegans model of AD, which expresses the pathologic Aβ42, Regitz et al. demonstrated that treatment with resveratrol, a mtUPR activator, decreased Aβ aggregation and mitigated worm paralysis [212]. Similarly, Pérez et al. observed the induction of the mtUPR in pitrylisin metallopeptidase 1 (PITRM1) mutant cortical neurons obtained from induced pluripotent stem cells, as shown by increased levels of ATF4, CHOP, Hsp60, ClpP, and LonP1. These mutant neurons exhibited increased levels of Aβ peptides without aggregation, possibly due to the protective role of the mtUPR. Inhibition of the mtUPR with ISRIB, an inhibitor of eif2α phosphorylation, resulted in the appearance of Aβ deposits and increased cell death [213]. Likewise, the treatment of SHSY5Y cells with Aβ caused the activation of Hsp60, ClpP, and OMI. In fact, the activation of OMI protease was also detected in various human brain regions postmortem [214]. In the brains of APPsw/PS1De9 double transgenic mice, mtUPR induction was also demonstrated [215]. Furthermore, the activation of the mtUPR via nicotinamide riboside or olaparib restored mitochondrial membrane potential and morphology, increased mtDNA content, reduced Aβ peptide aggregation, and improved muscle integrity and fitness in C. elegans [216].
Considering all the studies published to date and the activation of the mtUPR in several Alzheimer’s disease models, it is evident that the induction of this mitochondrial stress response with external agents could be an effective strategy for the treatment of AD.
3.4.2. Parkinson’s Disease
Parkinson’s Disease (PD) is the most prevalent movement condition and the second-most common neurodegenerative disease, affecting more than 2% of the population over the age of sixty worldwide [217]. This disorder is characterized by two hallmark features: (1) the presence of intraneuronal cytoplasmic aggregated α-synuclein protein inclusions known as Lewy bodies, and (2) a reduction in dopamine levels in the basal ganglia due to the death of dopaminergic neurons in the substantia nigra pars compacta, located in the midbrain [218]. Several studies have suggested that mitochondrial dysfunction plays a role in the pathophysiology of PD. Mutations in genes such as Parkin or PINK1 are responsible for early-onset autosomal recessive PD, highlighting the importance of mitochondria in the disease’s etiology [219,220]. Mutations in the DJ-1 gene, which controls the NRF2 transcription factor involved in mitochondrial biogenesis [221], and mutations in the OMI protease involved in the IMS-mtUPR [222] have also been linked to PD.
However, most PD cases are not genetic but sporadic, and in the sporadic form of the disease, mitochondria are still implicated. Exposure to compounds such as MPTP or rotenone, which are inhibitors of mitochondrial complex I, can induce a parkinsonian phenotype, lead to the degeneration of dopaminergic neurons, and result in the occurrence of α-synuclein inclusions [223,224,225]. Additionally, a reduction in complex I activity has been found in the brains of PD patients, establishing a link between complex I inhibition and sporadic PD [226]. Moreover, inherited mtDNA mutations can also cause parkinsonism [227], and PD patients have been found to have increased levels of somatic mtDNA deletions [228]. Furthermore, according to some findings, oxidative stress and the presence of ROS may be among the primary causes of PD, as increased levels of oxidized lipids, proteins, and DNA have been detected in the substantia nigra of PD patients. In addition to ROS, reactive nitrogen species (RNS) also play an important role in PD. Nitric oxide (NO) is generated by nitric oxide synthase (NOS) and is present in high concentrations in cells and in the extracellular space surrounding dopaminergic neurons, as demonstrated in postmortem brain tissue from PD patients [229]. NO can interfere with various enzymes and complexes I and IV, leading to increased ROS production. It can also cause lipid peroxidation and disrupt the proper functioning of proteins [230,231].
Based on these findings, a hypothesis regarding mitochondrial dysfunction in PD pathogenesis has been proposed. The inhibition of complex I disrupts electron flux through mtETC, leading to increased production of ROS and RNS. These reactive species damage different cellular components, including lipids, proteins, and DNAactivating proapoptotic pathways with the entry of Bax into the mitochondria. Bax subsequently causes the release of cytochrome c into the cytosol, ultimately activating the caspase signaling pathway and resulting in cell death via apoptosis [232] (see Figure 6).
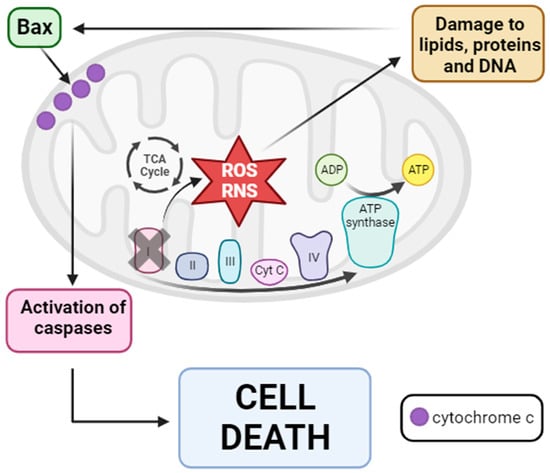
Figure 6.
Mitochondrial dysfunction hypothesis of PD pathogenesis. Inhibition of mitochondrial complex I provokes the disruption of mtETC with consequent ROS and RNS production. Then, these reactive species damage several cellular components such as lipids, proteins, and DNA, causing the entry of Bax from the cytosol into the OMM. Once in the OMM, Bax provokes the release of cytochrome c into the cytosol, leading to the activation of the caspase signaling pathway and finally cell death via apoptosis. Arrow: activation or induction of the subsequent process.
Given this hypothesis, it is not surprising that the activation of mitochondrial quality control mechanisms may act as a therapeutic target for Parkinson’s disease. Regarding the mtUPR, Cooper et al. showed that in mutant PINK1, Parkin, and DJ-1 C. elegans PD models, mtUPR activation occurred, which protected worms from the death of dopaminergic neurons [233]. Dastidar et al. reported that overexpression of 4E-BP1, an inhibitor of eukaryotic initiation factor 4E and protein translation, protected animals against neurotoxicity induced by rotenone and paraquat. This protection was attributed to the activation of the mtUPR, as indicated by increased levels of CHOP, ATF4, Hsp60, ClpP, and MnSOD [234]. When CHOP was silenced, neuroprotection did not occur, and increased neuronal death was observed [234]. Finally, Liu et al. proposed that, in a D. melanogaster PD model, treatment with Ginseng, a Chinese herbal medicine, alleviated dopaminergic neuron death, improved locomotor function, and extended lifespan. These authors suggested that the beneficial effect of Ginseng was mediated via mtUPR enhancement, as they observed a significant induction of mtUPR-related genes such as Hsp60, Hsp70, and ClpP [235].
Nonetheless, it is important to note that overactivation of the mtUPR, such as through the elimination of MTS of ATFS-1 in a C. elegans PD model, provoked the death of dopaminergic neurons in a non-apoptotic manner, worsening the disease phenotype [236]. For this reason, while mtUPR induction may be effective in the treatment of Parkinson’s disease, the levels of activation should be carefully controlled, as overactivation might have detrimental effects. Further research is needed to fully understand the optimal level of mtUPR activation for therapeutic benefit.
3.4.3. Huntington’s Disease
In Huntington’s Disease (HD), the role of mitochondrial dysfunction has also been proposed. HD is characterized by the progressive loss of GABAergic medium spiny neurons in the striatum, although degeneration is also observed in other brain regions such as the thalamus, the subthalamic nucleus, white matter, or the cerebellum [237]. This disorder affects 1 in 10,000 people worldwide [238]. Unlike Alzheimer’s and Parkinson’s diseases, where most cases are sporadic, the cause of HD is genetic [239]. This disease is caused by a CAG triplet expansion in the huntingtin gene, resulting in an expansion of polyglutamine in the N-terminal end of the protein. A number greater than 40 repeats is considered pathogenic, while the normal number is less than 36 [240].
It has been reported that mutant huntingtin (mtHtt) causes defects in mitochondria, leading to neurotoxicity, neuronal dysfunction, and finally cell death [241]. Indeed, it has been demonstrated that mtHtt interacts with several proteins. In the nucleus, mtHtt binds to p53, a tumor suppressor that, in response to stress, activates Bax and thus cell death via apoptosis [242]. The inhibition of p53 in D. melanogaster and M. musculus HD models prevented cellular dysfunction and neurodegeneration [243]. In addition, mtHtt interacts with CREB and CREB binding protein (CBP), inhibiting their function and therefore mitochondrial biogenesis [244]. In mitochondria, mtHtt activates the Drp1 protein, leading to mitochondrial fragmentation and general mitochondrial dysfunction [245]. Another protein with which mtHtt interacts is valosin-containing protein (VCP), an ATPase associated with OMM protein turnover and parkin-dependent mitophagy. As a result, mtHtt–VCP interaction promotes increased mitophagy and neuronal degeneration associated with mitochondrial dysfunction [246]. The involvement of mitochondrial dysfunction has been confirmed, as decreased mitochondrial complex I, II, III, and IV activity has been reported in the brains of HD patients [241,247], along with impaired mitochondrial respiration and ATP production [248]. Moreover, the inhibition of mitochondrial complex II by 3-nitropropionic acid or malonate leads to a pathological phenotype that resembles HD [249]. Impaired succinate dehydrogenase activity has also been observed in postmortem HD brains, along with reduced mitochondrial membrane potential in HD patients’ muscles and lymphoblasts [241]. This could be due to the interaction of mutant huntingtin with the OMM, which causes mitochondrial membrane potential reduction and potentiates mitochondrial permeability transition [250]. Finally, mtDNA deletions have been identified in HD patients during disease progression [251].
In summary, mtHtt interacts with a large number of proteins and mitochondrial structures such as the OMM. This interaction results in a reduction in mitochondrial biogenesis and membrane potential. It also leads to the opening of the mitochondrial permeability transition pore, calcium homeostasis failure, the induction of mitophagy, and mitochondrial fission. Additionally, there is a decrease in mitochondrial complex activity, elevated ROS production, and, ultimately, cell death, provoking the neurodegeneration characteristic of HD [252]. For this reason, the activation of mitochondrial quality control mechanisms and especially the mtUPR could be a potential treatment since mtHtt undergoes misfolding and proteolytic cleavage, resulting in N-terminal fragments that can form aggregates (see Figure 7).
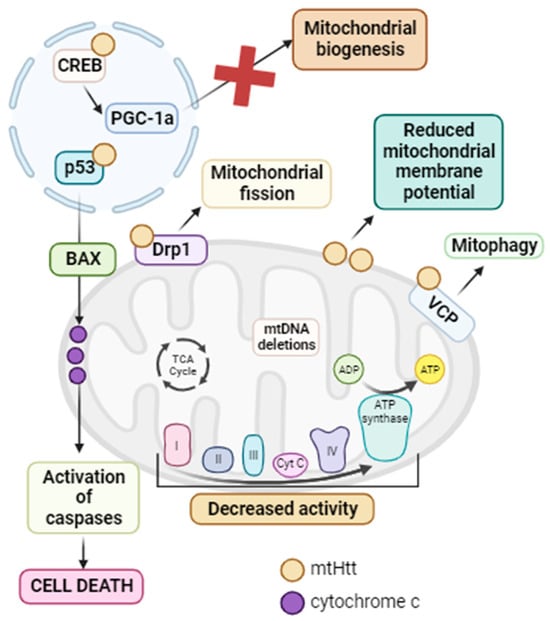
Figure 7.
Summary of mitochondrial dysfunction in HD. mtHtt interacts with a large number of proteins. In the nucleus, it interacts with p53, promoting the activation of Bax with the subsequent release of cytochrome c from the mitochondria and the activation of caspases, ultimately leading to cell death via apoptosis. Additionally, in this same cellular compartment, mtHtt interacts with CREB, inhibiting it and thus suppressing PGC-1α and mitochondrial biogenesis. In the mitochondria, mtHtt interacts with Drp1, causing its activation and consequent mitochondrial fragmentation. Moreover, mtHtt binds to the OMM, leading to a decrease in mitochondrial membrane potential. It also interacts with VCP, activating mitophagy. All of these processes, along with the decreased activity of mtETC complexes and the accumulation of mtDNA deletions, contribute to the mitochondrial dysfunction characteristic of HD. Arrow: activation or induction of the subsequent process.
Accordingly, Fu et al. related that ABCB10, a component of the mtUPR pathway, had decreased transcript and protein expression levels in an HD mouse model and in dermal fibroblasts of two HD patients [253]. Overexpression of ABCB10 in striatal cells of an HD mouse model induced CHOP expression and reduced fragmented mitochondria, ROS production, and cell death via apoptosis [253]. The protein and mRNA levels of two markers of the mtUPR, Hsp60 and ClpP, were significantly reduced in fibroblasts derived from two HD patients, suggesting that mtHtt inhibited mtUPR activation in sufferers of HD [253]. Moreover, Hernández et al. observed that the ATF5 transcription factor, the master regulator of the mtUPR, was sequestered in mtHtt aggregates in HD mice, C. elegans, and patients, mainly in the cortex and striatum in the case of mice and patients [254]. Almeida et al. also observed decreased levels of mtUPR markers in PC12 cells expressing mtHtt [255]. On the other hand, Naia et al. demonstrated that SIRT3 expression levels and activity were elevated in several in vitro and in vivo models of HD [256]. However, this upregulation is not sufficient to counteract the toxic effects of mtHtt. Overexpression of SIRT3 in HD mouse and D. melanogaster models, as well as in HD-patient-derived fibroblasts, resulted in increased mitochondrial membrane potential, the elevation of mitochondrial complex activity, and decreased mitochondrial fragmentation, leading to the improvement of HD pathophysiology [256]. Furthermore, these authors also tested the effect of resveratrol in an HD mouse model, with the result being the activation of mitochondrial biogenesis through SIRT1 induction [257]. In the striatal cells derived from this HD mouse model, the use of nicotinamide induced the activation of sirtuin mtUPR, provoking an increase in mitochondrial antioxidant capacity with a consequent increase in mitochondrial membrane potential [257]. Likewise, treatment with both resveratrol and nicotinamide in HD-derived lymphoblasts restored mitochondrial respiration, with increased mitochondrial biogenesis and ATP production [257]. The positive effect of resveratrol and nicotinamide was also tested in the HD mouse model, where the authors observed that the restoration of mitochondrial function alleviated neurodegeneration signs and improved motor function [257]. Furthermore, Fu et al. showed that the activation of mitochondrial SIRT3 via treatment with ε-viniferin in a PC12 HD cell model reduced oxidative stress, promoted mitochondrial biogenesis, and improved mitochondrial dysfunction [258].
In conclusion, mitochondrial dysfunction is strongly associated with the pathogenesis of HD. mtHtt interacts with many proteins, among which is ABCB10, involved in the mtUPR, or CREB implicated in mitochondrial biogenesis, resulting in an impairment in mitochondrial quality control mechanisms. Therefore, the use of compounds that induce these pathways may be a potential therapeutic option for the treatment of HD.
3.4.4. Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the progressive loss of upper and lower motor neurons, mainly in the spinal cord, brainstem, and motor cortex. With an incidence of approximately 2.5/per 100,000 people worldwide, ALS is the third-most-common neurodegenerative disease after AD and PD [259]. While the majority of ALS cases are sporadic, 10% have a genetic cause. Since the discovery of the first gene associated with ALS, superoxide dismutase 1 (SOD1) [260], more than 20 other genes have been reported to correlate with its pathogenesis, including matrin 3 (MATR3), coiled-coil-helix-coiled-coil-helix domain-containing 10 (CHCHD10), fused in sarcoma/translocated in liposarcoma (FUS), chromosome 9 open reading frame 72 (C9orf72), TAR DNA binding protein 43 (TDP-43), and OPTN, among others [261].
Several studies have proposed a strong association between mitochondrial function and ALS pathogenesis. In the case of familial ALS, mutations in the SOD1 gene alter its antioxidant capacity, leading to ROS accumulation that inactivates axonal transport, induces axonal degeneration, and causes SOD1 to bind to Bcl-2, inhibiting Bcl-2’s anti-apoptotic activity. This promotes cytochrome C release and mitochondrially initiated caspase activation. In addition, altered calcium homeostasis in mitochondria due to SOD1 mutations provokes the activation of apoptotic pathways and motor neuron death [262]. C9orf72 mutations are also associated with mitochondrial dysfunction since C9orf72 is an IMM-associated protein. C9orf72 regulates complex I assembly by interacting with the IMM-domain-containing 1 (TIMMDC1) protein. When C9orf72 is mutated, the result is reduced mitochondrial complex I activity, provoking mitochondrial dysfunction and ALS motor symptoms [263]. On the other hand, mutations in TDP-43 cause an alteration in mitochondrial dynamics, with an increased expression of FIS1 and Drp1 and decreased levels of Mfn1, leading to mitochondrial fragmentation, which affects mitochondrial function, including mtETC activity, ROS production, and calcium homeostasis, resulting in neuronal apoptosis [264]. In addition, mutations in OPTN cause a disruption in mitophagy since this protein is the primary receptor for PINK1/Parkin-mediated mitophagy. This causes an accumulation of damaged mitochondria and, once again, mitochondrial dysfunction, with a consequent cascade of signaling that eventually leads to neuronal death [265]. MATR3 mutations have been found in four families with ALS. This protein has RNA- and DNA-binding domains that regulate gene expression, and it forms a complex with TDP43 and FUS, contributing to the mitochondrial dysfunction described above [266]. Similarly, CHCHD10 mutations cause mitochondrial dynamics and cellular bioenergetics disturbances, as ALS patients with mutations in this gene present a fragmented mitochondrial network [267]. Moreover, the CHCHD10 protein interacts with TDP43 in the nucleus, but when it is mutated, the interaction does not occur, and TDP43 translocates to the cytosol, causing synaptic damage [268]. Finally, in the case of FUS mutations, the protein encoded by this gene forms aggregates in the cytosol of motor neurons. Additionally, it has been described that mutant FUS protein targets mRNAs encoding mitochondrial respiratory chain proteins in the nucleus and in the mtDNA, causing OXPHOS deficiency, ROS overproduction, and general mitochondrial dysfunction [269].
It is important to emphasize that in all these cases, there is an aggregation of the mutated protein inside the mitochondrion, which disrupts OXPHOS and leads to ROS accumulation, Ca2+ dyshomeostasis, mitochondrial dynamics alterations, mitophagy failure, and mitochondrial dysfunction. These alterations alter axonal transport, induce axonal degeneration, and ultimately lead to neuronal apoptosis [270].
Regarding sporadic cases, even though they do not follow a family history, mutations in some of the ALS-related genes have been found in most cases. In Table 2, we summarized the principal genes associated with ALS and their pathogenic mechanisms.
Table 2.
Summary of the main genes related to ALS and their pathogenic mechanisms. It is important to note that mitochondrial dysfunction is present in all cases.
It is therefore important to consider the role of mitochondria, and most notably mitochondrial dysfunction and oxidative stress, in the pathogenic mechanisms of ALS.
Interestingly, the mtUPR is also associated with ALS. Wang et al. observed mtUPR activation in TDP-43 ALS cellular and animal models. Moreover, they described that TDP-43 interacted with LonP1, leading to its degradation. LonP1 overexpression protected cells from TDP-43-induced neurotoxicity, reducing mitochondrial damage and neurodegeneration. In contrast, inhibiting this protease decreased cell viability, suggesting a protective role of the mtUPR in TDP-43 pathogenesis [271].
In addition, Deng et al. demonstrated that mutant FUS accumulated within mitochondria, where FUS aggregates interacted with the ATP5B subunit of the ATP synthase complex. This interaction reduced mitochondrial ATP synthesis in HEK293 cell lines expressing mutant FUS and in a FUS-ALS D. melanogaster model. As a result, mitochondrial impairment occurred, and the mtUPR was activated, evidenced by the upregulation of mtUPR-associated genes such as ATF5, LonP1, Hsp60, and Hsp70 in both ALS cellular and animal models [272]. However, in contrast to the TDP-43 mutation case, LonP1 overexpression in the FUS-ALS D. melanogaster model exacerbated retinal degeneration, with increased neurodegeneration, while silencing mtUPR-related genes ameliorated mutant FUS-induced neurodegeneration [272].
On the other hand, Zhou et al. reported that mtUPR activation in an ALS mouse model with an SOD1 gene mutation, achieved via treatment with nicotinamide precursors, decreased mitochondrial dysfunction and SOD1 aggregates and improved adult neurogenesis [273]. Furthermore, Straub et al. observed elevated mtUPR marker levels for both the transcriptional canonical and sirtuin mtUPR axes in fibroblasts derived from mutant CHCHD10 patients when cultured under galactose stress conditions [274]. Finally, Riar et al. described that in a mouse model of ALS carrying the G93A-mutation in the SOD1 protein, IMS-mtUPR axis activation was induced when mutant SOD1 accumulated in the IMS. Interestingly, the genetic ablation of ERα induced the activation of the transcriptional canonical mtUPR axis, suggesting that in the absence of ERα, cells induced the CHOP/ATF/ATF5 axis as a compensatory mechanism [275]. It is worth noting that the activation of both axes of the mtUPR was found to be sex-specific, with significantly higher levels of induction observed in female mice compared to male mice (for which the levels were low) [275].
In conclusion, the role of the mtUPR as a therapeutic target in ALS remains complex. Its activation shows varied effects, with some cases indicating positive outcomes due to the aggregation of mutated proteins, while others demonstrate deleterious effects, as observed with LonP1 overexpression. A potential avenue worth exploring is the induction of the sirtuin axis, as suggested by the positive effects seen in the treatment with nicotinamide precursors. However, it is important to note that this observation is based on limited studies, and further research is essential to elucidate the full therapeutic potential of mtUPR pathways in ALS.
3.4.5. Friedreich’s Ataxia (FA)
The implications of mitochondrial dysfunction and the mtUPR have also been described in Friedreich’s Ataxia (FA), a rare disease arising from a GAA repeat expansion in the frataxin (FXN) gene, which encodes the mitochondrial FXN protein involved in iron-sulfur cluster biosynthesis [276]. Consequently, decreased protein expression levels of FXN result in mitochondrial dysfunction, characterized by reduced OXPHOS, ROS overproduction, abnormal mitochondrial morphology, and Ca2+ homeostasis alterations [277]. It has been reported that transcriptional canonical mtUPR axis markers such as ATF4 and CHOP were upregulated in a mouse model of FA [278].
In summary, the interplay between mitochondrial dysfunction, mtUPR activation, and its potential benefit for FA remains intricate. Ageing and neurodegenerative diseases exhibit a strong association with mitochondrial dysfunction, leading to various pathological events culminating in cell apoptosis. In response, cells activate their mitochondrial quality control mechanisms, primarily the mtUPR, in an attempt to restore mitochondrial function. However, in the majority of cases, the induction of this stress response is not sufficient as the diseases continue to progress. Therefore, activation by external agents may hold therapeutic promise, allowing cells to regain their capacity to combat mitochondrial damage and resulting in improvements in symptoms and disease physiopathology. This therapeutic potential has been demonstrated in various animal and cellular models across different neurodegenerative diseases, as discussed throughout this review.
4. Conclusions
Mitochondrial function is essential for cell survival and overall fitness. In response to mitochondrial stress, cells activate distinct mitochondrial quality control processes based on the intensity of the stress. When faced with severe stress, the initial response involves inducing mitochondrial fission. If this fails to restore mitochondrial function, cells initiate mitophagy for the complete degradation of the damaged mitochondria. In case of moderate stress, mitochondria stimulate mitochondrial fusion and mtUPR mechanisms for the recovery of mitochondrial function. Four different axes of the mtUPR have been described, namely, transcriptional and translational mtUPR, sirtuin mtUPR, and IMS-mtUPR, which act in parallel and complementarily, working together to enhance the restoration of mitochondrial health.
On the other hand, the loss of mitochondrial homeostasis and proteostasis results in mitochondrial dysfunction, a core factor implicated in a wide range of pathologies. These include primary mitochondrial diseases, cardiovascular disorders, metabolic diseases, cancer, and neurodegenerative diseases, as well as a common physiological process: ageing. The activation of the different mtUPR axes has shown a beneficial effect in numerous models of these diseases. Notably, in cancer, the situation is the contrary, since the inhibition of this stress response is considered a therapeutic target.
Therefore, modulating the mtUPR has emerged as a potential therapeutic target for the treatment of complex diseases that compromise quality of life and currently lack curative treatments.
Author Contributions
Conceptualization and writing, P.C.-H., J.A.S.-A., D.G.-F. and R.P.-P.; review and editing, M.M.-C., D.R.-L., M.Á.-C., A.R.-G., M.T.-R., A.S.-C., A.L.-C. and J.M.R.-D. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by FIS PI19/00377 (2019) and FIS PI22/00142 (2022) grants, Instituto de Salud Carlos III, Ministerio de Sanidad, Spain, and the Fondo Europeo de Desarrollo Regional (FEDER Unión Europea), Spanish Ministry of Education, Culture, and Sport. This activity was co-financed by the European Regional Development Fund (ERDF) and by the Regional Ministry of Economic Transformation, Industry, Knowledge, and Universities of the Junta de Andalucía, within the framework of the ERDF Andalusia operational program 2014–2020 Thematic objective “01—Reinforcement of research, technological development and innovation” through the reference research project CTS-5725, PY18-850 and UPO-FEDER 2018 (UPO-1380614). We acknowledge the efforts of the patients’ associations, Fundación FEDER, and Fundación MERCK Salud. We also acknowledge the support from “Ayudas para la Formación de Profesorado Universitario” (FPU/MINECO) and “Ayudas B2 de Iniciación a la Investigación from Pablo de Olavide University”. A. López-Cabrera is a recipient of a contract from “Programa investigo” (Marco del Plan de Recuperación, Transformación y Resiliencia para Andalucía, funded by NextGenerationEU).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
We declare no conflict of interest.
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| AMBRA1 | autophagy and beclin 1 regulator 1 |
| AMPK | AMP-activated kinase |
| APP | amyloid precursor protein |
| ATF4 | Activating Transcription Factor 4 |
| ATF5 | Activating Transcription Factor 5 |
| ATFS-1 | activating transcription factor associated with stress-1 |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| C9orf72 | chromosome 9 open reading frame 72 |
| CaMK | calcium-calcium/calmodulin-dependent protein kinase |
| CHCHD10 | coiled-coil-helix-coiled-coil-helix domain containing 10 |
| CHOP | C/EBP Homologous Protein |
| CLPP-1 | caseinolytic protease P |
| CREB | cAMP response element-binding protein |
| DELE1 | DAP3 Binding Cell Death Enhancer 1 |
| Drp1 | dynamin-related protein 1 |
| DVE-1 | homeodomain-containing protein transcription factor “defective proventiculus” |
| eif2α | eukaryotic translation initiation factor 2α |
| ER | endoplasmic reticulum |
| ERα | estrogen receptor α |
| FA | Friedreich’s Ataxia |
| FIS1 | mitochondrial fission 1 protein |
| FUNDC1 | Fun14 domain-containing protein 1 |
| FUS | fused in sarcoma/translocated in liposarcoma |
| HAF-1 | inner-membrane-spanning ATP-binding casette transporter |
| HD | Huntington’s Disease |
| HSF1 | heat shock transcription factor 1 |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane space |
| ISR | Integrated Stress Response |
| LC3 | microtubule-associated protein 1A/1B light chain 3 |
| LonP1 | Lon protease-like 1 |
| MATR3 | matrin 3 |
| MDVs | mitochondrial-derived vesicles |
| MFF | mitochondrial fission factor |
| Mfn1 | mitofusin 1 |
| Mfn2 | mitofusin 2 |
| MID49 | mitochondrial dynamics protein 49 |
| MID51 | mitochondrial dynamics protein 51 |
| mPOS | mitochondrial precursor overaccumulation stress |
| mtDNA | mitochondrial DNA |
| mtETC | mitochondrial electron transport chain |
| MTS | mitochondrial targeting signal |
| mtUPR | mitochondrial unfolded protein response |
| nDNA | nuclear DNA |
| NLS | nuclear localization signal |
| NRF1 | nuclear respiratory factor 1 |
| NRF2 | nuclear respiratory factor 2 |
| OMM | outer mitochondrial membrane |
| Opa1 | Optic Atrophy 1 |
| OPTN | optineurin |
| OXPHOS | oxidative phosphorylation |
| PD | Parkinson’s Disease |
| PGC-1α | peroxisome proliferator-activated receptor γ co-activator 1α |
| PINK1 | PTEN-induced kinase 1 |
| PITRM1 | pitrylisin metallopeptidase 1 |
| PKA | protein kinase A |
| PLEKHM1 | pleckstrin homology domain-containing family M member 1 |
| ROS | reactive oxygen species |
| SIMH | stress-induced mitochondrial hyperfusion |
| SLP-2 | stromatin-like protein 2 |
| SOD1 | superoxide dismutase 1 |
| SOD2 | superoxide dismutase 2 |
| SSBP1 | single-stranded DNA binding protein 1 |
| TAX1BP1 | Tax1-binding protein 1 |
| TCA | tricarboxylic acid |
| TDP-43 | TAR DNA binding protein 43 |
| TFAM | mitochondrial transcription factor A |
| TIM | translocase of the inner membrane |
| TIMMDC1 | IMM-domain containing 1 |
| TOM | translocase of the outer membrane |
| UBL-5 | ubiquitin-like protein 5 |
| UPRam | Unfolded Protein Response activated by mistargeting of proteins |
| UPRER | Unfolded Protein Response of the endoplasmic reticulum |
| UV-C | ultraviolet C |
| VDAC1 | voltage/dependent anion channel 1 |
References
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Deshpande, O.A.; Mohiuddin, S.S. Biochemistry, Oxidative Phosphorylation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chandel, N.S. Mitochondria. Cold Spring Harb. Perspect. Biol. 2021, 13, a040543. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. eLlife 2020, 9, e58041. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Muhlenhoff, U. Iron-sulfur protein biogenesis in eukaryotes: Components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006, 22, 457–486. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Guda, P.; Guda, C.; Subramaniam, S. Reconstruction of pathways associated with amino acid metabolism in human mitochondria. Genom. Proteom. Bioinform. 2007, 5, 166–176. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Suhm, T.; Kaimal, J.M.; Dawitz, H.; Peselj, C.; Masser, A.E.; Hanzen, S.; Ambrozic, M.; Smialowska, A.; Bjorck, M.L.; Brzezinski, P.; et al. Mitochondrial Translation Efficiency Controls Cytoplasmic Protein Homeostasis. Cell Metab. 2018, 27, 1309–1322.e1306. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef]
- Lenkiewicz, A.M.; Krakowczyk, M.; Bragoszewski, P. Cytosolic Quality Control of Mitochondrial Protein Precursors-The Early Stages of the Organelle Biogenesis. Int. J. Mol. Sci. 2021, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial-derived vesicles: Recent insights. J. Cell Mol. Med. 2022, 26, 3323–3328. [Google Scholar] [CrossRef]
- Naresh, N.U.; Haynes, C.M. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033944. [Google Scholar] [CrossRef]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1alpha and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Zhao, Q.; Tian, Z.; Zhou, G.; Niu, Q.; Chen, J.; Li, P.; Dong, L.; Xia, T.; Zhang, S.; Wang, A. SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride. Theranostics 2020, 10, 4822–4838. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R. The role of calcium and calcium/calmodulin-dependent kinases in skeletal muscle plasticity and mitochondrial biogenesis. Proc. Nutr. Soc. 2004, 63, 279–286. [Google Scholar] [CrossRef]
- Mehta, S.L.; Mendelev, N.; Kumari, S.; Andy Li, P. Overexpression of human selenoprotein H in neuronal cells enhances mitochondrial biogenesis and function through activation of protein kinase A, protein kinase B, and cyclic adenosine monophosphate response element-binding protein pathway. Int. J. Biochem. Cell Biol. 2013, 45, 604–611. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J. Biol. Chem. 2006, 281, 324–333. [Google Scholar] [CrossRef]
- Ahting, U.; Thieffry, M.; Engelhardt, H.; Hegerl, R.; Neupert, W.; Nussberger, S. Tom40, the pore-forming component of the protein-conducting TOM channel in the outer membrane of mitochondria. J. Cell Biol. 2001, 153, 1151–1160. [Google Scholar] [CrossRef]
- Schendzielorz, A.B.; Schulz, C.; Lytovchenko, O.; Clancy, A.; Guiard, B.; Ieva, R.; van der Laan, M.; Rehling, P. Two distinct membrane potential-dependent steps drive mitochondrial matrix protein translocation. J. Cell Biol. 2017, 216, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Craig, E.A.; Zufall, N.; von Ahsen, O.; Pfanner, N.; Voos, W. The protein import motor of mitochondria: Unfolding and trapping of preproteins are distinct and separable functions of matrix Hsp70. Cell 1999, 97, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Meisinger, C.; Vogtle, F.N. Processing of mitochondrial presequences. Biochim. Biophys. Acta 2012, 1819, 1098–1106. [Google Scholar] [CrossRef]
- Brix, J.; Rudiger, S.; Bukau, B.; Schneider-Mergener, J.; Pfanner, N. Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22, and Tom70 in a presequence-carrying preprotein and a non-cleavable preprotein. J. Biol. Chem. 1999, 274, 16522–16530. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Grevel, A.; Pfanner, N.; Becker, T. Coupling of import and assembly pathways in mitochondrial protein biogenesis. Biol. Chem. 2019, 401, 117–129. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Bragoszewski, P.; Gornicka, A.; Sztolsztener, M.E.; Chacinska, A. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol. Cell Biol. 2013, 33, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 2015, 524, 481–484. [Google Scholar] [CrossRef]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.G.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPR(mt). Cell Rep. 2019, 28, 1659–1669.e5. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Nelson, G.M.; Watson, J.L.; Morf, J.; Dalglish, M.; Luh, L.M.; Weber, A.; Bertolotti, A. Protein Stability Buffers the Cost of Translation Attenuation following eIF2alpha Phosphorylation. Cell Rep. 2020, 32, 108154. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Arnoult, D.; Chen, H.; Chan, D.C.; Smith, C.L.; Youle, R.J. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 2004, 164, 493–499. [Google Scholar] [CrossRef]
- van der Bliek, A.M. Fussy mitochondria fuse in response to stress. EMBO J. 2009, 28, 1533–1534. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Frohlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta 2010, 1800, 250–256. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, K.; Li, J.; Cui, L.; Dong, J.; Li, J.; Meng, X.; Zhu, G.; Wang, H. PINK1/Parkin-mediated mitophagy enhances the survival of Staphylococcus aureus in bovine macrophages. J. Cell Mol. Med. 2023, 27, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Sulkshane, P.; Ram, J.; Thakur, A.; Reis, N.; Kleifeld, O.; Glickman, M.H. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021, 45, 102047. [Google Scholar] [CrossRef]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating Mitophagy in Mitochondrial Disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef]
- Denk, D.; Petrocelli, V.; Conche, C.; Drachsler, M.; Ziegler, P.K.; Braun, A.; Kress, A.; Nicolas, A.M.; Mohs, K.; Becker, C.; et al. Expansion of T memory stem cells with superior anti-tumor immunity by Urolithin A-induced mitophagy. Immunity 2022, 55, 2059–2073.e2058. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Hindle, A.; Kshirsagar, S.; Reddy, P.H. Are mitophagy enhancers therapeutic targets for Alzheimer’s disease? Biomed. Pharmacother. 2022, 149, 112918. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef]
- Liu, K.; Jing, M.J.; Liu, C.; Yan, D.Y.; Ma, Z.; Wang, C.; Deng, Y.; Liu, W.; Xu, B. Effect of trehalose on manganese-induced mitochondrial dysfunction and neuronal cell damage in mice. Basic. Clin. Pharmacol. Toxicol. 2019, 125, 536–547. [Google Scholar] [CrossRef]
- Lin, K.L.; Lin, K.J.; Wang, P.W.; Chuang, J.H.; Lin, H.Y.; Chen, S.D.; Chuang, Y.C.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic. Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef]
- Martin-Maestro, P.; Sproul, A.; Martinez, H.; Paquet, D.; Gerges, M.; Noggle, S.; Starkov, A.A. Autophagy Induction by Bexarotene Promotes Mitophagy in Presenilin 1 Familial Alzheimer’s Disease iPSC-Derived Neural Stem Cells. Mol. Neurobiol. 2019, 56, 8220–8236. [Google Scholar] [CrossRef]
- Wang, N.; Wang, H.; Pan, Q.; Kang, J.; Liang, Z.; Zhang, R. The Combination of beta-Asarone and Icariin Inhibits Amyloid-beta and Reverses Cognitive Deficits by Promoting Mitophagy in Models of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7158444. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Z.; Wang, H.; Zhang, D.; Han, T.; Chen, H.; Chen, J.; Chen, Z.; Xie, Y.; Wang, L.; et al. Melatonin promotes peripheral nerve repair through Parkin-mediated mitophagy. Free Radic. Biol. Med. 2022, 185, 52–66. [Google Scholar] [CrossRef]
- Cen, X.; Xu, X.; Xia, H. Targeting MCL1 to induce mitophagy is a potential therapeutic strategy for Alzheimer disease. Autophagy 2021, 17, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Ai, R.; Zhuang, X.X.; Anisimov, A.; Lu, J.H.; Fang, E.F. A synergized machine learning plus cross-species wet-lab validation approach identifies neuronal mitophagy inducers inhibiting Alzheimer disease. Autophagy 2022, 18, 939–941. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Wanet, A.; De Pauw, A.; Rommelaere, G.; Arnould, T.; Renard, P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J. Cell Physiol. 2012, 227, 2297–2310. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229–239. [Google Scholar] [CrossRef]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Berendzen, K.M.; Durieux, J.; Shao, L.W.; Tian, Y.; Kim, H.E.; Wolff, S.; Liu, Y.; Dillin, A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 2016, 166, 1553–1563.e1510. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gomez-Fernandez, D.; Pinero-Perez, R.; Reche-Lopez, D.; Alvarez-Cordoba, M.; Munuera-Cabeza, M.; Talaveron-Rey, M.; Povea-Cabello, S.; Suarez-Carrillo, A.; Romero-Gonzalez, A.; et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Holper, S.; Kruger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim. Biophys. Acta 2015, 1853, 2580–2591. [Google Scholar] [CrossRef]
- Kang, Z.; Chen, F.; Wu, W.; Liu, R.; Chen, T.; Xu, F. UPR(mt) and coordinated UPR(ER) in type 2 diabetes. Front. Cell Dev. Biol. 2022, 10, 974083. [Google Scholar] [CrossRef] [PubMed]
- Mesbah Moosavi, Z.S.; Hood, D.A. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2017, 312, C583–C594. [Google Scholar] [CrossRef]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Fessler, E.; Krumwiede, L.; Jae, L.T. DELE1 tracks perturbed protein import and processing in human mitochondria. Nat. Commun. 2022, 13, 1853. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Fujimoto, M.; Takii, R.; Takaki, E.; Hayashida, N.; Nakai, A. Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat. Commun. 2015, 6, 6580. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef]
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor. Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Watroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 5, 25. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef]
- Tao, R.; Vassilopoulos, A.; Parisiadou, L.; Yan, Y.; Gius, D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid. Redox Signal 2014, 20, 1646–1654. [Google Scholar] [CrossRef]
- Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. HTRA proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011, 12, 152–162. [Google Scholar] [CrossRef]
- Wang, Y.; Jasper, H.; Toan, S.; Muid, D.; Chang, X.; Zhou, H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021, 45, 102049. [Google Scholar] [CrossRef]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fan, Y.; Zong, R.; Tan, K. The mitochondrial unfolded protein response: A multitasking giant in the fight against human diseases. Ageing Res. Rev. 2022, 81, 101702. [Google Scholar] [CrossRef]
- Yi, H.S. Implications of Mitochondrial Unfolded Protein Response and Mitokines: A Perspective on Fatty Liver Diseases. Endocrinol. Metab. 2019, 34, 39–46. [Google Scholar] [CrossRef]
- Yi, H.S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Luo, S.; Chen, W.; He, L.; Liu, D.; Wang, X.; Xiao, L.; Sun, L. Mitochondrial unfolded protein response (mtUPR) and diseases. Curr. Med. Chem. 2023. E-pub Ahead of Print. [Google Scholar] [CrossRef]
- Lu, H.; Wang, X.; Li, M.; Ji, D.; Liang, D.; Liang, C.; Liu, Y.; Zhang, Z.; Cao, Y.; Zou, W. Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases. Cells 2022, 12, 20. [Google Scholar] [CrossRef]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; et al. UPR(mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-Gonzalez, A.; Gomez-Fernandez, D.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination With Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharmacol. 2022, 13, 862085. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. Mitochondria and Antibiotics: For Good or for Evil? Biomolecules 2021, 11, 1050. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, Z.; Yan, X.; Liu, J.; Li, F.; Liu, P.; Li, J.; Wei, Y.; Sun, Y.E.; Zhang, Y.; et al. ATF5, a putative therapeutic target for the mitochondrial DNA 3243A > G mutation-related disease. Cell Death Dis. 2021, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, X.; Zheng, S.; Zhu, A.; Wang, J. The Mitochondrial Unfolded Protein Response: A Novel Protective Pathway Targeting Cardiomyocytes. Oxid. Med. Cell Longev. 2022, 2022, 6430342. [Google Scholar] [CrossRef]
- Wang, Y.T.; Lim, Y.; McCall, M.N.; Huang, K.T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H472–H478. [Google Scholar] [CrossRef] [PubMed]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; Lopez, B.; Gonzalez, A.; Ravassa, S.; Diez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Xu, M.; Xue, R.Q.; Lu, Y.; Yong, S.Y.; Wu, Q.; Cui, Y.L.; Zuo, X.T.; Yu, X.J.; Zhao, M.; Zang, W.J. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc. Res. 2019, 115, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tan, Y.; Zhang, Z.; Feng, P.; Ding, W.; Wang, Q.; Liang, H.; Duan, W.; Wang, X.; Yu, S.; et al. Novel PGC-1alpha/ATF5 Axis Partly Activates UPR(mt) and Mediates Cardioprotective Role of Tetrahydrocurcumin in Pathological Cardiac Hypertrophy. Oxid. Med. Cell Longev. 2020, 2020, 9187065. [Google Scholar] [CrossRef]
- Sreekumar, R.; Halvatsiotis, P.; Schimke, J.C.; Nair, K.S. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 2002, 51, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Wardelmann, K.; Blumel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 2019, 21, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Chung, K.; Lee, H.; Lee, K.; Lim, J.H.; Song, J. Downregulation of mitochondrial lon protease impairs mitochondrial function and causes hepatic insulin resistance in human liver SK-HEP-1 cells. Diabetologia 2011, 54, 1437–1446. [Google Scholar] [CrossRef]
- Wu, G.; Xiong, Q.; Wei, X.; Wang, Y.; Hu, X.; He, G.; Liu, L.; Lai, Q.; Dai, Z.; Anushesh, D.; et al. Mitochondrial unfolded protein response gene CLPP changes mitochondrial dynamics and affects mitochondrial function. PeerJ 2019, 7, e7209. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Lauritzen, H.P.; Ussar, S.; Christensen, J.H.; Mori, M.A.; Bross, P.; Kahn, C.R. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J. Clin. Investig. 2013, 123, 4667–4680. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Seo, J.H.; Agarwal, E.; Wang, Y.; Kossenkov, A.V.; Tang, H.Y.; Speicher, D.W.; Altieri, D.C. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene 2019, 38, 6926–6939. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s disease: From genetic variants to the distinct pathological mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Luk, J.M.; Lee, N.P.; Peng, J.; Leng, X.; Guan, X.Y.; Lau, G.K.; Beretta, L.; Fan, S.T. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol. Cell. Proteom. 2008, 7, 315–325. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Xiao, H.; Cai, Y.; Zhang, N. ATF5 and HIF1α cooperatively activate HIF1 signaling pathway in esophageal cancer. Cell Commun. Signal. 2021, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Qian, D.; Wang, B.; Hu, M.; Lu, J.; Qi, Y.; Liu, D.X. ATF5 is overexpressed in epithelial ovarian carcinomas and interference with its function increases apoptosis through the downregulation of Bcl-2 in SKOV-3 Cells. Int. J. Gynecol. Pathol. 2012, 31, 532–537. [Google Scholar] [CrossRef]
- Kim, W.; Ryu, J.; Kim, J.E. CCAR2/DBC1 and Hsp60 positively regulate expression of survivin in neuroblastoma cells. Int. J. Mol. Sci. 2019, 20, 131. [Google Scholar] [CrossRef]
- Luo, J.; Zeng, B.; Tao, C.; Lu, M.; Ren, G. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ 2020, 2020, e8754. [Google Scholar] [CrossRef]
- Tang, H.; Li, J.; Liu, X.; Wang, G.; Luo, M.; Deng, H. Down-regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/mTOR Pathway. Sci. Rep. 2016, 6, 28388. [Google Scholar] [CrossRef] [PubMed]
- Feldheim, J.; Kessler, A.F.; Schmitt, D.; Wilczek, L.; Linsenmann, T.; Dahlmann, M.; Monoranu, C.M.; Ernestus, R.I.; Hagemann, C.; Löhr, M. Expression of activating transcription factor 5 (ATF5) is increased in astrocytomas of different WHO grades and correlates with survival of glioblastoma patients. OncoTargets Ther. 2018, 11, 8673–8684. [Google Scholar] [CrossRef]
- Li, X.S.; Xu, Q.; Fu, X.Y.; Luo, W.S. Heat shock protein 60 overexpression is associated with the progression and prognosis in gastric cancer. PLoS ONE 2014, 9, e107507. [Google Scholar] [CrossRef]
- Kumar, S.; O’Malley, J.; Chaudhary, A.K.; Inigo, J.R.; Yadav, N.; Kumar, R.; Chandra, D. Hsp60 and IL-8 axis promotes apoptosis resistance in cancer. Br. J. Cancer 2019, 121, 934–943. [Google Scholar] [CrossRef]
- Guo, J.; Li, X.; Zhang, W.; Chen, Y.; Zhu, S.; Chen, L.; Xu, R.; Lv, Y.; Wu, D.; Guo, M.; et al. HSP60-regulated Mitochondrial Proteostasis and Protein Translation Promote Tumor Growth of Ovarian Cancer. Sci. Rep. 2019, 9, 6792. [Google Scholar] [CrossRef] [PubMed]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Tartaro, D.L.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.S.; Porter, J.W. Mechanism of fatty acid synthesis. Life Sci. 1977, 20, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y.L. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Austin, G.L.; Young, L.E.A.; Johnson, L.A.; Sun, R. Mitochondrial metabolism in major neurological diseases. Cells 2018, 7, 229. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef]
- Starenki, D.; Sosonkina, N.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) promotes survival and proliferation of thyroid carcinoma cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef]
- Sun, J.; Che, S.L.; Piao, J.J.; Xu, M.; Chen, L.Y.; Lin, Z.H. Mortalin overexpression predicts poor prognosis in early stage of non–small cell lung cancer. Tumor Biol. 2017, 39, 1010428317695918. [Google Scholar] [CrossRef]
- Liu, L.X.; Lu, J.C.; Zeng, H.Y.; Cai, J.B.; Zhang, P.F.; Guo, X.J.; Huang, X.Y.; Dong, R.Z.; Zhang, C.; Kang, Q.; et al. Mortalin stabilizes CD151-depedent tetraspanin-enriched microdomains and implicates in the progression of hepatocellular carcinoma. J. Cancer 2019, 10, 6199–6206. [Google Scholar] [CrossRef]
- Vocka, M.; Langer, D.; Fryba, V.; Petrtyl, J.; Hanus, T.; Kalousova, M.; Zima, T.; Petruzelka, L. Novel serum markers HSP60, CHI3L1, and IGFBP-2 in metastatic colorectal cancer. Oncol. Lett. 2019, 18, 6284–6292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, H.; Zheng, C.; Gao, J.; Fu, Q.; Hu, N.; Shao, X.; Zhou, Y.; Xiong, J.; Nie, K.; et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth article. Cell Death Dis. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, L.; Yang, Y.; Han, Y.; Wang, Y.; Liu, W.; Zuo, J. Oncogenic role of mortalin contributes to ovarian tumorigenesis by activating the MAPK–ERK pathway. J. Cell. Mol. Med. 2016, 20, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Meng, W.; Zhou, Z.; Li, Y.; Zhou, B.; Wang, R.; Zhan, L. Overexpression of activating transcription factor 5 in human rectal cancer. Exp. Ther. Med. 2011, 2, 827–831. [Google Scholar] [CrossRef]
- Hua, Z.Y.; Hansen, J.N.; He, M.; Dai, S.K.; Choi, Y.; Fulton, M.D.; Lloyd, S.M.; Szemes, M.; Sen, J.; Ding, H.F.; et al. PRMT1 promotes neuroblastoma cell survival through ATF5. Oncogenesis 2020, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.A.; Eidelman, A.S.; Ewer, J.C.; Ricca, J.M.; Serrano, A.; Tucker, K.C.; Vail, C.M.; Kurt, R.A. A role for HMGB1, HSP60 and Myd88 in growth of murine mammary carcinoma in vitro. Cell. Immunol. 2013, 282, 136–145. [Google Scholar] [CrossRef]
- Nukuda, A.; Endoh, H.; Yasuda, M.; Mizutani, T.; Kawabata, K.; Haga, H. Role of ATF5 in the invasive potential of diverse human cancer cell lines. Biochem. Biophys. Res. Commun. 2016, 474, 509–514. [Google Scholar] [CrossRef]
- Monaco, S.E.; Angelastro, J.M.; Szabolcs, M.; Greene, L.A. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int. J. Cancer 2007, 120, 1883–1890. [Google Scholar] [CrossRef]
- Greene, L.A.; Lee, H.Y.; Angelastro, J.M. The transcription factor ATF5: Role in neurodevelopment and neural tumors. J. Neurochem. 2009, 108, 11–22. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Yoon, A.R.; Wadhwa, R.; Kaul, S.C.; Yun, C.O. Why is Mortalin a Potential Therapeutic Target for Cancer? Front. Cell Dev. Biol. 2022, 10, 1219. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.R.; Chen, H.H.; Ko, C.H.; Chan, M.H. Neuroprotective activity of honokiol and magnolol in cerebellar granule cell damage. Eur. J. Pharmacol. 2006, 537, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.U.; Trumpff, C.; Yang, H.S.; Lee, A.J.; Picard, M.; Bennett, D.A.; De Jager, P.L. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol. Neurodegener. 2021, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Mancuso, M.; Siciliano, G.; Filosto, M.; Murri, L. Mitochondrial dysfunction and Alzheimer’s disease: New developments. J. Alzheimers Dis. 2006, 9, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.; Schiavi, A.; Ventura, N. Mitochondrial autophagy promotes healthy aging. Cell Cycle 2016, 15, 1805–1806. [Google Scholar] [CrossRef]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef]
- Hashimoto, T.; Horikawa, M.; Nomura, T.; Sakamoto, K. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology 2010, 11, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Ozkurede, U.; Miller, R.A. Improved mitochondrial stress response in long-lived Snell dwarf mice. Aging Cell 2019, 18, e13030. [Google Scholar] [CrossRef] [PubMed]
- Baqri, R.M.; Pietron, A.V.; Gokhale, R.H.; Turner, B.A.; Kaguni, L.S.; Shingleton, A.W.; Kunes, S.; Miller, K.E. Mitochondrial chaperone TRAP1 activates the mitochondrial UPR and extends healthspan in Drosophila. Mech. Ageing Dev. 2014, 141–142, 35–45. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef]
- Ohyagi, Y.; Yamada, T.; Nishioka, K.; Clarke, N.J.; Tomlinson, A.J.; Naylor, S.; Nakabeppu, Y.; Kira, J.; Younkin, S.G. Selective increase in cellular A beta 42 is related to apoptosis but not necrosis. Neuroreport 2000, 11, 167–171. [Google Scholar] [CrossRef]
- Busciglio, J.; Pelsman, A.; Wong, C.; Pigino, G.; Yuan, M.; Mori, H.; Yankner, B.A. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 2002, 33, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Delanty, N. Oxidative injury in diseases of the central nervous system: Focus on Alzheimer’s disease. Am. J. Med. 2000, 109, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev. 2005, 49, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S499–S512. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Regitz, C.; Fitzenberger, E.; Mahn, F.L.; Dussling, L.M.; Wenzel, U. Resveratrol reduces amyloid-beta (Abeta(1)(-)(4)(2))-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur. J. Nutr. 2016, 55, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2021, 26, 5733–5750. [Google Scholar] [CrossRef] [PubMed]
- Darreh-Shori, T.; Rezaeianyazdi, S.; Lana, E.; Mitra, S.; Gellerbring, A.; Karami, A.; Bogdanovic, N.; Lithner, C.U.; Winblad, B.; Behbahani, H. Increased Active OMI/HTRA2 Serine Protease Displays a Positive Correlation with Cholinergic Alterations in the Alzheimer’s Disease Brain. Mol. Neurobiol. 2019, 56, 4601–4619. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ding, M.; Xie, Z.; Liu, X.; Yang, H.; Jin, S.; Xu, S.; Zhu, Z.; Wang, Y.; Wang, D.; et al. Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Front. Cell Neurosci. 2019, 13, 568. [Google Scholar] [CrossRef]
- Romani, M.; Sorrentino, V.; Oh, C.M.; Li, H.; de Lima, T.I.; Zhang, H.; Shong, M.; Auwerx, J. NAD(+) boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep. 2021, 34, 108660. [Google Scholar] [CrossRef]
- Jagadeesan, A.J.; Murugesan, R.; Vimala Devi, S.; Meera, M.; Madhumala, G.; Vishwanathan Padmaja, M.; Ramesh, A.; Banerjee, A.; Sushmitha, S.; Khokhlov, A.N.; et al. Current trends in etiology, prognosis and therapeutic aspects of Parkinson’s disease: A review. Acta Biomed. 2017, 88, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Muller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The rotenone model of Parkinson’s disease: Genes, environment and mitochondria. Parkinsonism Relat. Disord. 2003, 9 (Suppl. S2), S59–S64. [Google Scholar] [CrossRef]
- Spivey, A. Rotenone and paraquat linked to Parkinson’s disease: Human exposure study supports years of animal studies. Environ. Health Perspect. 2011, 119, A259. [Google Scholar] [CrossRef]
- Vos, M. Mitochondrial Complex I deficiency: Guilty in Parkinson’s disease. Signal Transduct. Target. Ther. 2022, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Tieu, K.; Ischiropoulos, H.; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Pham, M.T.; Mitchell, M.B.; Yeom, S.G.; Jordan, S.; Chang, A.; Sopher, B.L.; La Spada, A.R. 4E-BP1 Protects Neurons from Misfolded Protein Stress and Parkinson’s Disease Toxicity by Inducing the Mitochondrial Unfolded Protein Response. J. Neurosci. 2020, 40, 8734–8745. [Google Scholar] [CrossRef]
- Liu, M.; Yu, S.; Wang, J.; Qiao, J.; Liu, Y.; Wang, S.; Zhao, Y. Ginseng protein protects against mitochondrial dysfunction and neurodegeneration by inducing mitochondrial unfolded protein response in Drosophila melanogaster PINK1 model of Parkinson’s disease. J. Ethnopharmacol. 2020, 247, 112213. [Google Scholar] [CrossRef]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Marcus, R. What Is Huntington Disease? JAMA 2023, 330, 1014. [Google Scholar] [CrossRef]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Milakovic, T.; Johnson, G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P. Effects of chronic MPTP and 3-nitropropionic acid in nonhuman primates. Curr. Opin. Neurol. 1995, 8, 469–473. [Google Scholar] [CrossRef]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Horton, T.M.; Graham, B.H.; Corral-Debrinski, M.; Shoffner, J.M.; Kaufman, A.E.; Beal, M.F.; Wallace, D.C. Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 1995, 45, 1879–1883. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Liu, F.; Liu, C.; Jin, B.; Jiang, Y.; Tang, M.; Qi, X.; Guo, X. Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1428–1435. [Google Scholar] [CrossRef]
- Hernandez, I.H.; Torres-Peraza, J.; Santos-Galindo, M.; Ramos-Moron, E.; Fernandez-Fernandez, M.R.; Perez-Alvarez, M.J.; Miranda-Vizuete, A.; Lucas, J.J. The neuroprotective transcription factor ATF5 is decreased and sequestered into polyglutamine inclusions in Huntington’s disease. Acta Neuropathol. 2017, 134, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.M.; Oliveira, A.; Oliveira, J.M.A.; Pinho, B.R. Stress response mechanisms in protein misfolding diseases: Profiling a cellular model of Huntington’s disease. Arch. Biochem. Biophys. 2023, 745, 109711. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Carmo, C.; Campesan, S.; Fao, L.; Cotton, V.E.; Valero, J.; Lopes, C.; Rosenstock, T.R.; Giorgini, F.; Rego, A.C. Mitochondrial SIRT3 confers neuroprotection in Huntington’s disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic. Biol. Med. 2021, 163, 163–179. [Google Scholar] [CrossRef]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laco, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative Mitochondrial-Based Protective Effects of Resveratrol and Nicotinamide in Huntington’s Disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Jin, J.; Cichewicz, R.H.; Hageman, S.A.; Ellis, T.K.; Xiang, L.; Peng, Q.; Jiang, M.; Arbez, N.; Hotaling, K.; et al. trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J. Biol. Chem. 2012, 287, 24460–24472. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Chia, R.; Chio, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e539. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1(G93A) Mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, E.A. Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex specific activation of the ERalpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet. 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikova, M.M.; Humphries, C.L.; Shinsky, E.M. Friedreich’s ataxia: New insights. Emerg. Top. Life Sci. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Stepanova, A.; Magrane, J. Mitochondrial dysfunction in neurons in Friedreich’s ataxia. Mol. Cell Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef]
- Lu, C.; Cortopassi, G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 2007, 457, 111–122. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).