
APE1/Ref-1 as a Therapeutic Target for Inflammatory Bowel Disease
 , and
, and
Abstract
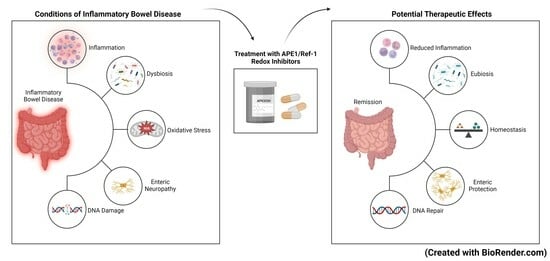
{kind=link}
{kind=link}
1. Introduction
1.1. ENS and IBD
1.2. Current Treatments for IBD
1.3. Oxidative Stress and IBD
2. APE1/Ref-1
APE1/Ref-1 in Intestinal Inflammation
3. Targeting APE1/Ref-1 as a Therapeutic Approach
4. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Matricon, J.; Barnich, N.; Ardid, D. Immunopathogenesis of inflammatory bowel disease. Self Nonself 2010, 1, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Investig. 2007, 117, 514–521. [Google Scholar] [CrossRef]
- Subasinghe, D.; Nawarathna, N.M.M.; Samarasekera, D.N. Disease characteristics of inflammatory bowel disease (IBD). J. Gastrointest. Surg. 2011, 15, 1562–1567. [Google Scholar] [CrossRef]
- Tan, Z.; Zhu, S.; Liu, C.; Meng, Y.; Li, J.; Zhang, J.; Dong, W. Causal link between inflammatory bowel disease and fistula: Evidence from mendelian randomization study. J. Clin. Med. 2023, 12, 2482. [Google Scholar] [CrossRef]
- Wang, R.; Li, Z.; Liu, S.; Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: A systematic analysis based on the Global Burden of Disease Study 2019. Br. Med. J. 2023, 13, 65–138. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Epidemiology and risk factors for ibd. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Muzammil, M.A.; Fariha, F.; Patel, T.; Sohail, R.; Kumar, M.; Khan, E.; Khanam, B.; Kumar, S.; Khatri, M.; Varrassi, G.; et al. Advancements in inflammatory bowel disease: A narrative review of diagnostics, management, epidemiology, prevalence, patient outcomes, quality of life, and clinical presentation. Cureus 2023, 15, 411–420. [Google Scholar] [CrossRef]
- Knowles, C.H.; Lindberg, G.; Panza, E.; Giorgio, R.D. New perspectives in the diagnosis and management of enteric neuropathies. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.B.; Mazmanian, S.K. The enteric network: Interactions between the immune and nervous systems of the gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef]
- Chesné, J.; Cardoso, V.; Veiga-Fernandes, H. Neuro-immune regulation of mucosal physiology. Mucosal Immunol. 2019, 12, 10–20. [Google Scholar] [CrossRef]
- Verheijden, S.; Boeckxstaens, G.E. Neuroimmune interaction and the regulation of intestinal immune homeostasis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2018, 314, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.B. The enteric nervous system I: Organisation and classification. Pharmacol. Toxicol. 2003, 92, 105–113. [Google Scholar] [CrossRef]
- Wang, H.; Foong, J.P.P.; Harris, N.L.; Bornstein, J.C. Enteric neuroimmune interactions coordinate intestinal responses in health and disease. Mucosal Immunol. 2022, 15, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.; Yang, D.; Vella, M.; Chiu, I.M. The intestinal neuro-immune axis: Crosstalk between neurons, immune cells, and microbes. Mucosal Immunol. 2021, 14, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.R.; Poole, D.P.; Thacker, M.; Furness, J.B. The involvement of nitric oxide synthase neurons in enteric neuropathies. Neurogastroenterol. Motil. 2011, 23, 980–988. [Google Scholar] [CrossRef]
- Sahakian, L.; Filippone, R.T.; Stavely, R.; Robinson, A.M.; Yan, X.S.; Abalo, R.; Eri, R.; Bornstein, J.C.; Kelley, M.R.; Nurgali, K. Inhibition of APE1/Ref-1 redox signaling alleviates intestinal dysfunction and damage to myenteric neurons in a mouse model of spontaneous chronic colitis. Inflamm. Bowel Dis. 2021, 27, 388–406. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Rahman, A.A.; Carbone, S.E.; Randall-Demllo, S.; Filippone, R.; Bornstein, J.C.; Eri, R.; Nurgali, K. Alterations of colonic function in the Winnie mouse model of spontaneous chronic colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, 85–102. [Google Scholar] [CrossRef]
- Robinson, A.M.; Stavely, R.; Miller, S.; Eri, R.; Nurgali, K. Mesenchymal stem cell treatment for enteric neuropathy in the Winnie mouse model of spontaneous chronic colitis. Cell Tissue Res. 2022, 389, 41–70. [Google Scholar] [CrossRef]
- Oehmichen, M.; Reifferscheid, P. Intramural ganglion cell degeneration in inflammatory bowel disease. Digestion 1977, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, N.; Segnani, C.; Ippolito, C.; De Giorgio, R.; Colucci, R.; Faussone-Pellegrini, M.S.; Chiarugi, M.; Campani, D.; Castagna, M.; Mattii, L.; et al. Immunohistochemical analysis of myenteric ganglia and interstitial cells of Cajal in ulcerative colitis. J. Cell. Mol. Med. 2012, 16, 318–327. [Google Scholar] [CrossRef]
- Riemann, J.F.; Schmidt, H. Ultrastructural changes in the gut autonomic nervous system following laxative abuse and in other conditions. Scand. J. Gastroenterol. 1982, 71, 111–124. [Google Scholar]
- Krauter, E.M.; Strong, D.S.; Brooks, E.M.; Linden, D.R.; Sharkey, K.A.; Mawe, G.M. Changes in colonic motility and the electrophysiological properties of myenteric neurons persist following recovery from trinitrobenzene sulfonic acid colitis in the guinea pig. Neurogastroenterol. Motil. 2007, 19, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A. Neuroinflammation in inflammatory bowel disease. J. Neuroinflammation 2010, 7, 37. [Google Scholar] [CrossRef]
- Lomax, A.E.; O’Hara, J.R.; Hyland, N.P.; Mawe, G.M.; Sharkey, K.A. Persistent alterations to enteric neural signaling in the guinea pig colon following the resolution of colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2007, 292, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.A.M.; Gulbransen, B.D. The antioxidant glutathione protects against enteric neuron death in situ, but its depletion is protective during colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2017, 314, G39–G52. [Google Scholar] [CrossRef]
- Villanacci, V.; Bassotti, G.; Nascimbeni, R.; Antonelli, E.; Cadei, M.; Fisogni, S. Enteric nervous system abnormalities in inflammatory bowel diseases. Neurogastroenterol. Motil. 2008, 20, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Nezami, B.G.; Srinivasan, S. Enteric nervous system in the small intestine: Pathophysiology and clinical implications. Curr. Gastroenterol. Rep. 2010, 12, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.K.; Ding, N.S.; Niewiadomski, O. Management of Inflammatory Bowel Disease. Med. J. Aust. 2018, 209, 318–323. [Google Scholar] [CrossRef]
- Mijan, M.A.; Lim, B.O. Diets, functional foods, and nutraceuticals as alternative therapies for inflammatory bowel disease: Present status and future trends. World J. Gastroenterol. 2018, 24, 2673–2685. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Panaccione, R.; Ghosh, S.; Rioux, K. Optimizing clinical use of mesalazine (5-aminosalicylic acid) in inflammatory bowel disease. Ther. Adv. Gastroenterol. 2011, 4, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, J.; Matsuoka, K.; Yoshida, A.; Naganuma, M.; Hisamatsu, T.; Yajima, T.; Inoue, N.; Okamoto, S.; Iwao, Y.; Ogata, H.; et al. 5-Aminosalicylic acid aggravates colitis mimicking exacerbation of ulcerative colitis. Intest. Res. 2018, 16, 635–640. [Google Scholar] [CrossRef]
- Barrett, K.; Saxena, S.; Pollok, R. Using corticosteroids appropriately in inflammatory bowel disease: A guide for primary care. Br. J. Gen. Pract. 2018, 68, 497–498. [Google Scholar] [CrossRef] [PubMed]
- Harlan, W.R.; Meyer, A.; Fisher, J. Inflammatory bowel disease: Epidemiology, evaluation, treatment, and health maintenance. North. Carol. Med. J. 2016, 77, 198–201. [Google Scholar] [CrossRef][Green Version]
- Bernstein, C.N. Treatment of IBD: Where we are and where we are going. Am. J. Gastroenterol. 2014, 110, 114. [Google Scholar] [CrossRef] [PubMed]
- Cheah, M.; Khanna, R. Chapter 1—Current medical therapies for ulcerative colitis. In Pouchitis and Ileal Pouch Disorders; Shen, B., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–15. [Google Scholar] [CrossRef]
- Miyatani, Y.; Kobayashi, T. Evidence-based approach to the discontinuation of immunomodulators or biologics in inflammatory bowel disease. Digestion 2023, 104, 66–73. [Google Scholar] [CrossRef]
- Hanauer, S.B.; Sandborn, W.J.; Lichtenstein, G.R. Evolving considerations for thiopurine therapy for inflammatory bowel diseases: A clinical practice update. Gastroenterology 2019, 156, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Chowers, Y. Review article: Loss of response to anti-tnf treatments in Crohn’s disease. Aliment. Pharmacol. Ther. 2011, 33, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in inflammatory bowel disease: Pathogenic role and potential therapeutic targets. Int. J. Mol. Sci. 2022, 23, 3464. [Google Scholar] [CrossRef]
- Wu, Z.A.; Wang, H.X. A Systematic Review of the Interaction Between Gut Microbiota and Host Health from a Symbiotic Perspective. SN Compr. Clin. Med. 2018, 1, 224–235. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Blaser, M.J.; Ley, R.E.; Knight, R. Development of the Human Gastrointestinal Microbiota and Insights From High-Throughput Sequencing. Gastroenterology 2011, 140, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kanai, T. The gut microbiota and inflammatory bowel disease. Semin. Immunopathol. 2015, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; Van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, W.; Smarr, L.; Nelson, K.; Yooseph, S.; Torralba, M. Large memory high performance computing enables comparison across human gut microbiome of patients with autoimmune diseases and healthy subjects. In Proceedings of the Conference on Extreme Science and Engineering Discovery Environment: Gateway to Discovery, San Diego, CA, USA, 22–25 July 2013. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; Leleiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Grigoryan, Z.; Shen, M.J.; Twardus, S.W.; Beuttler, M.M.; Chen, L.A.; Bateman-House, A. Fecal microbiota transplantation: Uses, questions and ethics. Med. Microbiol. 2020, 6, 100027. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chang, E.B. Inflammatory bowel diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology 2021, 160, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.-K.; Park, D.I. Novel treatments for inflammatory bowel disease. Korean J. Intern. Med. 2018, 33, 20–27. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of oxidative stress in inflammatory bowel disease and potential antioxidant therapies. Oxidative Med. Cell. Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Shan, J.-L.; He, H.-T.; Li, M.-X.; Zhu, J.-W.; Cheng, Y.; Hu, N.; Wang, G.; Wang, D.; Yang, X.-Q.; He, Y.; et al. APE1 promotes antioxidant capacity by regulating Nrf-2 function through a redox-dependent mechanism. Free Radic. Biol. Med. 2015, 78, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, 453–462. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-f. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Lih-Brody, L.; Powell, S.R.; Collier, K.P.; Reddy, G.M.; Cerchia, R.; Kahn, E.; Weissman, G.S.; Katz, S.; Floyd, R.A.; McKinley, M.J.; et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig. Dis. Sci. 1996, 41, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Khan, M.A.; Ambrose, M.; Nikolayeva, O.; Xu-Welliver, M.; Kartalou, M.; Hussain, S.P.; Roth, R.B.; Zhou, X.; Mechanic, L.E.; et al. The adaptive imbalance in base excision–repair enzymes generates microsatellite instability in chronic inflammation. J. Clin. Investig. 2003, 112, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Tiwari, S.K.; Gunisetty, S.; Anjum, F.; Nallari, P.; Habeeb, M.A.; Khan, A.A. Functional polymorphisms in XRCC-1 and APE-1 contribute to increased apoptosis and risk of ulcerative colitis. Inflamm. Res. 2012, 61, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.; Parker, R.D.; Abdollahi, M. Oxidative stress and pathogenesis of inflammatory bowel disease: An epiphenomenon or the cause? Dig. Dis. Sci. 2007, 52, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Chami, B.; Martin, N.J.J.; Dennis, J.M.; Witting, P.K. Myeloperoxidase in the inflamed colon: A novel target for treating inflammatory bowel disease. Arch. Biochem. Biophys. 2018, 645, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Oshitani, N.; Sawa, Y.; Hara, J.; Adachi, K.; Nakamura, S.; Matsumoto, T.; Arakawa, T.; Kuroki, T. Functional and phenotypical activation of leucocytes in inflamed human colonic mucosa. J. Gastroenterol. Hepatol. 2008, 12, 809–814. [Google Scholar] [CrossRef]
- Andresen, L.; Jørgensen, V.L.; Perner, A.; Hansen, A.; Eugen-Olsen, J.; Rask-Madsen, J. Activation of nuclear factor kappaB in colonic mucosa from patients with collagenous and ulcerative colitis. Gut 2005, 54, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, L.-w.; Sun, W.-j.; Qin, S.-h.; Qin, L.-z.; Wang, X. Expressions of E-cadherin, p120ctn, β-catenin and NF-κB in ulcerative colitis. J. Huazhong Univ. Sci. Technol. 2015, 35, 368–373. [Google Scholar] [CrossRef]
- Sharma, R.; Young, C.; Neu, J. Molecular modulation of intestinal epithelial barrier: Contribution of microbiota. J. Biomed. Biotechnol. 2010, 2010, 305879. [Google Scholar] [CrossRef]
- Kinnebrew, M.A.; Pamer, E.G. Innate immune signaling in defense against intestinal microbes. Immunol. Rev. 2012, 245, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, S.; Cao, Y.; Tian, X.; Zeng, R.; Liao, D.-F.; Cao, D. Oxidative stress and carbonyl lesions in ulcerative colitis and associated colorectal cancer. Oxidative Med. Cell. Longev. 2016, 2016, 9875298. [Google Scholar] [CrossRef] [PubMed]
- Aldars-García, L.; Marin, A.C.; Chaparro, M.; Gisbert, J.P. The Interplay between immune system and microbiota in inflammatory bowel disease: A narrative review. Int. J. Mol. Sci. 2021, 22, 3076. [Google Scholar] [CrossRef]
- Juge, N. Relationship between mucosa-associated gut microbiota and human diseases. Biochem. Soc. Trans. 2022, 50, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: Updated experimental and clinical evidence. Exp. Biol. Med. 2012, 237, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 2014, 147, 1055. [Google Scholar] [CrossRef]
- Henson, M.A.; Phalak, P. Microbiota dysbiosis in inflammatory bowel diseases: In silico investigation of the oxygen hypothesis. BMC Syst. Biol. 2017, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Leclerc, M.; Martin, R.; Chain, F.; Lenoir, M.; Raguideau, S.; Hudault, S.; Bridonneau, C.; Northen, T.; Bowen, B.; et al. Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. mBio 2015, 6, e00300-15. [Google Scholar] [CrossRef] [PubMed]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef]
- McQuade, R.M.; Carbone, S.E.; Stojanovska, V.; Rahman, A.; Gwynne, R.M.; Robinson, A.M.; Goodman, C.A.; Bornstein, J.C.; Nurgali, K. Role of oxidative stress in oxaliplatin-induced enteric neuropathy and colonic dysmotility in mice. Br. J. Pharmacol. 2016, 173, 3502–3521. [Google Scholar] [CrossRef]
- Nurgali, K.; Qu, Z.; Hunne, B.; Thacker, M.; Pontell, L.; Furness, J.B. Morphological and functional changes in guinea-pig neurons projecting to the ileal mucosa at early stages after inflammatory damage. J. Physiol. 2011, 589, 325–339. [Google Scholar] [CrossRef]
- Chandrasekharan, B.; Anitha, M.; Blatt, R.; Shahnavaz, N.; Kooby, D.; Staley, C.; Mwangi, S.; Jones, D.P.; Sitaraman, S.V.; Srinivasan, S. Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol. Motil. 2011, 23, 131–138. [Google Scholar] [CrossRef]
- Qu, Z.-D.; Thacker, M.; Castelucci, P.; Bagyánszki, M.; Epstein, M.L.; Furness, J.B. Immunohistochemical analysis of neuron types in the mouse small intestine. Cell Tissue Res. 2008, 334, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Miller, S.; Payne, N.; Boyd, R.; Sakkal, S.; Nurgali, K. Neuroprotective potential of mesenchymal stem cell-based therapy in acute stages of TNBS-induced colitis in guinea-pigs. PLoS ONE 2015, 10, e0139023. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Sakkal, S.; Park, A.; Jovanovska, V.; Payne, N.; Carbone, S.E. Mesenchymal stem cells and conditioned medium avert enteric neuropathy and colon dysfunction in guinea pig TNBS-induced colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, G1115–G1129. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.; Sidpra, D.; Jevon, G.; Buchan, A.M.; Jacobson, K. Differential responses of VIPergic and nitrergic neurons in paediatric patients with Crohn’s disease. Auton. Neurosci. 2007, 134, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.H.; Li, Q.; Sarna, S.K. Paradoxical regulation of ChAT and nNOS expression in animal models of Crohn’s colitis and ulcerative colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, 295–302. [Google Scholar] [CrossRef]
- Bagyanszki, M.; Bodi, N. Diabetes-related alterations in the enteric nervous system and its microenvironment. World J. Diabetes 2012, 3, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Boughton-Smith, N.K.; Evans, S.M.; Whittle, B.J.R.; Moncada, S.; Hawkey, C.J.; Cole, A.T.; Balsitis, M. Nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Lancet 1993, 342, 2–340. [Google Scholar] [CrossRef]
- Furness, J.B.; Li, Z.S.; Young, H.M.; Förstermann, U. Nitric oxide synthase in the enteric nervous system of the guinea-pig: A quantitative description. Cell Tissue Res. 1994, 277, 139–149. [Google Scholar] [CrossRef] [PubMed]
- D’Incà, R.; Cardin, R.; Benazzato, L.; Angriman, I.; Martines, D.; Sturniolo Giacomo, C. Oxidative DNA damage in the mucosa of ulcerative colitis increases with disease duration and dysplasia. Inflamm. Bowel Dis. 2006, 10, 23–27. [Google Scholar] [CrossRef]
- Fang, J.; Seki, T.; Tsukamoto, T.; Qin, H.; Yin, H.; Liao, L.; Nakamura, H.; Maeda, H. Protection from inflammatory bowel disease and colitis-associated carcinogenesis with 4-vinyl-2,6-dimethoxyphenol (canolol) involves suppression of oxidative stress and inflammatory cytokines. Carcinogenesis 2013, 34, 2833–2841. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–619. [Google Scholar] [CrossRef]
- Kelley, M.R.; Georgiadis, M.M.; Fishel, M.L. APE1/Ref-1 Role in redox signaling: Translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012, 5, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.J.; Robson, C.N.; Black, E.; Gillespie, D.; Hickson, I.D. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol. 1993, 13, 5370–5376. [Google Scholar]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef]
- Georgiadis, M.M.; Luo, M.; Gaur, R.K.; Delaplane, S.; Li, X.; Kelley, M.R. Evolution of the redox function in mammalian Apurinic/apyrimidinic endonuclease. Mutat. Res. 2008, 643, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res./DNA Repair 2001, 485, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, 125. [Google Scholar] [CrossRef]
- Franchi, L.P.; de Freitas Lima, J.E.B.; Piva, H.L.; Tedesco, A.C. The redox function of apurinic/apyrimidinic endonuclease 1 as key modulator in photodynamic therapy. J. Photochem. Photobiol. B Biol. 2020, 211, 111–992. [Google Scholar] [CrossRef] [PubMed]
- Mantha, A.K.; Sarkar, B.; Tell, G. A short review on the implications of base excision repair pathway for neurons: Relevance to neurodegenerative diseases. Mitochondrion 2014, 16, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.R.; Limp-Foster, M.; Kelley, M.R. Going APE over ref-1. Mutat. Res./DNA Repair 2000, 461, 83–108. [Google Scholar] [CrossRef]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 2000, 403, 451. [Google Scholar] [CrossRef]
- Krokan, H.E.; Standal, R.; Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997, 325, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.M.; Monick, M.M.; Carter, A.B.; Peterson, M.W.; Hunninghake, G.W. Oxidant-mediated increases in redox factor-1 nuclear protein and activator protein-1 DNA binding in asbestos-treated macrophages. J. Immunol. 2002, 168, 5675. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Yang, S.; Misner, B.J.; Chiu, R.J.; Meyskens, J.F.L. Redox effector factor-1, combined with reactive oxygen species, plays an important role in the transformation of JB6 cells. Carcinogenesis 2007, 28, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Luo, M.; Kelley, M.R. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: Enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 2004, 3, 679–686. [Google Scholar] [CrossRef]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L.; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the multifunctional DNA repair and redox signaling protein APE1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of APE1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef]
- Sardar Pasha, S.P.B.; Sishtla, K.; Sulaiman, R.S.; Park, B.; Shetty, T.; Shah, F.; Fishel, M.L.; Wikel, J.H.; Kelley, M.R.; Corson, T.W. Ref-1/APE1 inhibition with novel small molecules blocks ocular neovascularization. J. Pharmacol. Exp. Ther. 2018, 367, 108–118. [Google Scholar] [CrossRef]
- Mijit, M.; Caston, R.; Gampala, S.; Fishel, M.L.; Fehrenbacher, J.; Kelley, M.R. APE1/Ref-1—One target with multiple indications: Emerging aspects and new directions. J. Cell. Signal. 2021, 2, 151–161. [Google Scholar]
- Fung, H.; Demple, B. A vital role for APE1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 2005, 17, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. NPJ Precis. Oncol. 2017, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- McNeill, D.R.; Wilson, D.M., III. A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol. Cancer Res. 2007, 5, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Vasko, M.R.; Guo, C.; Thompson, E.L.; Kelley, M.R. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair 2011, 10, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Jiang, Y.; Guo, C.; Reed, A.; Meng, H.; Vasko, M.R. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS ONE 2014, 9, 106. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Zhang, C.; Zhu, H.; Xu, Q.; Bu, Y.; Lei, Y. Redox imbalance in the development of colorectal cancer. J. Cancer 2017, 8, 1586–1597. [Google Scholar] [CrossRef]
- Chang, I.Y.; Kim, J.N.; Maeng, Y.H.; Yoon, S.P. Apurinic/apyrimidinic endonuclease 1, the sensitive marker for DNA deterioration in dextran sulfate sodium-induced acute colitis. Redox Rep. 2013, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sarkar, B.; Cholia, R.P.; Gautam, N.; Dhiman, M.; Mantha, A.K. APE1/Ref-1 as an emerging therapeutic target for various human diseases: Phytochemical modulation of its functions. Exp. Mol. Med. 2014, 46, 106. [Google Scholar] [CrossRef] [PubMed]
- Jedinak, A.; Dudhgaonkar, S.; Kelley, M.R.; Sliva, D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer. Res. 2011, 31, 379–385. [Google Scholar]
- O’Hara, A.M.; Bhattacharyya, A.; Mifflin, R.C.; Smith, M.F.; Ryan, K.A.; Scott, K.G.E.; Naganuma, M.; Casola, A.; Izumi, T.; Mitra, S.; et al. Interleukin-8 induction by helicobacter pylori; in gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J. Immunol. 2006, 177, 7990. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Roychoudhury, S.; Kling, M.J.; Song, H.; Biswas, P.; Shukla, A.; Band, H.; Joshi, S.; Bhakat, K.K. The extracellular role of DNA damage repair protein APE1 in regulation of IL-6 expression. Cell Signal 2017, 39, 18–31. [Google Scholar] [CrossRef]
- Xie, J.Y.; Li, M.X.; Xiang, D.B.; Mou, J.H.; Qing, Y.; Zeng, L.L.; Yang, Z.Z.; Guan, W.; Wang, D. Elevated expression of APE1/Ref-1 and its regulation on IL-6 and IL-8 in bone marrow stromal cells of multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2010, 10, 385–393. [Google Scholar] [CrossRef]
- Han, J.; Theiss, A.L. STAT3: Friend or foe in colitis and colitis-associated cancer? Inflamm. Bowel Dis. 2014, 20, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.; Terui, K.; Zhang, H.Q.; Enosawa, S.; Ogawa, W.; Inoue, H.; Okuyama, T.; Takeda, K.; Akira, S.; Ogino, T.; et al. STAT3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J. Clin. Investig. 2003, 112, 989–998. [Google Scholar] [CrossRef]
- Loeb, K.R.; Loeb, L.A. Genetic instability and the mutator phenotype : Studies in ulcerative colitis. Am. J. Pathol. 1999, 154, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, T.A.; Crispin, D.A.; Bronner, M.P.; Cherian, S.P.; Hueffed, M.; Rabinovitch, P.S.; Rubin, C.E.; Haggitt, R.C.; Boland, C.R. Microsatellite instability in nonneoplastic mucosa from patients with chronic ulcerative colitis. Cancer Res. 1996, 56, 1237. [Google Scholar]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.M.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Loeb, L.A. Tumbling down a different pathway to genetic instability. J. Clin. Investig. 2003, 112, 1793–1795. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seril, D.N.; Liao, J.; Yang, G.-Y.; Yang, C.S. Oxidative stress and ulcerative colitis-associated carcinogenesis: Studies in humans and animal models. Carcinogenesis 2003, 24, 353–362. [Google Scholar] [CrossRef]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Nyland, R.L.; Luo, M.; Kelley, M.R.; Borch, R.F. Design and synthesis of novel quinone inhibitors targeted to the redox function of Apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (APE1/Ref-1). J. Med. Chem. 2010, 53, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.A.; Gampala, S.; Armstrong, L.; Messmann, R.A.; Fishel, M.L.; Kelley, M.R. The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease. Drug Discov. Today 2021, 26, 218–228. [Google Scholar] [CrossRef]
- Lally, D.; Brigell, M.; Withers, B.; Kolli, A.; Rahmani, K.; Sooch, M.; Lazar, A.; Patel, R.; Kelley, M.R.; Boyer, D.; et al. Masked safety data from ZETA-1, an ongoing 24-week Phase 2 clinical trial of APX3330, an oral therapeutic being developed for the treatment of diabetic retinopathy. In Proceedings of the Retina World Congress 2022, Fort Lauderdale, FL, USA, 12–15 May 2022. [Google Scholar]
- Zhong, C.; Xu, M.; Wang, Y.; Xu, J.; Yuan, Y. An APE1 inhibitor reveals critical roles of the redox function of APE1 in KSHV replication and pathogenic phenotypes. PLoS Pathog. 2017, 13, e1006289. [Google Scholar] [CrossRef]
- Shimizu, N.; Sugimoto, K.; Tang, J.; Nishi, T.; Sato, I.; Hiramoto, M.; Aizawa, S.; Hatakeyama, M.; Ohba, R.; Hatori, H.; et al. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000, 18, 877. [Google Scholar] [CrossRef]
- Zou, G.-M.; Maitra, A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol. Cancer Ther. 2008, 7, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Dawodu, O.I.; Johnson, S.M.; Vilseck, J.Z.; Kelley, M.R.; Ziarek, J.J.; Georgiadis, M.M. Chemically induced partial unfolding of the multifunctional Apurinic/apyrimidinic endonuclease 1. bioRxiv 2023. [Google Scholar] [CrossRef]
- Shah, Y.M. The role of hypoxia in intestinal inflammation. Mol. Cell. Pediatr. 2016, 3, 1. [Google Scholar] [CrossRef]
- Xue, X.; Ramakrishnan, S.; Anderson, E.; Taylor, M.; Zimmermann, E.M.; Spence, J.R.; Huang, S.; Greenson, J.K.; Shah, Y.M. Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 2013, 145, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Shahda, S.; Lakhani, N.J.; O’Neil, B.; Rasco, D.W.; Wan, J.; Mosley, A.L.; Liu, H.; Kelley, M.R.; Messmann, R.A. A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3097. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahakian, L.; Robinson, A.M.; Sahakian, L.; Stavely, R.; Kelley, M.R.; Nurgali, K. APE1/Ref-1 as a Therapeutic Target for Inflammatory Bowel Disease. Biomolecules 2023, 13, 1569. https://doi.org/10.3390/biom13111569
Sahakian L, Robinson AM, Sahakian L, Stavely R, Kelley MR, Nurgali K. APE1/Ref-1 as a Therapeutic Target for Inflammatory Bowel Disease. Biomolecules. 2023; 13(11):1569. https://doi.org/10.3390/biom13111569
Chicago/Turabian StyleSahakian, Lauren, Ainsley M. Robinson, Linda Sahakian, Rhian Stavely, Mark R. Kelley, and Kulmira Nurgali. 2023. "APE1/Ref-1 as a Therapeutic Target for Inflammatory Bowel Disease" Biomolecules 13, no. 11: 1569. https://doi.org/10.3390/biom13111569
APA StyleSahakian, L., Robinson, A. M., Sahakian, L., Stavely, R., Kelley, M. R., & Nurgali, K. (2023). APE1/Ref-1 as a Therapeutic Target for Inflammatory Bowel Disease. Biomolecules, 13(11), 1569. https://doi.org/10.3390/biom13111569