
Engagement of Mesenchymal Stromal Cells in the Remodeling of the Bone Marrow Microenvironment in Hematological Cancers

 , ,
, ,  ,
,  ,
,  and
and {kind=link}
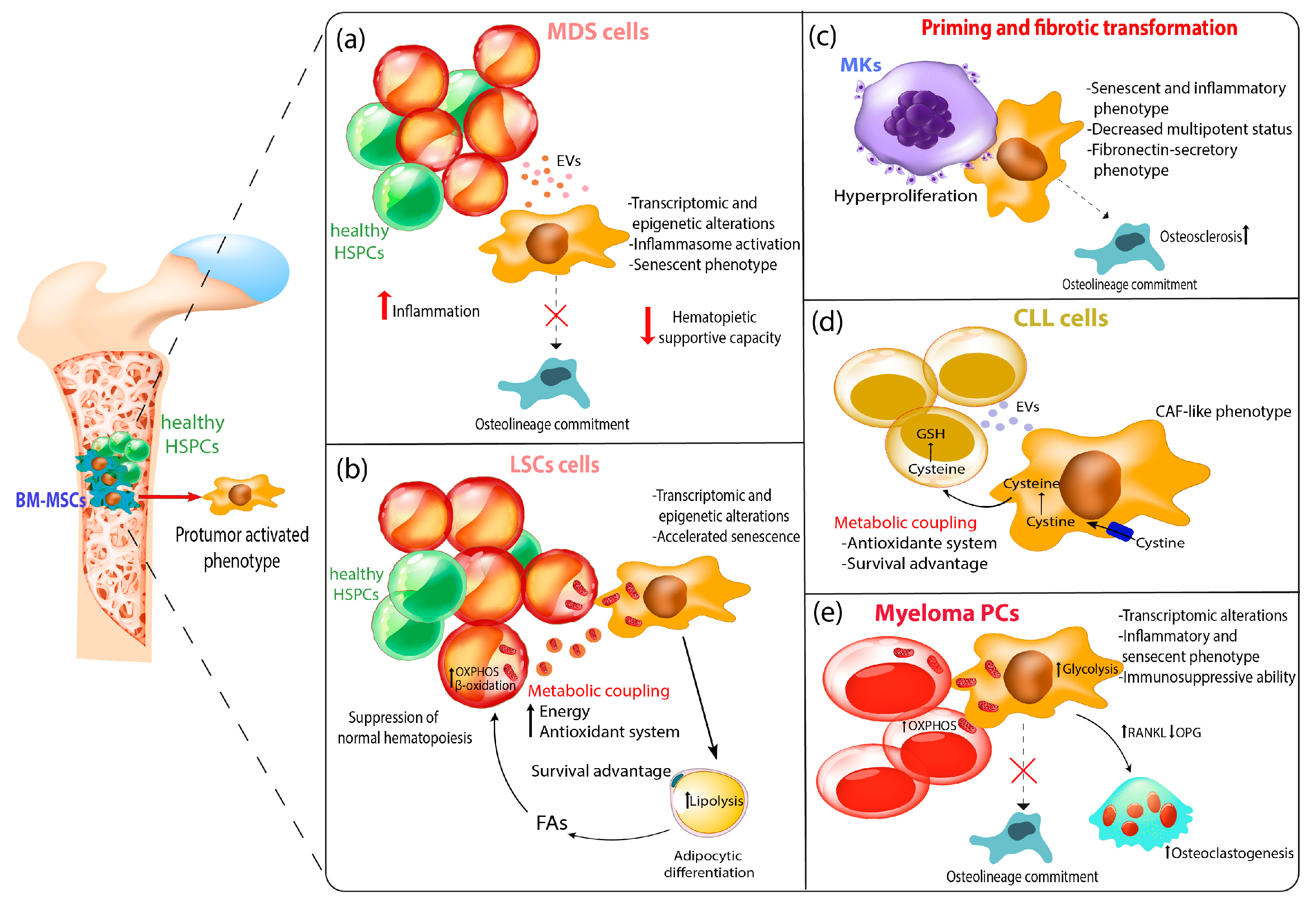{kind=link}
Abstract
1. Introduction
2. Role of MSCs in Hematological Cancers
2.1. Role of MSCs in Myelodysplastic Syndromes
2.2. Role of MSCs in Acute Leukemia
2.3. Role of MSCs in Myeloproliferative Neoplasms
2.4. Role of MSCs in in Chronic Lymphocytic Leukemia
2.5. Role of MSCs in in Multiple Myeloma
3. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro Gomes, A.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.C.; Robin, C.; Dzierzak, E. Mesenchymal progenitor cells localize within hematopoietic sites throughout ontogeny. Development 2005, 132, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Fallati, A.; Di Marzo, N.; D’Amico, G.; Dander, E. Mesenchymal Stromal Cells (MSCs): An Ally of B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells in Disease Maintenance and Progression within the Bone Marrow Hematopoietic Niche. Cancers 2022, 14, 3303. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Arai, F.; Iwasaki, H.; Hosokawa, K.; Kobayashi, I.; Gomei, Y.; Matsumoto, Y.; Yoshihara, H.; Suda, T. Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood 2010, 116, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Tormin, A.; Li, O.; Brune, J.C.; Walsh, S.; Schutz, B.; Ehinger, M.; Ditzel, N.; Kassem, M.; Scheding, S. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 2011, 117, 5067–5077. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef][Green Version]
- Bowers, M.; Zhang, B.; Ho, Y.; Agarwal, P.; Chen, C.C.; Bhatia, R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015, 125, 2678–2688. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Fei, C.; Guo, J.; Gu, S.; Zhu, Y.; Xu, F.; Zhang, Z.; Wu, L.; Li, X.; et al. Down-regulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica 2015, 100, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Aithal, A.P.; Bairy, L.K.; Seetharam, R.N. Safety and therapeutic potential of human bone marrow-derived mesenchymal stromal cells in regenerative medicine. Stem Cell Investig. 2021, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aparicio, P.F.; Vernot, J.P. Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities. J. Pers. Med. 2022, 12, 716. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The bone-marrow niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef]
- Weng, Z.; Wang, Y.; Ouchi, T.; Liu, H.; Qiao, X.; Wu, C.; Zhao, Z.; Li, L.; Li, B. Mesenchymal Stem/Stromal Cell Senescence: Hallmarks, Mechanisms, and Combating Strategies. Stem Cells Transl. Med. 2022, 11, 356–371. [Google Scholar] [CrossRef]
- Hu, D.; Yuan, S.; Zhong, J.; Liu, Z.; Wang, Y.; Liu, L.; Li, J.; Wen, F.; Liu, J.; Zhang, J. Cellular senescence and hematological malignancies: From pathogenesis to therapeutics. Pharmacol. Ther. 2021, 223, 107817. [Google Scholar] [CrossRef]
- Chen, X.; Li, N.; Weng, J.; Du, X. Senescent Mesenchymal Stem Cells in Myelodysplastic Syndrome: Functional Alterations, Molecular Mechanisms, and Therapeutic Strategies. Front. Cell Dev. Biol. 2020, 8, 617466. [Google Scholar] [CrossRef]
- Vanegas, N.P.; Ruiz-Aparicio, P.F.; Uribe, G.I.; Linares-Ballesteros, A.; Vernot, J.P. Leukemia-Induced Cellular Senescence and Stemness Alterations in Mesenchymal Stem Cells Are Reversible upon Withdrawal of B-Cell Acute Lymphoblastic Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 8166. [Google Scholar] [CrossRef]
- O’Hagan-Wong, K.; Nadeau, S.; Carrier-Leclerc, A.; Apablaza, F.; Hamdy, R.; Shum-Tim, D.; Rodier, F.; Colmegna, I. Increased IL-6 secretion by aged human mesenchymal stromal cells disrupts hematopoietic stem and progenitor cells’ homeostasis. Oncotarget 2016, 7, 13285–13296. [Google Scholar] [CrossRef]
- Adams, P.D.; Jasper, H.; Rudolph, K.L. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell 2015, 16, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Turkmen, S.; Benlasfer, O.; Schumann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chi, Y.; Wei, Y.; Zhang, W.; Wang, F.; Zhang, L.; Zou, L.; Song, B.; Zhao, X.; Han, Z. Bone marrow-derived mesenchymal stem/stromal cells in patients with acute myeloid leukemia reveal transcriptome alterations and deficiency in cellular vitality. Stem Cell Res. Ther. 2021, 12, 365. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Sui, B.D.; Zheng, C.X.; Li, M.; Jin, Y.; Hu, C.H. Epigenetic Regulation of Mesenchymal Stem Cell Homeostasis. Trends Cell Biol. 2020, 30, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, T.D.; Chen, S.; Bartenstein, M.; Barlowe, A.T.; Von Ahrens, D.; Choudhary, G.S.; Tivnan, P.; Amin, E.; Marcondes, A.M.; Sanders, M.A.; et al. Epigenetically Aberrant Stroma in MDS Propagates Disease via Wnt/beta-Catenin Activation. Cancer Res. 2017, 77, 4846–4857. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, A.; Li, T.; de la Calle-Fabregat, C.; Rodriguez-Ubreva, J.; Ciudad, L.; Catala-Moll, F.; Godoy-Tena, G.; Martin-Sanchez, M.; San-Segundo, L.; Muntion, S.; et al. Targeting aberrant DNA methylation in mesenchymal stromal cells as a treatment for myeloma bone disease. Nat. Commun. 2021, 12, 421. [Google Scholar] [CrossRef]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Correction: Bone marrow MSCs in MDS: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2019, 33, 1542. [Google Scholar] [CrossRef]
- Huang, J.; Liu, Z.; Sun, Y.; Zhong, Q.; Xu, L.; Ou, R.; Li, C.; Chen, R.; Yao, M.; Zhang, Q.; et al. Use of methylation profiling to identify significant differentially methylated genes in bone marrow mesenchymal stromal cells from acute myeloid leukemia. Int. J. Mol. Med. 2018, 41, 679–686. [Google Scholar] [CrossRef]
- Frassanito, M.A.; De Veirman, K.; Desantis, V.; Di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I.; et al. Halting pro-survival autophagy by TGFbeta inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016, 30, 640–648. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhang, J.; Wang, H.; Lu, W.; Liu, S.; Yu, Y.; Weng, W.; Ding, Z.; Zhu, Q.; Shi, J. Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J. Exp. Clin. Cancer Res. 2016, 35, 147. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Zi, F.M.; He, J.S.; Li, Y.; Wu, C.; Wu, W.J.; Yang, Y.; Wang, L.J.; He, D.H.; Yang, L.; Zhao, Y.; et al. Fibroblast activation protein protects bortezomib-induced apoptosis in multiple myeloma cells through beta-catenin signaling pathway. Cancer Biol. Ther. 2014, 15, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.W.; Shi, J.; Chen, J.; Wang, B.; Yu, Y.H.; Qin, X.; Zhou, X.C.; Cai, Y.J.; Li, Z.Q.; Zhang, F.; et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 2014, 25, 778–793. [Google Scholar] [CrossRef]
- Pan, C.; Fang, Q.; Liu, P.; Ma, D.; Cao, S.; Zhang, L.; Chen, Q.; Hu, T.; Wang, J. Mesenchymal Stem Cells With Cancer-Associated Fibroblast-Like Phenotype Stimulate SDF-1/CXCR4 Axis to Enhance the Growth and Invasion of B-Cell Acute Lymphoblastic Leukemia Cells Through Cell-to-Cell Communication. Front. Cell Dev. Biol. 2021, 9, 708513. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Oda, T.; Mori, N.; Kida, Y.S. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio 2020, 10, 2268–2281. [Google Scholar] [CrossRef]
- Rubinstein-Achiasaf, L.; Morein, D.; Ben-Yaakov, H.; Liubomirski, Y.; Meshel, T.; Elbaz, E.; Dorot, O.; Pichinuk, E.; Gershovits, M.; Weil, M.; et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers 2021, 13, 1472. [Google Scholar] [CrossRef]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef]
- Longhitano, L.; Li Volti, G.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G.A. The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; De Las Rivas, J.; Ocio, E.M.; Diaz-Rodriguez, E.; Montero, J.C.; Martin, M.; Blanco, J.F.; Sanchez-Guijo, F.M.; Pandiella, A.; San Miguel, J.F.; et al. Transcriptomic profile induced in bone marrow mesenchymal stromal cells after interaction with multiple myeloma cells: Implications in myeloma progression and myeloma bone disease. Oncotarget 2014, 5, 8284–8305. [Google Scholar] [CrossRef]
- Giallongo, C.; Romano, A.; Parrinello, N.L.; La Cava, P.; Brundo, M.V.; Bramanti, V.; Stagno, F.; Vigneri, P.; Chiarenza, A.; Palumbo, G.A.; et al. Mesenchymal Stem Cells (MSC) Regulate Activation of Granulocyte-Like Myeloid Derived Suppressor Cells (G-MDSC) in Chronic Myeloid Leukemia Patients. PLoS ONE 2016, 11, e0158392. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Sui, K.; Tang, J.; Yu, H.; Yang, C.; Zhang, H.; Li, S.C.; Zhong, J.F.; Wang, Z.; Zhang, X. BCR-ABL1-driven exosome-miR130b-3p-mediated gap-junction Cx43 MSC intercellular communications imply therapies of leukemic subclonal evolution. Theranostics 2023, 13, 3943–3963. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.I.; Minskaia, E.; Fernandes-Platzgummer, A.; Vieira, A.I.S.; da Silva, C.L.; Cabral, J.M.S.; Lacerda, J.F. Mesenchymal stromal cells induce regulatory T cells via epigenetic conversion of human conventional CD4 T cells in vitro. Stem Cells 2020, 38, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, L.; Guo, Y.; Xu, X.; Liu, Z.; Yan, Z.; Fu, R. The immunological role of mesenchymal stromal cells in patients with myelodysplastic syndrome. Front. Immunol. 2022, 13, 1078421. [Google Scholar] [CrossRef] [PubMed]
- Holthof, L.C.; van der Schans, J.J.; Katsarou, A.; Poels, R.; Gelderloos, A.T.; Drent, E.; van Hal-van Veen, S.E.; Li, F.; Zweegman, S.; van de Donk, N.; et al. Bone Marrow Mesenchymal Stromal Cells Can Render Multiple Myeloma Cells Resistant to Cytotoxic Machinery of CAR T Cells through Inhibition of Apoptosis. Clin. Cancer Res. 2021, 27, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Tibullo, D.; Camiolo, G.; Parrinello, N.L.; Romano, A.; Puglisi, F.; Barbato, A.; Conticello, C.; Lupo, G.; Anfuso, C.D.; et al. TLR4 signaling drives mesenchymal stromal cells commitment to promote tumor microenvironment transformation in multiple myeloma. Cell Death Dis. 2019, 10, 704, Erratum in Cell Death Dis. 2019, 10, 820. [Google Scholar] [CrossRef]
- Li, W.; Ren, G.; Huang, Y.; Su, J.; Han, Y.; Li, J.; Chen, X.; Cao, K.; Chen, Q.; Shou, P.; et al. Mesenchymal stem cells: A double-edged sword in regulating immune responses. Cell Death Differ. 2012, 19, 1505–1513. [Google Scholar] [CrossRef]
- Cao, Y.J.; Zheng, Y.H.; Li, Q.; Zheng, J.; Ma, L.T.; Zhao, C.J.; Li, T. MSC Senescence-Related Genes Are Associated with Myeloma Prognosis and Lipid Metabolism-Mediated Resistance to Proteasome Inhibitors. J. Oncol. 2022, 2022, 4705654. [Google Scholar] [CrossRef]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Xu, M.; Deng, Z.; Zhao, Y.; Yang, M.; Liu, Y.; Yuan, R.; Sun, Y.; Zhang, H.; et al. Regulation of Inflammatory Cytokine Storms by Mesenchymal Stem Cells. Front. Immunol. 2021, 12, 726909. [Google Scholar] [CrossRef] [PubMed]
- Loussouarn, C.; Pers, Y.M.; Bony, C.; Jorgensen, C.; Noel, D. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Regulate the Mitochondrial Metabolism via Transfer of miRNAs. Front. Immunol. 2021, 12, 623973. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.A.; Fahey, M.J.; Pugliese, B.R.; Irwin, R.M.; Antonyak, M.A.; Delco, M.L. Human mesenchymal stromal cells release functional mitochondria in extracellular vesicles. Front. Bioeng. Biotechnol. 2022, 10, 870193. [Google Scholar] [CrossRef] [PubMed]
- Phetfong, J.; Tawonsawatruk, T.; Kamprom, W.; Ontong, P.; Tanyong, D.; Borwornpinyo, S.; Supokawej, A. Bone marrow-mesenchymal stem cell-derived extracellular vesicles affect proliferation and apoptosis of leukemia cells in vitro. FEBS Open Bio 2022, 12, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Dulcamare, I.; Tibullo, D.; Del Fabro, V.; Vicario, N.; Parrinello, N.; Romano, A.; Scandura, G.; Lazzarino, G.; Conticello, C.; et al. CXCL12/CXCR4 axis supports mitochondrial trafficking in tumor myeloma microenvironment. Oncogenesis 2022, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Vilaplana-Lopera, N.; Cuminetti, V.; Almaghrabi, R.; Papatzikas, G.; Rout, A.K.; Jeeves, M.; Gonzalez, E.; Alyahyawi, Y.; Cunningham, A.; Erdem, A.; et al. Crosstalk between AML and stromal cells triggers acetate secretion through the metabolic rewiring of stromal cells. eLife 2022, 11, e75908. [Google Scholar] [CrossRef] [PubMed]
- Matamala Montoya, M.; van Slobbe, G.J.J.; Chang, J.C.; Zaal, E.A.; Berkers, C.R. Metabolic changes underlying drug resistance in the multiple myeloma tumor microenvironment. Front. Oncol. 2023, 13, 1155621. [Google Scholar] [CrossRef]
- Chiu, M.; Toscani, D.; Marchica, V.; Taurino, G.; Costa, F.; Bianchi, M.G.; Andreoli, R.; Franceschi, V.; Storti, P.; Burroughs-Garcia, J.; et al. Myeloma Cells Deplete Bone Marrow Glutamine and Inhibit Osteoblast Differentiation Limiting Asparagine Availability. Cancers 2020, 12, 3267. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Puglisi, F.; Barbato, A.; Vicario, N.; Cambria, D.; Parrinello, N.L.; Romano, A.; Conticello, C.; Forte, S.; et al. Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers 2020, 12, 1999. [Google Scholar] [CrossRef]
- Nemkov, T.; D’Alessandro, A.; Reisz, J.A. Metabolic underpinnings of leukemia pathology and treatment. Cancer Rep. 2019, 2, e1139. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Dabbah, M.; Jarchowsky-Dolberg, O.; Attar-Schneider, O.; Tartakover Matalon, S.; Pasmanik-Chor, M.; Drucker, L.; Lishner, M. Multiple myeloma BM-MSCs increase the tumorigenicity of MM cells via transfer of VLA4-enriched microvesicles. Carcinogenesis 2020, 41, 100–110. [Google Scholar] [CrossRef]
- Dabbah, M.; Attar-Schneider, O.; Tartakover Matalon, S.; Shefler, I.; Jarchwsky Dolberg, O.; Lishner, M.; Drucker, L. Microvesicles derived from normal and multiple myeloma bone marrow mesenchymal stem cells differentially modulate myeloma cells’ phenotype and translation initiation. Carcinogenesis 2017, 38, 708–716. [Google Scholar] [CrossRef]
- Dotson, J.L.; Lebowicz, Y. Myelodysplastic Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Stahl, M.; Bewersdorf, J.P.; Xie, Z.; Porta, M.G.D.; Komrokji, R.; Xu, M.L.; Abdel-Wahab, O.; Taylor, J.; Steensma, D.P.; Starczynowski, D.T.; et al. Classification, risk stratification and response assessment in myelodysplastic syndromes/neoplasms (MDS): A state-of-the-art report on behalf of the International Consortium for MDS (icMDS). Blood Rev. 2023; in press. [Google Scholar] [CrossRef]
- Calvi, L.M.; Li, A.J.; Becker, M.W. What is the role of the microenvironment in MDS? Best. Pract. Res. Clin. Haematol. 2019, 32, 101113. [Google Scholar] [CrossRef]
- Lopez-Villar, O.; Garcia, J.L.; Sanchez-Guijo, F.M.; Robledo, C.; Villaron, E.M.; Hernandez-Campo, P.; Lopez-Holgado, N.; Diez-Campelo, M.; Barbado, M.V.; Perez-Simon, J.A.; et al. Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q- syndrome. Leukemia 2009, 23, 664–672. [Google Scholar] [CrossRef]
- Aanei, C.M.; Flandrin, P.; Eloae, F.Z.; Carasevici, E.; Guyotat, D.; Wattel, E.; Campos, L. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2012, 21, 1604–1615. [Google Scholar] [CrossRef]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef]
- Jann, J.C.; Mossner, M.; Riabov, V.; Altrock, E.; Schmitt, N.; Flach, J.; Xu, Q.; Nowak, V.; Oblander, J.; Palme, I.; et al. Bone marrow derived stromal cells from myelodysplastic syndromes are altered but not clonally mutated in vivo. Nat. Commun. 2021, 12, 6170. [Google Scholar] [CrossRef]
- Fei, C.; Zhao, Y.; Guo, J.; Gu, S.; Li, X.; Chang, C. Senescence of bone marrow mesenchymal stromal cells is accompanied by activation of p53/p21 pathway in myelodysplastic syndromes. Eur. J. Haematol. 2014, 93, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, G.; Mattiucci, D.; Mariani, M.; Ciarlantini, M.; Traini, S.; Mancini, S.; Olivieri, A.; Leoni, P.; Poloni, A. DNA demethylating therapy reverts mesenchymal stromal cells derived from high risk myelodysplastic patients to a normal phenotype. Br. J. Haematol. 2017, 177, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Wenk, C.; Garz, A.K.; Grath, S.; Huberle, C.; Witham, D.; Weickert, M.; Malinverni, R.; Niggemeyer, J.; Kyncl, M.; Hecker, J.; et al. Direct modulation of the bone marrow mesenchymal stromal cell compartment by azacitidine enhances healthy hematopoiesis. Blood Adv. 2018, 2, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef]
- Lambert, C.; Wu, Y.; Aanei, C. Bone Marrow Immunity and Myelodysplasia. Front. Oncol. 2016, 6, 172. [Google Scholar] [CrossRef] [PubMed]
- Kobune, M.; Iyama, S.; Kikuchi, S.; Horiguchi, H.; Sato, T.; Murase, K.; Kawano, Y.; Takada, K.; Ono, K.; Kamihara, Y.; et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J. 2012, 2, e87. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Dulcamare, I.; Giallongo, S.; Duminuco, A.; Pieragostino, D.; Cufaro, M.C.; Amorini, A.M.; Lazzarino, G.; Romano, A.; Parrinello, N.; et al. MacroH2A1.1 as a crossroad between epigenetics, inflammation and metabolism of mesenchymal stromal cells in myelodysplastic syndromes. Cell Death Dis. 2023, 14, 686. [Google Scholar] [CrossRef]
- Chen, S.; Zambetti, N.A.; Bindels, E.M.; Kenswill, K.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; Sanders, M.A.; Cremers, E.M.; Westers, T.M.; et al. Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue-context-dependent activation of inflammatory programs. Leukemia 2016, 30, 1938–1942. [Google Scholar] [CrossRef][Green Version]
- Pleyer, L.; Valent, P.; Greil, R. Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis-Masters of Survival and Clonality? Int. J. Mol. Sci. 2016, 17, 1009. [Google Scholar] [CrossRef]
- Calkoen, F.G.; Vervat, C.; van Pel, M.; de Haas, V.; Vijfhuizen, L.S.; Eising, E.; Kroes, W.G.; t Hoen, P.A.; van den Heuvel-Eibrink, M.M.; Egeler, R.M.; et al. Despite differential gene expression profiles pediatric MDS derived mesenchymal stromal cells display functionality in vitro. Stem Cell Res. 2015, 14, 198–210. [Google Scholar] [CrossRef]
- Kim, M.; Hwang, S.; Park, K.; Kim, S.Y.; Lee, Y.K.; Lee, D.S. Increased expression of interferon signaling genes in the bone marrow microenvironment of myelodysplastic syndromes. PLoS ONE 2015, 10, e0120602. [Google Scholar] [CrossRef]
- Hayashi, Y.; Kawabata, K.C.; Tanaka, Y.; Uehara, Y.; Mabuchi, Y.; Murakami, K.; Nishiyama, A.; Kiryu, S.; Yoshioka, Y.; Ota, Y.; et al. MDS cells impair osteolineage differentiation of MSCs via extracellular vesicles to suppress normal hematopoiesis. Cell Rep. 2022, 39, 110805. [Google Scholar] [CrossRef]
- Muntion, S.; Ramos, T.L.; Diez-Campelo, M.; Roson, B.; Sanchez-Abarca, L.I.; Misiewicz-Krzeminska, I.; Preciado, S.; Sarasquete, M.E.; de Las Rivas, J.; Gonzalez, M.; et al. Microvesicles from Mesenchymal Stromal Cells Are Involved in HPC-Microenvironment Crosstalk in Myelodysplastic Patients. PLoS ONE 2016, 11, e0146722. [Google Scholar] [CrossRef]
- Jager, P.; Geyh, S.; Twarock, S.; Cadeddu, R.P.; Rabes, P.; Koch, A.; Maus, U.; Hesper, T.; Zilkens, C.; Rautenberg, C.; et al. Acute myeloid leukemia-induced functional inhibition of healthy CD34+ hematopoietic stem and progenitor cells. Stem Cells 2021, 39, 1270–1284. [Google Scholar] [CrossRef]
- Pan, C.; Liu, P.; Ma, D.; Zhang, S.; Ni, M.; Fang, Q.; Wang, J. Bone marrow mesenchymal stem cells in microenvironment transform into cancer-associated fibroblasts to promote the progression of B-cell acute lymphoblastic leukemia. Biomed. Pharmacother. 2020, 130, 110610. [Google Scholar] [CrossRef]
- Leotta, S.; Condorelli, A.; Sciortino, R.; Milone, G.A.; Bellofiore, C.; Garibaldi, B.; Schinina, G.; Spadaro, A.; Cupri, A.; Milone, G. Prevention and Treatment of Acute Myeloid Leukemia Relapse after Hematopoietic Stem Cell Transplantation: The State of the Art and Future Perspectives. J. Clin. Med. 2022, 11, 253. [Google Scholar] [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef]
- Feng, X.; Sun, R.; Lee, M.; Chen, X.; Guo, S.; Geng, H.; Muschen, M.; Choi, J.; Pereira, J.P. Cell circuits between leukemic cells and mesenchymal stem cells block lymphopoiesis by activating lymphotoxin beta receptor signaling. eLife 2023, 12, e83533. [Google Scholar] [CrossRef]
- Binato, R.; de Almeida Oliveira, N.C.; Du Rocher, B.; Abdelhay, E. The molecular signature of AML mesenchymal stromal cells reveals candidate genes related to the leukemogenic process. Cancer Lett. 2015, 369, 134–143. [Google Scholar] [CrossRef]
- Lim, M.; Pang, Y.; Ma, S.; Hao, S.; Shi, H.; Zheng, Y.; Hua, C.; Gu, X.; Yang, F.; Yuan, W.; et al. Altered mesenchymal niche cells impede generation of normal hematopoietic progenitor cells in leukemic bone marrow. Leukemia 2016, 30, 154–162. [Google Scholar] [CrossRef]
- Falconi, G.; Galossi, E.; Fabiani, E.; Pieraccioli, M.; Travaglini, S.; Hajrullaj, H.; Cerretti, R.; Palmieri, R.; Latagliata, R.; Maurillo, L.; et al. Impairment of FOXM1 expression in mesenchymal cells from patients with myeloid neoplasms, de novo and therapy-related, may compromise their ability to support hematopoiesis. Sci. Rep. 2022, 12, 21231. [Google Scholar] [CrossRef]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Borella, G.; Da Ros, A.; Borile, G.; Porcu, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting the plasticity of mesenchymal stromal cells to reroute the course of acute myeloid leukemia. Blood 2021, 138, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Glait-Santar, C.; Desmond, R.; Feng, X.; Bat, T.; Chen, J.; Heuston, E.; Mizukawa, B.; Mulloy, J.C.; Bodine, D.M.; Larochelle, A.; et al. Functional Niche Competition Between Normal Hematopoietic Stem and Progenitor Cells and Myeloid Leukemia Cells. Stem Cells 2015, 33, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Luis, T.C.; Singbrant, S.; Lo Celso, C.; Mendez-Ferrer, S. The evolving view of the hematopoietic stem cell niche. Exp. Hematol. 2017, 50, 22–26. [Google Scholar] [CrossRef]
- Le, Y.; Fraineau, S.; Chandran, P.; Sabloff, M.; Brand, M.; Lavoie, J.R.; Gagne, R.; Rosu-Myles, M.; Yauk, C.L.; Richardson, R.B.; et al. Adipogenic Mesenchymal Stromal Cells from Bone Marrow and Their Hematopoietic Supportive Role: Towards Understanding the Permissive Marrow Microenvironment in Acute Myeloid Leukemia. Stem Cell Rev. Rep. 2016, 12, 235–244. [Google Scholar] [CrossRef]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2020, 34, 391–403. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, e90036. [Google Scholar] [CrossRef]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Q.; Cang, H.; Wang, Z.; Hu, X.; Pan, R.; Yang, Y.; Chen, Y. Acute Myeloid Leukemia Cells Educate Mesenchymal Stromal Cells toward an Adipogenic Differentiation Propensity with Leukemia Promotion Capabilities. Adv. Sci. 2022, 9, 2105811. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Lee, H.R.; Yang, S.J.; Choi, H.K.; Kim, J.A.; Oh, I.H. The Chromatin Remodeling Complex CHD1 Regulates the Primitive State of Mesenchymal Stromal Cells to Control Their Stem Cell Supporting Activity. Stem Cells Dev. 2021, 30, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Gareau, Y.; Lehnertz, B.; Gingras, S.; Spinella, J.F.; Corneau, S.; Mayotte, N.; Girard, S.; Frechette, M.; Blouin-Chagnon, V.; et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 2019, 36, 84–99 e88. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Forte, D.; Garcia-Fernandez, M.; Sanchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martin-Perez, D.; Lopez, J.A.; Costa, A.S.H.; Tronci, L.; Nikitopoulou, E.; et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab. 2020, 32, 829–843 e829. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Mistry, J.J.; Moore, J.A.; Kumar, P.; Marlein, C.R.; Hellmich, C.; Pillinger, G.; Jibril, A.; Di Palma, F.; Collins, A.; Bowles, K.M.; et al. Daratumumab inhibits acute myeloid leukaemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells. Haematologica 2021, 106, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed]
- Tucci, J.; Chen, T.; Margulis, K.; Orgel, E.; Paszkiewicz, R.L.; Cohen, M.D.; Oberley, M.J.; Wahhab, R.; Jones, A.E.; Divakaruni, A.S.; et al. Adipocytes Provide Fatty Acids to Acute Lymphoblastic Leukemia Cells. Front. Oncol. 2021, 11, 665763. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Dempsey, C.; Chadwick, A.; Harrison, S.; Liu, J.; Di, Y.; McGinn, O.J.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P.; et al. Metabolic reprogramming of bone marrow stromal cells by leukemic extracellular vesicles in acute lymphoblastic leukemia. Blood 2016, 128, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. [Google Scholar] [CrossRef]
- Rambaldi, B.; Diral, E.; Donsante, S.; Di Marzo, N.; Mottadelli, F.; Cardinale, L.; Dander, E.; Isimbaldi, G.; Pioltelli, P.; Biondi, A.; et al. Heterogeneity of the bone marrow niche in patients with myeloproliferative neoplasms: ActivinA secretion by mesenchymal stromal cells correlates with the degree of marrow fibrosis. Ann. Hematol. 2021, 100, 105–116. [Google Scholar] [CrossRef]
- Xie, J.; Chen, J.; Wang, B.; He, X.; Huang, H. Bone mesenchymal stromal cells exhibit functional inhibition but no chromosomal aberrations in chronic myelogenous leukemia. Oncol. Lett. 2019, 17, 999–1007. [Google Scholar] [CrossRef]
- Avanzini, M.A.; Bernardo, M.E.; Novara, F.; Mantelli, M.; Poletto, V.; Villani, L.; Lenta, E.; Ingo, D.M.; Achille, V.; Bonetti, E.; et al. Functional and genetic aberrations of in vitro-cultured marrow-derived mesenchymal stromal cells of patients with classical Philadelphia-negative myeloproliferative neoplasms. Leukemia 2014, 28, 1742–1745. [Google Scholar] [CrossRef]
- Zhao, Z.G.; Xu, W.; Sun, L.; Li, W.M.; Li, Q.B.; Zou, P. The characteristics and immunoregulatory functions of regulatory dendritic cells induced by mesenchymal stem cells derived from bone marrow of patient with chronic myeloid leukaemia. Eur. J. Cancer 2012, 48, 1884–1895. [Google Scholar] [CrossRef]
- Jafarzadeh, N.; Safari, Z.; Pornour, M.; Amirizadeh, N.; Forouzandeh Moghadam, M.; Sadeghizadeh, M. Alteration of cellular and immune-related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J. Cell. Physiol. 2019, 234, 3697–3710. [Google Scholar] [CrossRef]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, A.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 2019, 24, 769–784 e766. [Google Scholar] [CrossRef] [PubMed]
- Aggoune, D.; Sorel, N.; Bonnet, M.L.; Goujon, J.M.; Tarte, K.; Herault, O.; Domenech, J.; Rea, D.; Legros, L.; Johnson-Ansa, H.; et al. Bone marrow mesenchymal stromal cell (MSC) gene profiling in chronic myeloid leukemia (CML) patients at diagnosis and in deep molecular response induced by tyrosine kinase inhibitors (TKIs). Leuk. Res. 2017, 60, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed]
- La Spina, E.; Giallongo, S.; Giallongo, C.; Vicario, N.; Duminuco, A.; Parenti, R.; Giuffrida, R.; Longhitano, L.; Li Volti, G.; Cambria, D.; et al. Mesenchymal stromal cells in tumor microenvironment remodeling of BCR-ABL negative myeloproliferative diseases. Front. Oncol. 2023, 13, 1141610. [Google Scholar] [CrossRef] [PubMed]
- Abbonante, V.; Gruppi, C.; Catarsi, P.; Avanzini, M.A.; Tira, M.E.; Barosi, G.; Rosti, V.; Balduini, A. Altered fibronectin expression and deposition by myeloproliferative neoplasm-derived mesenchymal stromal cells. Br. J. Haematol. 2016, 172, 140–144. [Google Scholar] [CrossRef]
- Badalucco, S.; Di Buduo, C.A.; Campanelli, R.; Pallotta, I.; Catarsi, P.; Rosti, V.; Kaplan, D.L.; Barosi, G.; Massa, M.; Balduini, A. Involvement of TGFbeta1 in autocrine regulation of proplatelet formation in healthy subjects and patients with primary myelofibrosis. Haematologica 2013, 98, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.K.; Ziegler, S.; Leisten, I.; Ferreira, M.S.; Schumacher, A.; Rath, B.; Fahrenkamp, D.; Muller-Newen, G.; Crysandt, M.; Wilop, S.; et al. Activated fibronectin-secretory phenotype of mesenchymal stromal cells in pre-fibrotic myeloproliferative neoplasms. J. Hematol. Oncol. 2014, 7, 92. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Merchant, D.; Mahmud, N.; Ishii, T.; Zhao, Y.; Hu, W.; Bruno, E.; Barosi, G.; Xu, M.; Hoffman, R. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007, 110, 986–993. [Google Scholar] [CrossRef]
- Avanzini, M.A.; Abbonante, V.; Catarsi, P.; Dambruoso, I.; Mantelli, M.; Poletto, V.; Lenta, E.; Guglielmelli, P.; Croce, S.; Cobianchi, L.; et al. The spleen of patients with myelofibrosis harbors defective mesenchymal stromal cells. Am. J. Hematol. 2018, 93, 615–622. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Liu, Y.C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef]
- Bedekovics, J.; Kiss, A.; Beke, L.; Karolyi, K.; Mehes, G. Platelet derived growth factor receptor-beta (PDGFRbeta) expression is limited to activated stromal cells in the bone marrow and shows a strong correlation with the grade of myelofibrosis. Virchows Arch. 2013, 463, 57–65. [Google Scholar] [CrossRef]
- Longhitano, L.; Tibullo, D.; Vicario, N.; Giallongo, C.; La Spina, E.; Romano, A.; Lombardo, S.; Moretti, M.; Masia, F.; Coda, A.R.D.; et al. IGFBP-6/sonic hedgehog/TLR4 signalling axis drives bone marrow fibrotic transformation in primary myelofibrosis. Aging 2021, 13, 25055–25071. [Google Scholar] [CrossRef]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Busche, G.; Fleck, D.; et al. Gli1(+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 2017, 20, 785–800 e788. [Google Scholar] [CrossRef]
- Lataillade, J.J.; Pierre-Louis, O.; Hasselbalch, H.C.; Uzan, G.; Jasmin, C.; Martyre, M.C.; Le Bousse-Kerdiles, M.C.; French, I.; the European, E.N.o.M. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood 2008, 112, 3026–3035. [Google Scholar] [CrossRef] [PubMed]
- Martinaud, C.; Desterke, C.; Konopacki, J.; Pieri, L.; Torossian, F.; Golub, R.; Schmutz, S.; Anginot, A.; Guerton, B.; Rochet, N.; et al. Osteogenic Potential of Mesenchymal Stromal Cells Contributes to Primary Myelofibrosis. Cancer Res. 2015, 75, 4753–4765. [Google Scholar] [CrossRef] [PubMed]
- Leimkuhler, N.B.; Gleitz, H.F.E.; Ronghui, L.; Snoeren, I.A.M.; Fuchs, S.N.R.; Nagai, J.S.; Banjanin, B.; Lam, K.H.; Vogl, T.; Kuppe, C.; et al. Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell 2021, 28, 637–652 e638. [Google Scholar] [CrossRef] [PubMed]
- Nasnas, P.; Cerchione, C.; Musuraca, G.; Martinelli, G.; Ferrajoli, A. How I Manage Chronic Lymphocytic Leukemia. Hematol. Rep. 2023, 15, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Gattei, V.; Bulian, P.; Del Principe, M.I.; Zucchetto, A.; Maurillo, L.; Buccisano, F.; Bomben, R.; Dal-Bo, M.; Luciano, F.; Rossi, F.M.; et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood 2008, 111, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.N.; Grabovsky, V.; Wang, W.; Desch, P.; Rubenzer, G.; Wollner, S.; Binsky, I.; Vallon-Eberhard, A.; Sapoznikov, A.; Burger, M.; et al. Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res. 2009, 69, 3121–3130. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Nowakowski, G.S.; Knox, T.R.; Boysen, J.C.; Maas, M.L.; Schwager, S.M.; Wu, W.; Wellik, L.E.; Dietz, A.B.; Ghosh, A.K.; et al. Bi-directional activation between mesenchymal stem cells and CLL B-cells: Implication for CLL disease progression. Br. J. Haematol. 2009, 147, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Crompot, E.; Van Damme, M.; Pieters, K.; Vermeersch, M.; Perez-Morga, D.; Mineur, P.; Maerevoet, M.; Meuleman, N.; Bron, D.; Lagneaux, L.; et al. Extracellular vesicles of bone marrow stromal cells rescue chronic lymphocytic leukemia B cells from apoptosis, enhance their migration and induce gene expression modifications. Haematologica 2017, 102, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Vangapandu, H.V.; Havranek, O.; Ayres, M.L.; Kaipparettu, B.A.; Balakrishnan, K.; Wierda, W.G.; Keating, M.J.; Davis, R.E.; Stellrecht, C.M.; Gandhi, V. B-cell Receptor Signaling Regulates Metabolism in Chronic Lymphocytic Leukemia. Mol. Cancer Res. 2017, 15, 1692–1703. [Google Scholar] [CrossRef]
- von Heydebrand, F.; Fuchs, M.; Kunz, M.; Voelkl, S.; Kremer, A.N.; Oostendorp, R.A.J.; Wilke, J.; Leitges, M.; Egle, A.; Mackensen, A.; et al. Protein kinase C-beta-dependent changes in the glucose metabolism of bone marrow stromal cells of chronic lymphocytic leukemia. Stem Cells 2021, 39, 819–830. [Google Scholar] [CrossRef]
- Vangapandu, H.V.; Ayres, M.L.; Bristow, C.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Stellrecht, C.M.; Gandhi, V. The Stromal Microenvironment Modulates Mitochondrial Oxidative Phosphorylation in Chronic Lymphocytic Leukemia Cells. Neoplasia 2017, 19, 762–771. [Google Scholar] [CrossRef]
- Ding, W.; Knox, T.R.; Tschumper, R.C.; Wu, W.; Schwager, S.M.; Boysen, J.C.; Jelinek, D.F.; Kay, N.E. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: Implications for an angiogenic switch. Blood 2010, 116, 2984–2993. [Google Scholar] [CrossRef]
- Kini, A.R.; Kay, N.E.; Peterson, L.C. Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia 2000, 14, 1414–1418. [Google Scholar] [CrossRef]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef]
- Korde, N.; Kristinsson, S.Y.; Landgren, O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): Novel biological insights and development of early treatment strategies. Blood 2011, 117, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Brigle, K.; Rogers, B. Pathobiology and Diagnosis of Multiple Myeloma. Semin. Oncol. Nurs. 2017, 33, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Rasch, S.; Lund, T.; Asmussen, J.T.; Lerberg Nielsen, A.; Faebo Larsen, R.; Osterheden Andersen, M.; Abildgaard, N. Multiple Myeloma Associated Bone Disease. Cancers 2020, 12, 2113. [Google Scholar] [CrossRef]
- Gunn, W.G.; Conley, A.; Deininger, L.; Olson, S.D.; Prockop, D.J.; Gregory, C.A. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: A potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells 2006, 24, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.A.; Mundy, G.R.; Lwin, S.T.; Edwards, C.M. Bone marrow stromal cells create a permissive microenvironment for myeloma development: A new stromal role for Wnt inhibitor Dkk1. Cancer Res. 2012, 72, 2183–2189. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Sanchez-Guijo, F.; Del Canizo, M.C.; San Miguel, J.F.; Garayoa, M. Multiple myeloma mesenchymal stromal cells: Contribution to myeloma bone disease and therapeutics. World J. Stem Cells 2014, 6, 322–343. [Google Scholar] [CrossRef]
- Todoerti, K.; Lisignoli, G.; Storti, P.; Agnelli, L.; Novara, F.; Manferdini, C.; Codeluppi, K.; Colla, S.; Crugnola, M.; Abeltino, M.; et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp. Hematol. 2010, 38, 141–153. [Google Scholar] [CrossRef]
- Schinke, C.; Qu, P.; Mehdi, S.J.; Hoering, A.; Epstein, J.; Johnson, S.K.; van Rhee, F.; Zangari, M.; Thanendrarajan, S.; Barlogie, B.; et al. The Pattern of Mesenchymal Stem Cell Expression Is an Independent Marker of Outcome in Multiple Myeloma. Clin. Cancer Res. 2018, 24, 2913–2919. [Google Scholar] [CrossRef]
- Fernando, R.C.; Mazzotti, D.R.; Azevedo, H.; Sandes, A.F.; Rizzatti, E.G.; de Oliveira, M.B.; Alves, V.L.F.; Eugenio, A.I.P.; de Carvalho, F.; Dalboni, M.A.; et al. Transcriptome Analysis of Mesenchymal Stem Cells from Multiple Myeloma Patients Reveals Downregulation of Genes Involved in Cell Cycle Progression, Immune Response, and Bone Metabolism. Sci. Rep. 2019, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, L.; DoSouto Ferreira, L.; Joubert, M.V.; Avet-Loiseau, H.; Martinet, L.; Corre, J.; Couderc, B. Imprinting of Mesenchymal Stromal Cell Transcriptome Persists even after Treatment in Patients with Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 3854. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.M.E.; Kellermayer, Z.; Papazian, N.; Tahri, S.; Hofste Op Bruinink, D.; Hoogenboezem, R.; Sanders, M.A.; van de Woestijne, P.C.; Bos, P.K.; Khandanpour, C.; et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat. Immunol. 2021, 22, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhao, Y.; Fei, C.; Zhao, S.; Zheng, Q.; Su, J.; Wu, D.; Li, X.; Chang, C. Dicer1 downregulation by multiple myeloma cells promotes the senescence and tumor-supporting capacity and decreases the differentiation potential of mesenchymal stem cells. Cell Death Dis. 2018, 9, 512. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Meuleman, N.; Stamatopoulos, B.; De Bruyn, C.; Pieters, K.; Bron, D.; Lagneaux, L. Evidences of early senescence in multiple myeloma bone marrow mesenchymal stromal cells. PLoS ONE 2013, 8, e59756. [Google Scholar] [CrossRef] [PubMed]
- Yaccoby, S.; Wezeman, M.J.; Zangari, M.; Walker, R.; Cottler-Fox, M.; Gaddy, D.; Ling, W.; Saha, R.; Barlogie, B.; Tricot, G.; et al. Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica 2006, 91, 192–199. [Google Scholar]
- Zhan, X.; Yu, W.; Franqui-Machin, R.; Bates, M.L.; Nadiminti, K.; Cao, H.; Amendt, B.A.; Jethava, Y.; Frech, I.; Zhan, F.; et al. Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget 2017, 8, 111213–111224. [Google Scholar] [CrossRef]
- Romano, A.; Conticello, C.; Cavalli, M.; Vetro, C.; La Fauci, A.; Parrinello, N.L.; Di Raimondo, F. Immunological dysregulation in multiple myeloma microenvironment. Biomed. Res. Int. 2014, 2014, 198539. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Parrinello, N.L.; La Cava, P.; Di Rosa, M.; Bramanti, V.; Di Raimondo, C.; Conticello, C.; Chiarenza, A.; Palumbo, G.A.; et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, 85764–85775. [Google Scholar] [CrossRef]
- Chen, D.; Tang, P.; Liu, L.; Wang, F.; Xing, H.; Sun, L.; Jiang, Z. Bone marrow-derived mesenchymal stem cells promote cell proliferation of multiple myeloma through inhibiting T cell immune responses via PD-1/PD-L1 pathway. Cell Cycle 2018, 17, 858–867. [Google Scholar] [CrossRef]
- Liu, Z.; Mi, F.; Han, M.; Tian, M.; Deng, L.; Meng, N.; Luo, J.; Fu, R. Bone marrow-derived mesenchymal stem cells inhibit CD8(+) T cell immune responses via PD-1/PD-L1 pathway in multiple myeloma. Clin. Exp. Immunol. 2021, 205, 53–62. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Deng, L.; Jia, Y.; Liu, H.; Ding, K.; Wang, W.; Zhang, H.; Fu, R. CD155/TIGIT signalling plays a vital role in the regulation of bone marrow mesenchymal stem cell-induced natural killer-cell exhaustion in multiple myeloma. Clin. Transl. Med. 2022, 12, e861. [Google Scholar] [CrossRef]
- Plakhova, N.; Panagopoulos, V.; Vandyke, K.; Zannettino, A.C.W.; Mrozik, K.M. Mesenchymal stromal cell senescence in haematological malignancies. Cancer Metastasis Rev. 2023, 42, 277–296. [Google Scholar] [CrossRef] [PubMed]
- van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak, V. From MGUS to Multiple Myeloma, a Paradigm for Clonal Evolution of Premalignant Cells. Cancer Res. 2018, 78, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuan, X.; Munoz, N.; Logan, T.M.; Ma, T. Commitment to Aerobic Glycolysis Sustains Immunosuppression of Human Mesenchymal Stem Cells. Stem Cells Transl. Med. 2019, 8, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Matula, Z.; Mikala, G.; Lukacsi, S.; Matko, J.; Kovacs, T.; Monostori, E.; Uher, F.; Valyi-Nagy, I. Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers 2021, 13, 3461. [Google Scholar] [CrossRef]
- Matula, Z.; Uher, F.; Valyi-Nagy, I.; Mikala, G. The Effect of Belantamab Mafodotin on Primary Myeloma-Stroma Co-Cultures: Asymmetrical Mitochondrial Transfer between Myeloma Cells and Autologous Bone Marrow Stromal Cells. Int. J. Mol. Sci. 2023, 24, 5303. [Google Scholar] [CrossRef]
- Barbato, A.; Giallongo, C.; Giallongo, S.; Romano, A.; Scandura, G.; Concetta, S.; Zuppelli, T.; Lolicato, M.; Lazzarino, G.; Parrinello, N.; et al. Lactate trafficking inhibition restores sensitivity to proteasome inhibitors and orchestrates immuno-microenvironment in multiple myeloma. Cell Prolif. 2023, 56, e13388. [Google Scholar] [CrossRef] [PubMed]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 2020, 10, 604143. [Google Scholar] [CrossRef]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdiles, M.C. Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediat. Inflamm. 2015, 2015, 415024. [Google Scholar] [CrossRef]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef]
- Cancilla, D.; Rettig, M.P.; DiPersio, J.F. Targeting CXCR4 in AML and ALL. Front. Oncol. 2020, 10, 1672. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, P.; Scaglione, S.; Quarto, R.; Narcisi, R.; Parodi, M.; Balleari, E.; Barbieri, F.; Pattarozzi, A.; Florio, T.; Ferrini, S.; et al. An interaction between hepatocyte growth factor and its receptor (c-MET) prolongs the survival of chronic lymphocytic leukemic cells through STAT3 phosphorylation: A potential role of mesenchymal cells in the disease. Haematologica 2011, 96, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, C.; Wojtowicz, E.; Moore, J.A.; Mistry, J.J.; Jibril, A.; Johnson, B.B.; Smith, J.G.W.; Beraza, N.; Bowles, K.M.; Rushworth, S.A. p16INK4A-dependent senescence in the bone marrow niche drives age-related metabolic changes of hematopoietic progenitors. Blood Adv. 2023, 7, 256–268. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giallongo, S.; Duminuco, A.; Dulcamare, I.; Zuppelli, T.; La Spina, E.; Scandura, G.; Santisi, A.; Romano, A.; Di Raimondo, F.; Tibullo, D.; et al. Engagement of Mesenchymal Stromal Cells in the Remodeling of the Bone Marrow Microenvironment in Hematological Cancers. Biomolecules 2023, 13, 1701. https://doi.org/10.3390/biom13121701
Giallongo S, Duminuco A, Dulcamare I, Zuppelli T, La Spina E, Scandura G, Santisi A, Romano A, Di Raimondo F, Tibullo D, et al. Engagement of Mesenchymal Stromal Cells in the Remodeling of the Bone Marrow Microenvironment in Hematological Cancers. Biomolecules. 2023; 13(12):1701. https://doi.org/10.3390/biom13121701
Chicago/Turabian StyleGiallongo, Sebastiano, Andrea Duminuco, Ilaria Dulcamare, Tatiana Zuppelli, Enrico La Spina, Grazia Scandura, Annalisa Santisi, Alessandra Romano, Francesco Di Raimondo, Daniele Tibullo, and et al. 2023. "Engagement of Mesenchymal Stromal Cells in the Remodeling of the Bone Marrow Microenvironment in Hematological Cancers" Biomolecules 13, no. 12: 1701. https://doi.org/10.3390/biom13121701
APA StyleGiallongo, S., Duminuco, A., Dulcamare, I., Zuppelli, T., La Spina, E., Scandura, G., Santisi, A., Romano, A., Di Raimondo, F., Tibullo, D., Palumbo, G. A., & Giallongo, C. (2023). Engagement of Mesenchymal Stromal Cells in the Remodeling of the Bone Marrow Microenvironment in Hematological Cancers. Biomolecules, 13(12), 1701. https://doi.org/10.3390/biom13121701