
A Kinetic Transition Network Model Reveals the Diversity of Protein Dimer Formation Mechanisms


Abstract
:1. Introduction
2. Methods
2.1. Studied Structures
2.2. Wako–Saito–Muñoz–Eaton Model
2.3. Transition Network
2.4. Transition Path Theory
3. Results
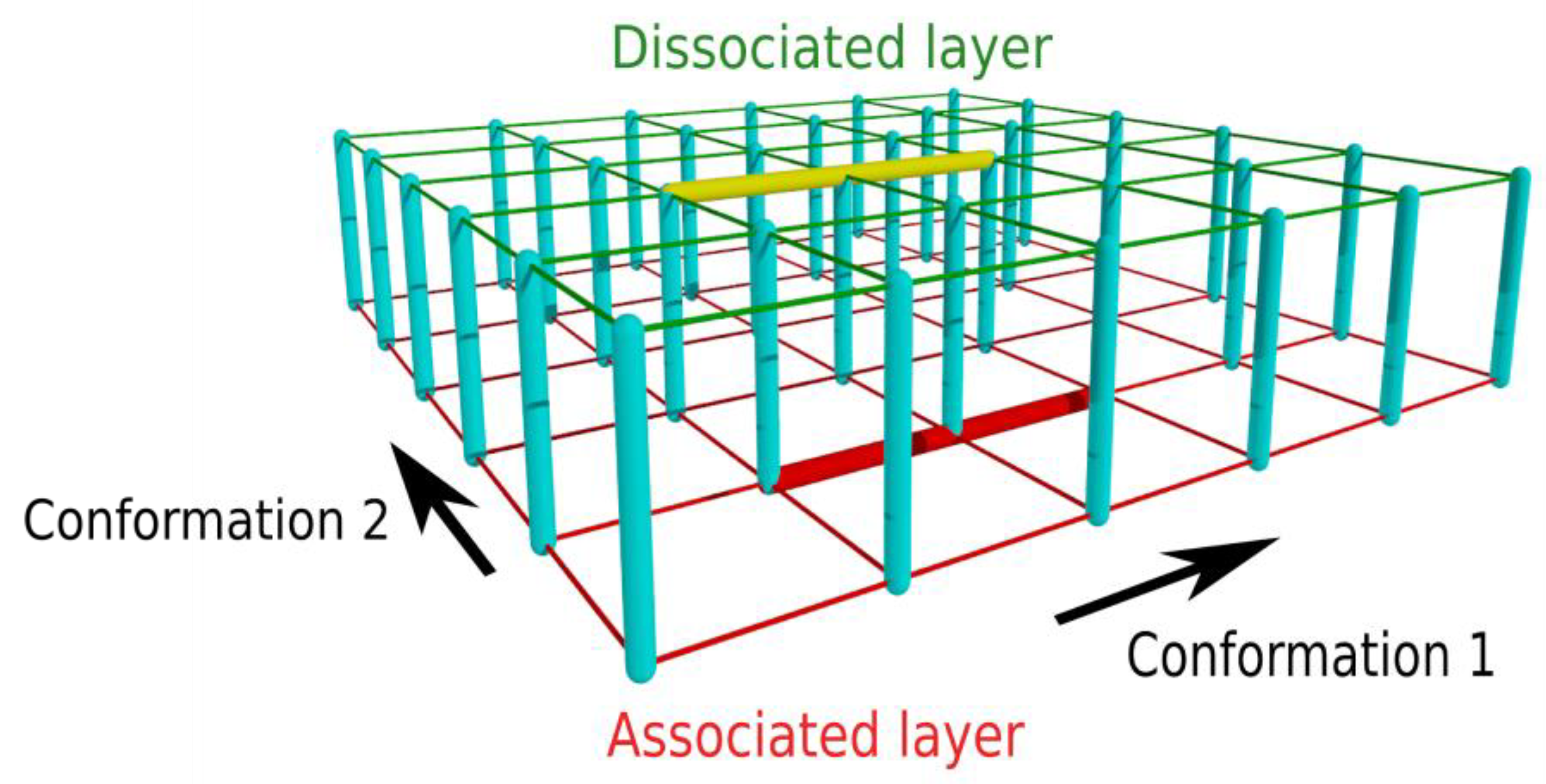
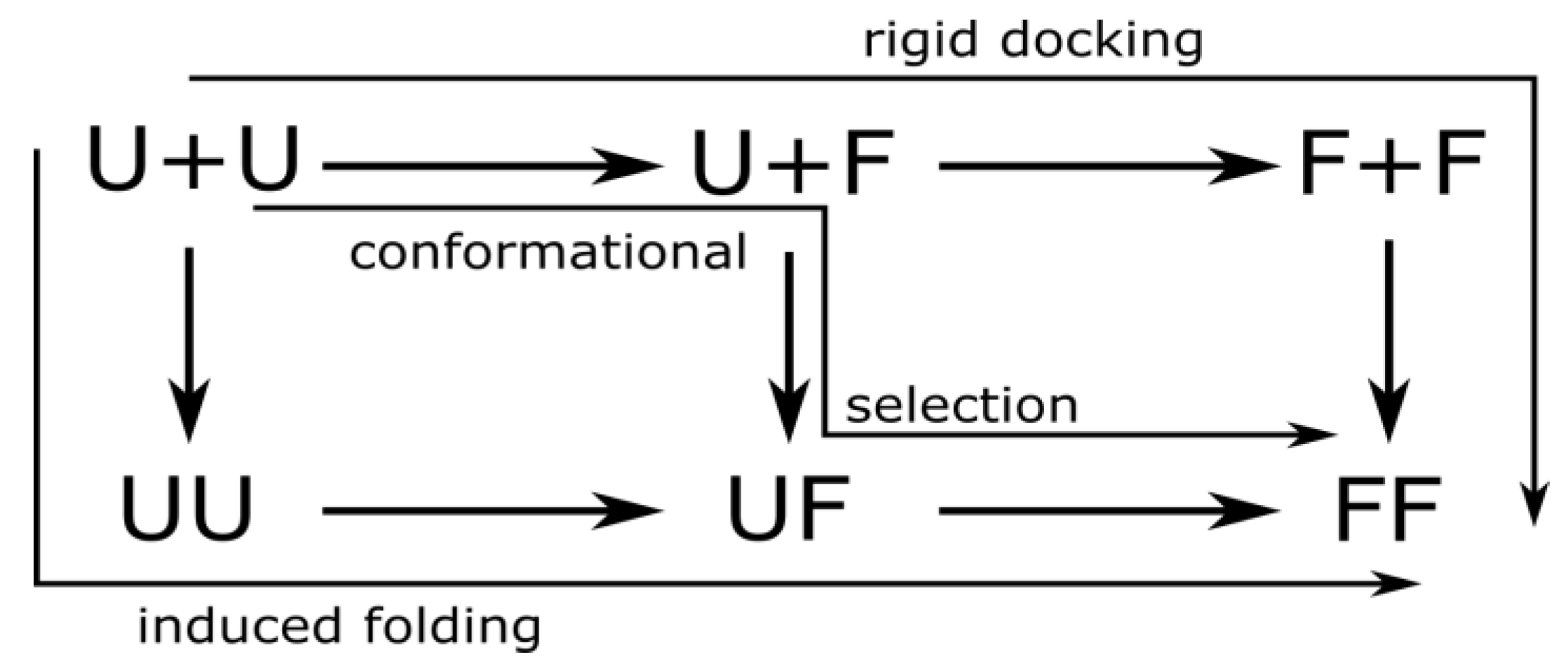
3.1. Two-Layer Network Model
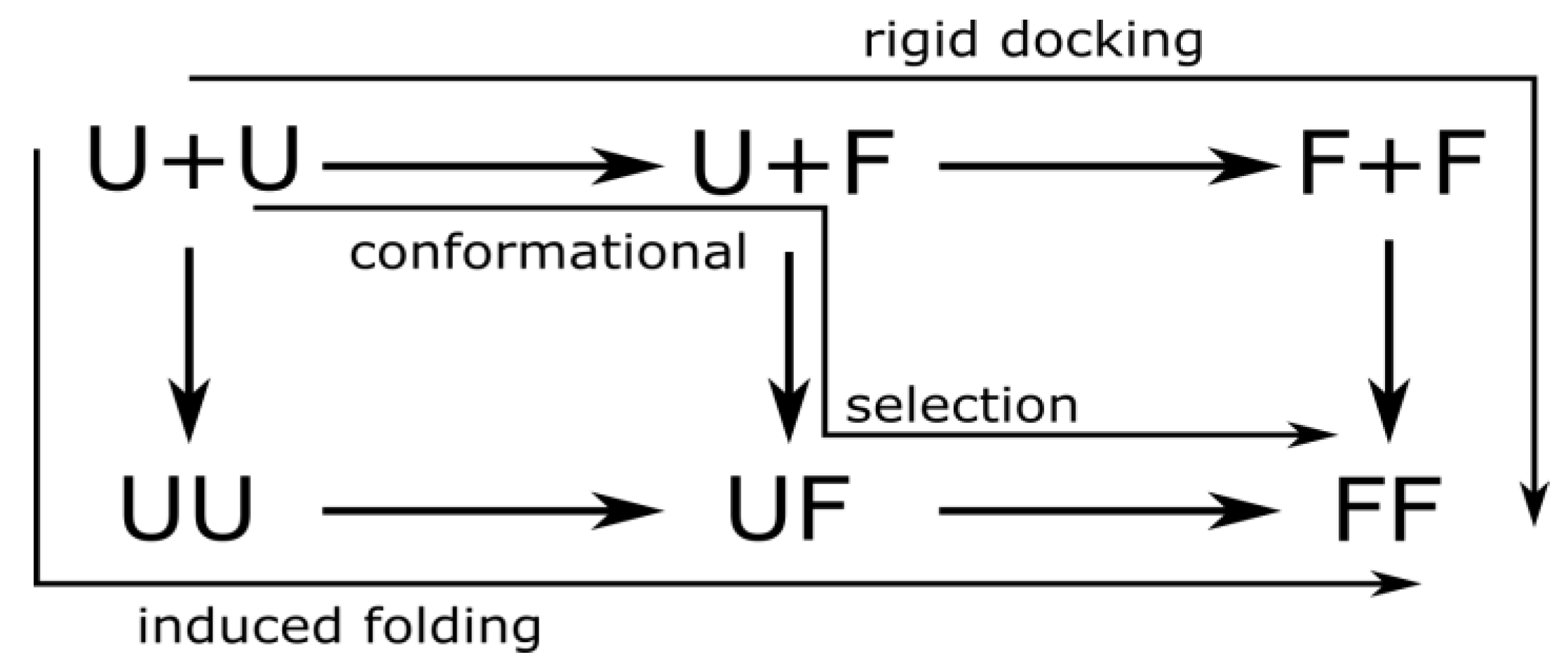
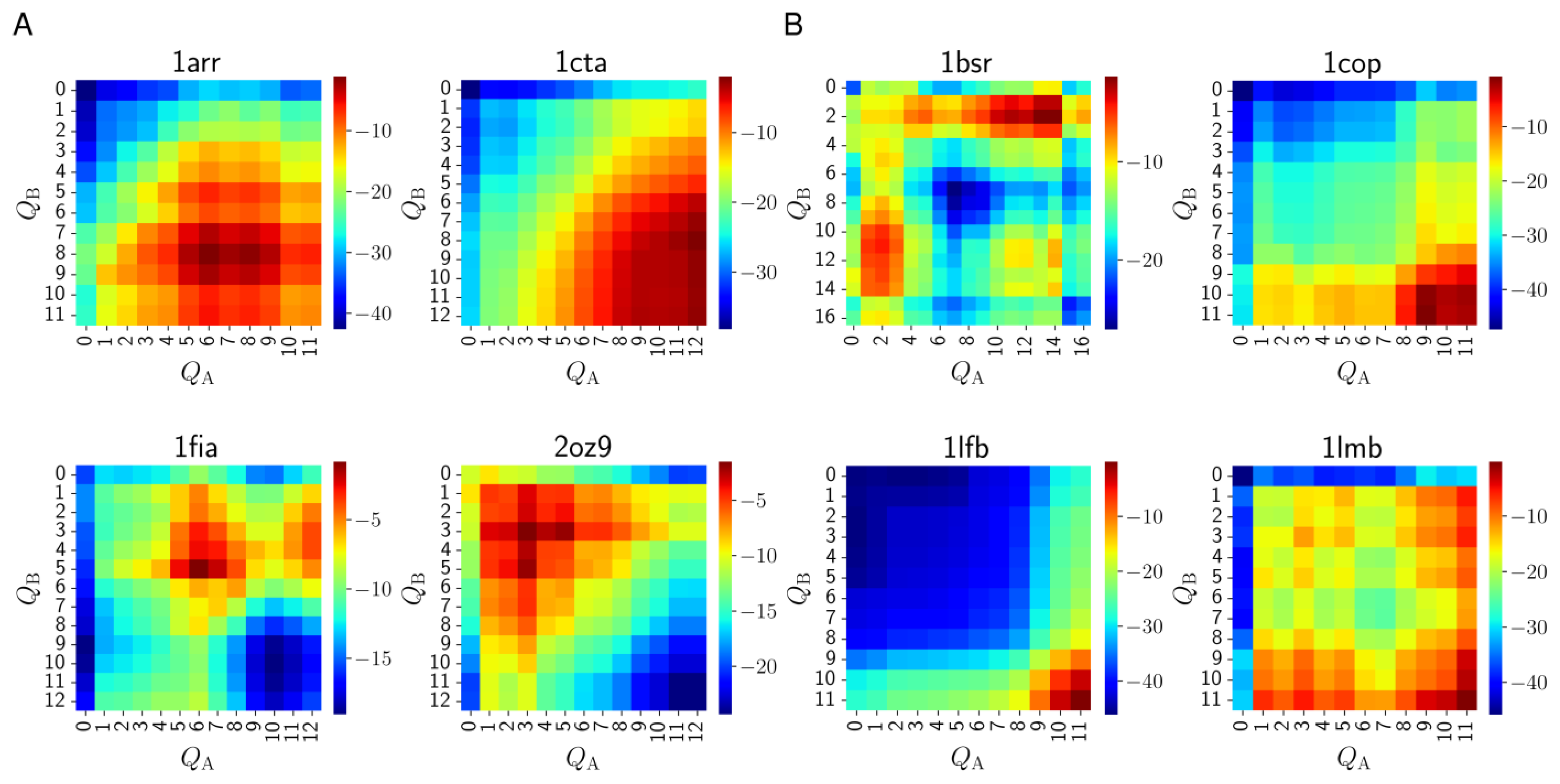
3.2. Mechanisms of Dimer Formation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
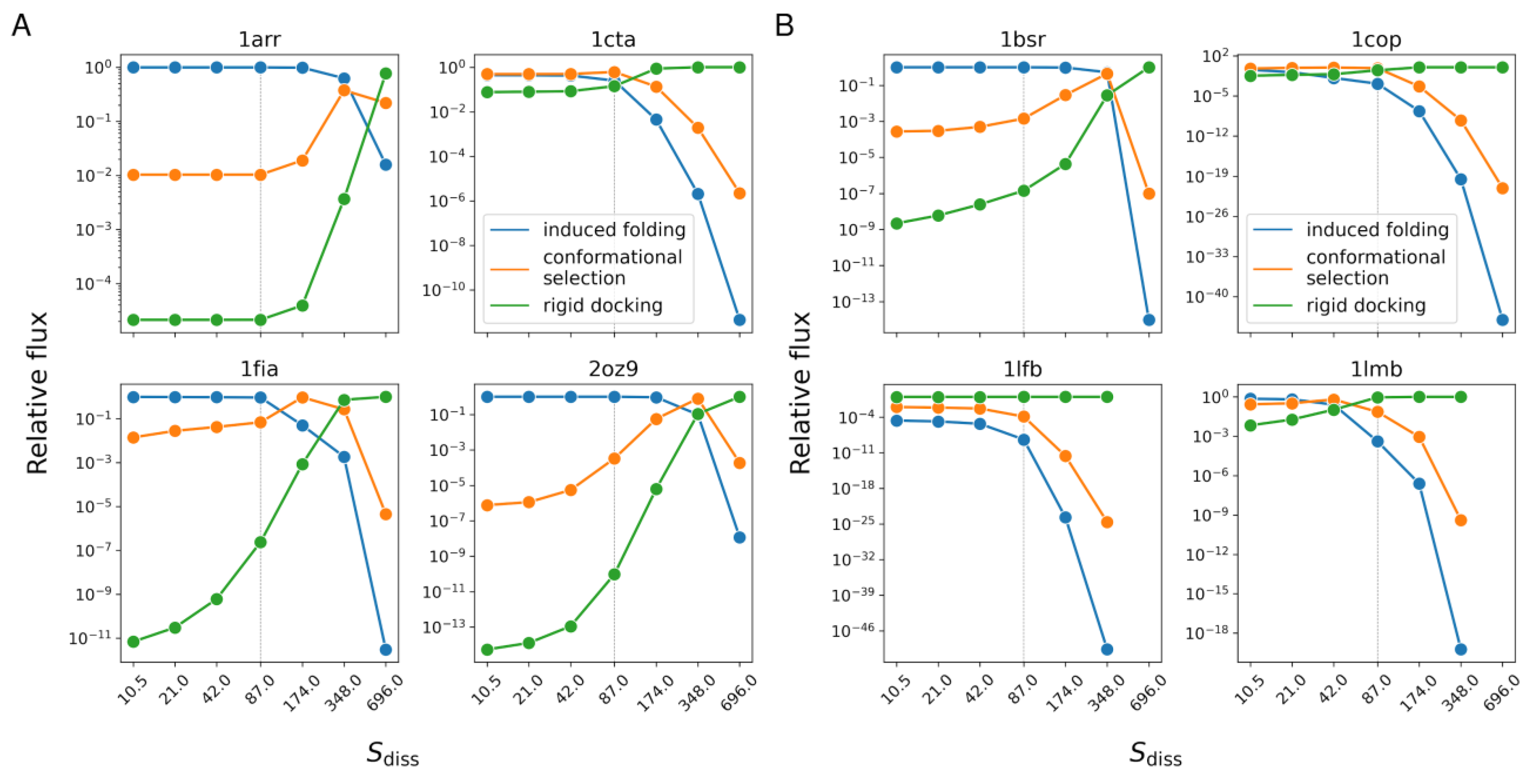
| Protein Name/PDB ID (States) | Induced Folding | Conformational Selection | Rigid Docking |
|---|---|---|---|
| Arc repressor/1arr (2) | 0.9897 | 1.029 × 10−2 | 2.100 × 10−5 |
| Troponin C site III/1cta (2) | 0.2489 | 0.6102 | 0.141 |
| Factor for inversion stimulation/1fia (2) | 0.9311 | 6.889 × 10−2 | 2.398 × 10−7 |
| Trp repressor/2oz9 (2) | 0.9997 | 3.313 × 10−4 | 9.611 × 10−11 |
| BS-RNase/1bsr (3) | 0.9986 | 1.424 × 10−3 | 1.429 × 10−7 |
| λ Cro repressor/1cop (3) | 1.376 × 10−3 | 0.6994 | 0.2993 |
| LFB1 transcription factor/1lfb (3) | 4.096 × 10−9 | 1.341 × 10−4 | 0.9999 |
| λ repressor/1lmb (3) | 4.080 × 10−4 | 7.295 × 10−2 | 0.9266 |
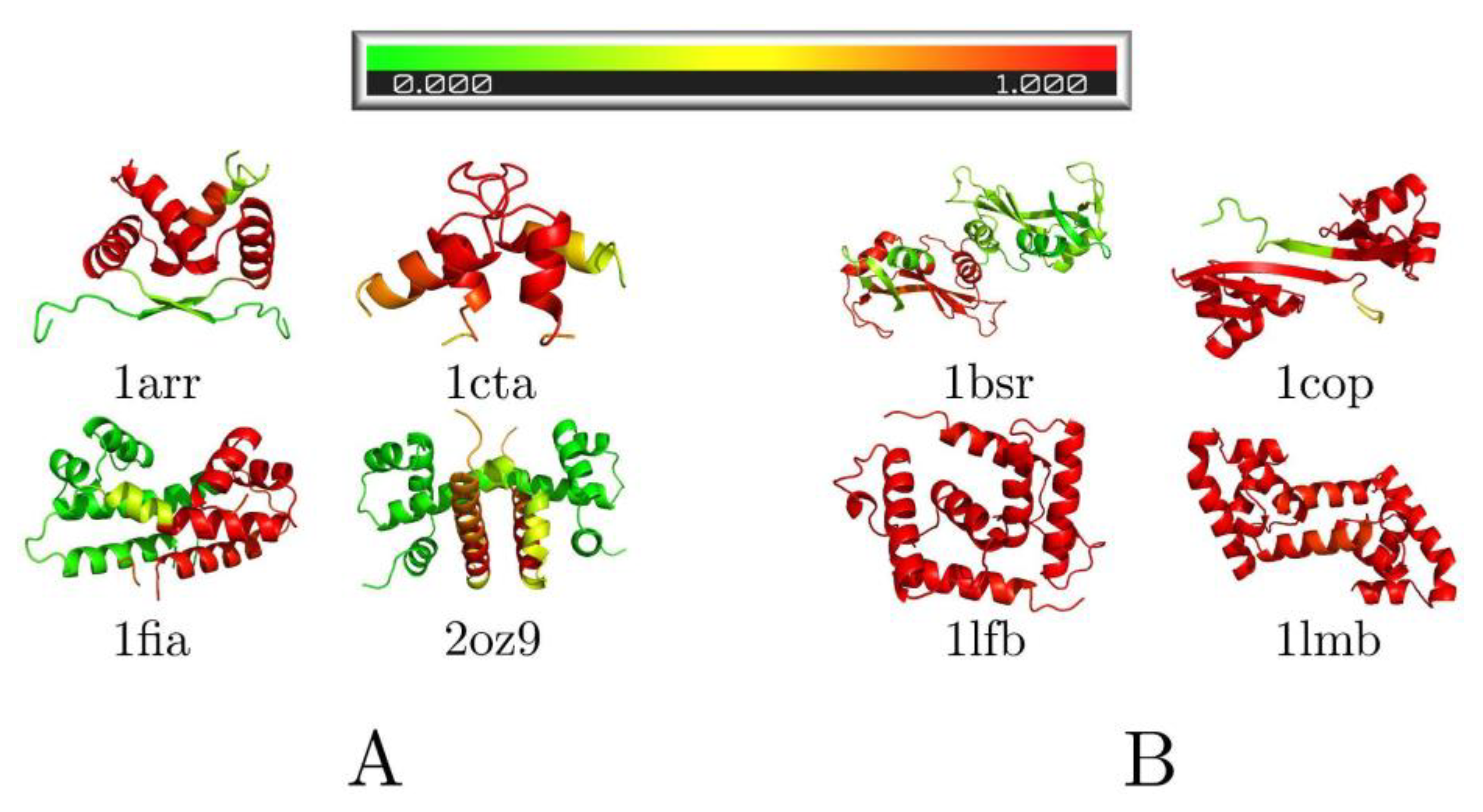
3.3. Folding Degree of Binding Chains
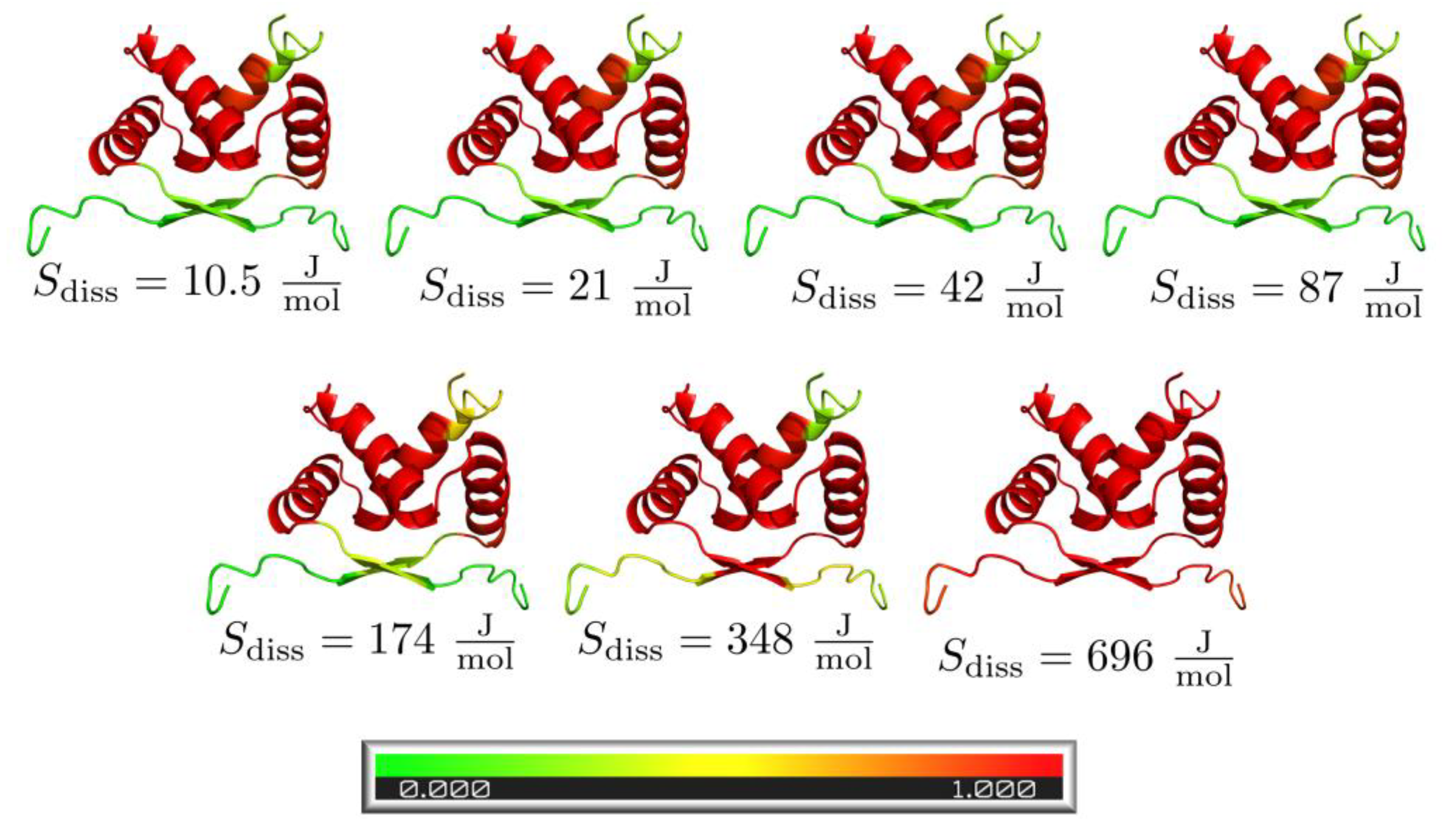
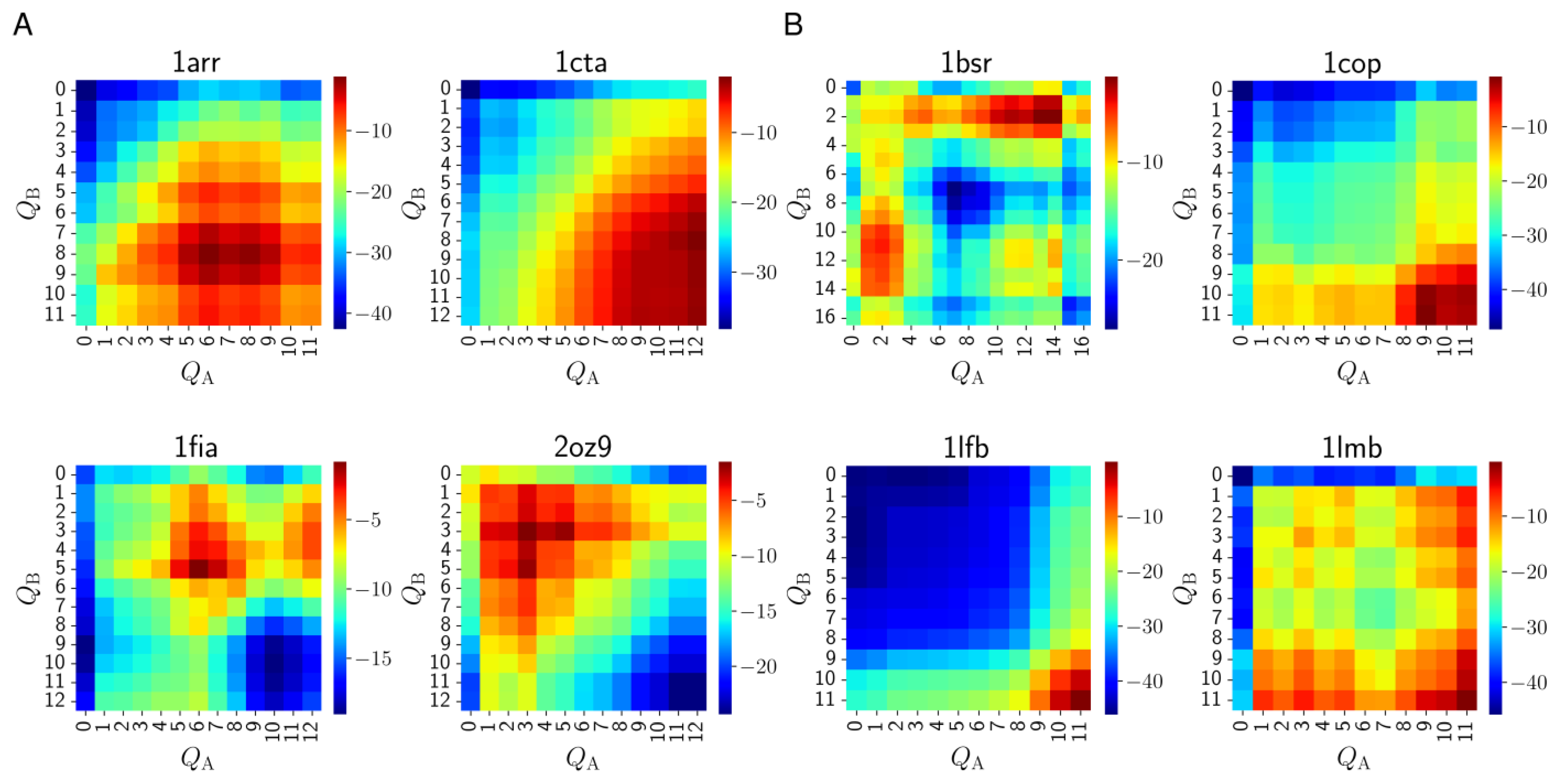
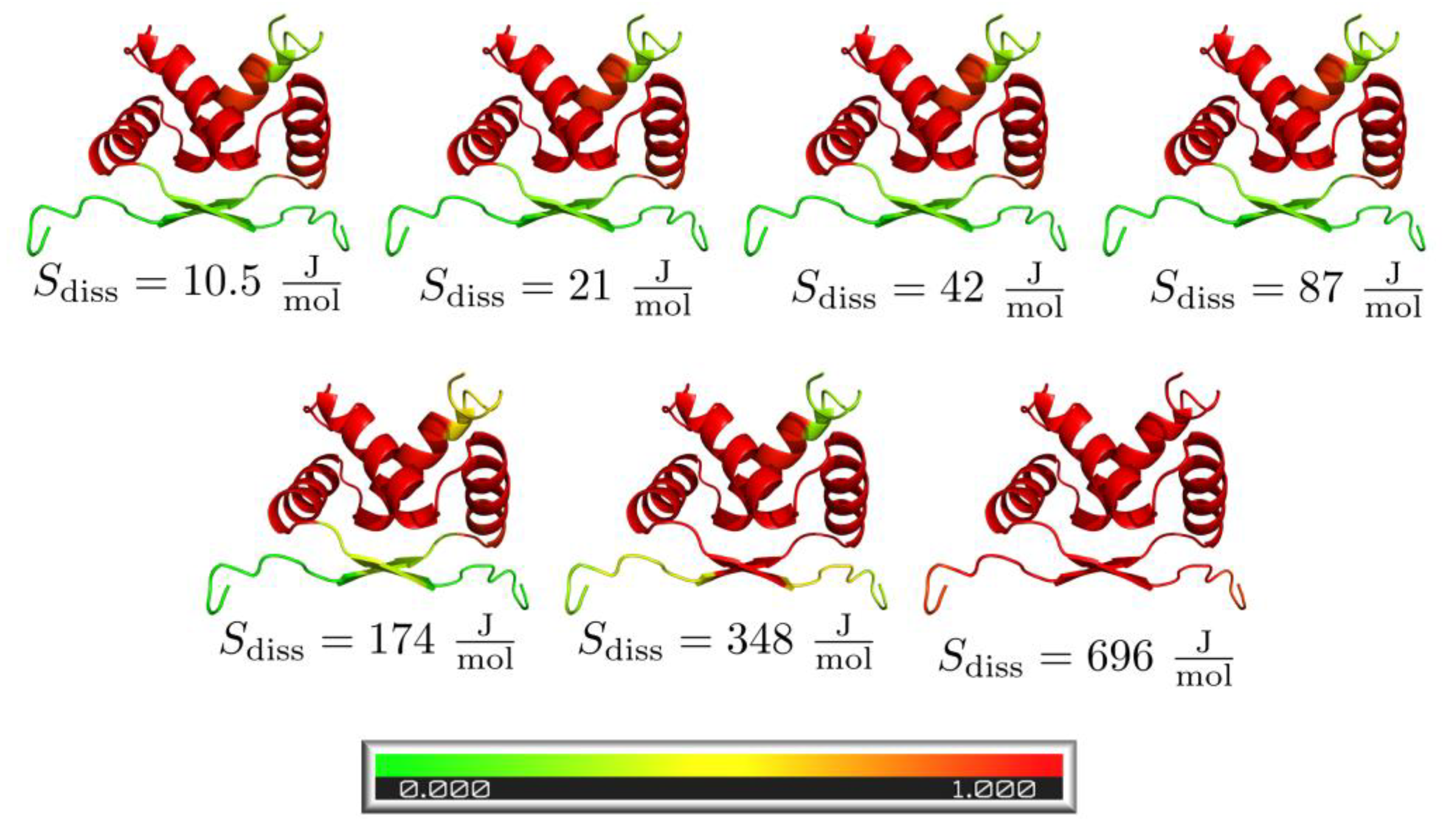
3.4. Pre-Folded Segments
3.5. Relative Weights of Mechanisms vs. Concentration
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Fischer, E. Einfluss der Konfiguration auf die Wirkung der Enzyme. Berichte Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Wolynes, P.G.; Onuchic, J.N. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Papoian, G.A.; Onuchic, J.N.; Wolynes, P.G. Energy Landscape Analysis of Protein Dimers. Isr. J. Chem. 2004, 44, 281–297. [Google Scholar] [CrossRef]
- Levy, Y.; Cho, S.S.; Onuchic, J.N.; Wolynes, P.G. A survey of flexible protein binding mechanisms and their transition states using native topology based energy landscapes. J. Mol. Biol. 2005, 346, 1121–1145. [Google Scholar] [CrossRef]
- Noé, F.; Fischer, S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008, 18, 154–162. [Google Scholar] [CrossRef]
- Voelz, V.A.; Bowman, G.R.; Beauchamp, K.; Pande, V.S. Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1-39). J. Am. Chem. Soc. 2010, 132, 1526–1528. [Google Scholar] [CrossRef]
- Schwantes, C.R.; Pande, V.S. Improvements in Markov State Model Construction Reveal Many Non-Native Interactions in the Folding of NTL9. J. Chem. Theory Comput. 2013, 9, 2000–2009. [Google Scholar] [CrossRef]
- Bowman, G.R.; Voelz, V.A.; Pande, V.S. Atomistic folding simulations of the five-helix bundle protein λ(6−85). J. Am. Chem. Soc. 2011, 133, 664–667. [Google Scholar] [CrossRef]
- Noé, F.; Schütte, C.; Vanden-Eijnden, E.; Reich, L.; Weikl, T.R. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc. Natl. Acad. Sci. USA 2009, 106, 19011–19016. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Kumar, S.; Tsai, C.J.; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng. 1999, 12, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.A.; Bowman, G.R.; Sosa-Peinado, A.; Huang, X. A role for both conformational selection and induced fit in ligand binding by the LAO protein. PLoS Comput. Biol. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Silva, D.A.; Meng, L.; Yue, A.; Huang, X. Quantitatively characterizing the ligand binding mechanisms of choline binding protein using Markov state model analysis. PLoS Comput. Biol. 2014, 10, e1003767. [Google Scholar] [CrossRef]
- Zhou, G.; Pantelopulos, G.A.; Mukherjee, S.; Voelz, V.A. Bridging Microscopic and Macroscopic Mechanisms of p53-MDM2 Binding with Kinetic Network Models. Biophys. J. 2017, 113, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.W.; Vishal, V.; Krafft, G.A.; Pande, V.S. Simulating oligomerization at experimental concentrations and long timescales: A Markov state model approach. J. Chem. Phys. 2008, 129, 214707. [Google Scholar] [CrossRef]
- Noé, F.; Wu, H.; Prinz, J.H.; Plattner, N. Projected and hidden Markov models for calculating kinetics and metastable states of complex molecules. J. Chem. Phys. 2013, 139, 184114. [Google Scholar] [CrossRef]
- Hilser, V.J.; Freire, E. Structure-based calculation of the equilibrium folding pathway of proteins. Correlation with hydrogen exchange protection factors. J. Mol. Biol. 1996, 262, 756–772. [Google Scholar] [CrossRef]
- Zamparo, M.; Pelizzola, A. Kinetics of the Wako-Saitô-Muñoz-Eaton model of protein folding. Phys. Rev. Lett. 2006, 97, 068106. [Google Scholar] [CrossRef]
- Muñoz, V.; Eaton, W.A. A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11311–11316. [Google Scholar] [CrossRef]
- Jacobs, D.J. Ensemble-based methods for describing protein dynamics. Curr. Opin. Pharmacol. 2010, 10, 760–769. [Google Scholar] [CrossRef]
- Ooka, K.; Liu, R.; Arai, M. The Wako-Saitô-Muñoz-Eaton Model for Predicting Protein Folding and Dynamics. Molecules 2022, 27, 4460. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Metzner, P.; Schütte, C.; Vanden-Eijnden, E. Transition Path Theory for Markov Jump Processes. Multiscale Model. Simul. 2009, 7, 1192–1219. [Google Scholar] [CrossRef]
- E, W.; Vanden-Eijnden, E. Transition-path theory and path-finding algorithms for the study of rare events. Annu. Rev. Phys. Chem. 2010, 61, 391–420. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Privalov, P.L. The entropy cost of protein association. J. Mol. Biol. 1997, 273, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Henry, E.R.; Eaton, W.A. Combinatorial modeling of protein folding kinetics: Free energy profiles and rates. Chem. Phys. 2004, 307, 163–185. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Györffy, D.; Závodszky, P.; Szilágyi, A. “Pull moves” for rectangular lattice polymer models are not fully reversible. IEEEACM Trans. Comput. Biol. Bioinform. 2012, 9, 1847–1849. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, J.N.; Wolynes, P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004, 14, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, R.; Tamburrini, M.; Piccialli, G.; Di Donato, A.; Parente, A.; D’Alessio, G. The dual-mode quaternary structure of seminal RNase. Proc. Natl. Acad. Sci. USA 1992, 89, 1870–1874. [Google Scholar] [CrossRef]
- Merlino, A.; Ercole, C.; Picone, D.; Pizzo, E.; Mazzarella, L.; Sica, F. The buried diversity of bovine seminal ribonuclease: Shape and cytotoxicity of the swapped non-covalent form of the enzyme. J. Mol. Biol. 2008, 376, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Burgering, M.J.; Hald, M.; Boelens, R.; Breg, J.N.; Kaptein, R. Hydrogen exchange studies of the Arc repressor: Evidence for a monomeric folding intermediate. Biopolymers 1995, 35, 217–226. [Google Scholar] [CrossRef]
- Peng, X.; Jonas, J.; Silva, J.L. Molten-globule conformation of Arc repressor monomers determined by high-pressure 1H NMR spectroscopy. Proc. Natl. Acad. Sci. USA 1993, 90, 1776–1780. [Google Scholar] [CrossRef]
- Hammes, G.G.; Chang, Y.-C.; Oas, T.G. Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 13737–13741. [Google Scholar] [CrossRef]
- Daniels, K.G.; Tonthat, N.K.; McClure, D.R.; Chang, Y.-C.; Liu, X.; Schumacher, M.A.; Fierke, C.A.; Schmidler, S.C.; Oas, T.G. Ligand concentration regulates the pathways of coupled protein folding and binding. J. Am. Chem. Soc. 2014, 136, 822–825. [Google Scholar] [CrossRef]
- Mori, Y.; Mizukami, T.; Segawa, S.; Roder, H.; Maki, K. Folding of Staphylococcal Nuclease Induced by Binding of Chemically Modified Substrate Analogues Sheds Light on Mechanisms of Coupled Folding/Binding Reactions. Biochemistry 2023, 62, 1670–1678. [Google Scholar] [CrossRef]
- Sen, S.; Udgaonkar, J.B. Binding-induced folding under unfolding conditions: Switching between induced fit and conformational selection mechanisms. J. Biol. Chem. 2019, 294, 16942–16952. [Google Scholar] [CrossRef]
- Cai, L.; Zhou, H.-X. Theory and simulation on the kinetics of protein-ligand binding coupled to conformational change. J. Chem. Phys. 2011, 134, 105101. [Google Scholar] [CrossRef]
- Greives, N.; Zhou, H.-X. Both protein dynamics and ligand concentration can shift the binding mechanism between conformational selection and induced fit. Proc. Natl. Acad. Sci. USA 2014, 111, 10197–10202. [Google Scholar] [CrossRef] [PubMed]
- Dogan, J.; Gianni, S.; Jemth, P. The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. PCCP 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed]
- Gianni, S.; Dogan, J.; Jemth, P. Coupled binding and folding of intrinsically disordered proteins: What can we learn from kinetics? Curr. Opin. Struct. Biol. 2016, 36, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Gianni, S.; Dogan, J.; Jemth, P. Distinguishing induced fit from conformational selection. Biophys. Chem. 2014, 189, 33–39. [Google Scholar] [CrossRef]
- Lau, K.F.; Dill, K.A. A lattice statistical mechanics model of the conformational and sequence spaces of proteins. Macromolecules 1989, 22, 3986–3997. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Györffy, D.; Závodszky, P.; Szilágyi, A. A Kinetic Transition Network Model Reveals the Diversity of Protein Dimer Formation Mechanisms. Biomolecules 2023, 13, 1708. https://doi.org/10.3390/biom13121708
Györffy D, Závodszky P, Szilágyi A. A Kinetic Transition Network Model Reveals the Diversity of Protein Dimer Formation Mechanisms. Biomolecules. 2023; 13(12):1708. https://doi.org/10.3390/biom13121708
Chicago/Turabian StyleGyörffy, Dániel, Péter Závodszky, and András Szilágyi. 2023. "A Kinetic Transition Network Model Reveals the Diversity of Protein Dimer Formation Mechanisms" Biomolecules 13, no. 12: 1708. https://doi.org/10.3390/biom13121708
APA StyleGyörffy, D., Závodszky, P., & Szilágyi, A. (2023). A Kinetic Transition Network Model Reveals the Diversity of Protein Dimer Formation Mechanisms. Biomolecules, 13(12), 1708. https://doi.org/10.3390/biom13121708