
Emerging Roles for Dendritic Cells in Heart Failure

 ,
, {kind=link}
Abstract
:1. Introduction
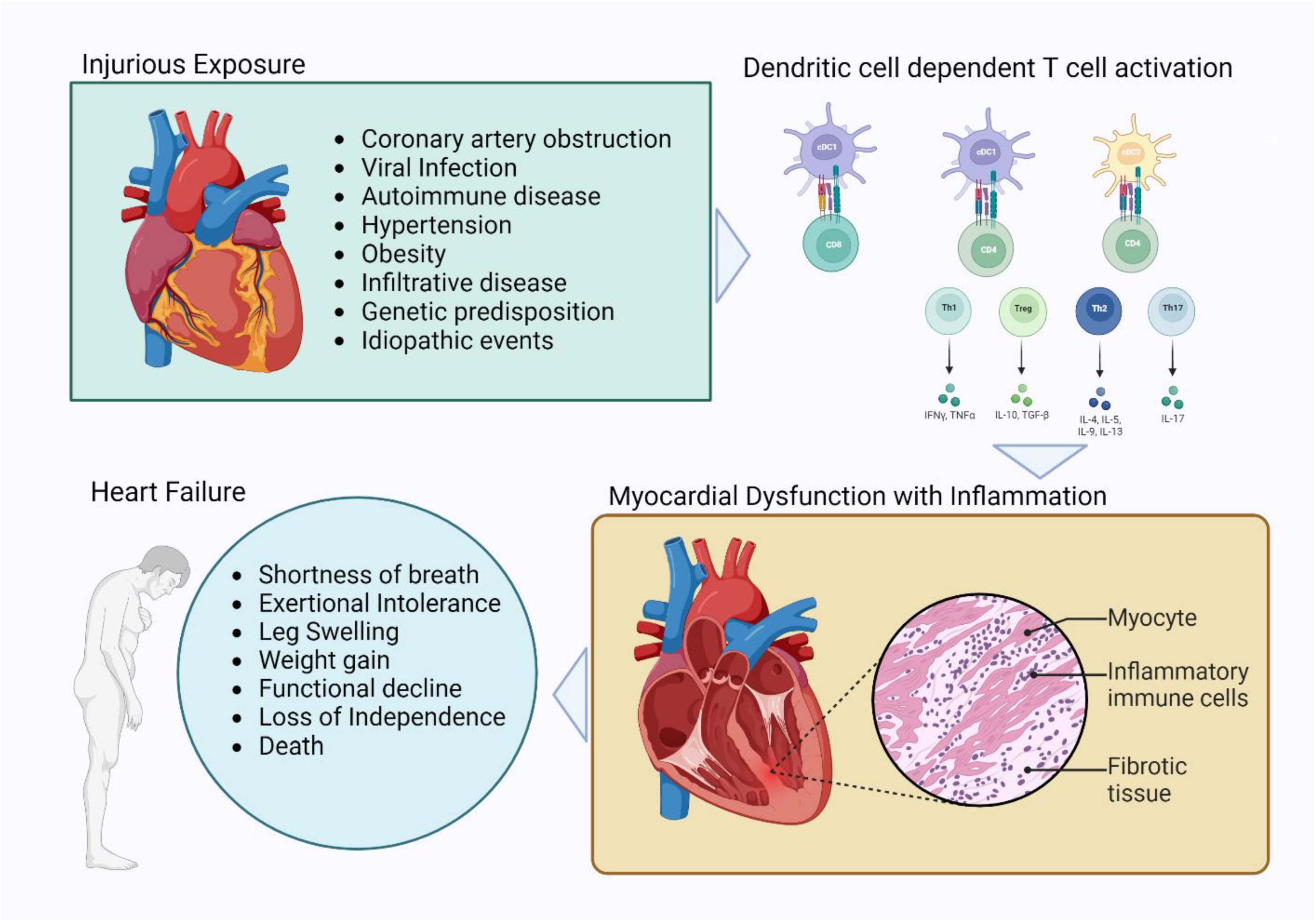
1.1. Cardiovascular Disease, Heart Failure, and Inflammation
1.2. Dendritic Cells
1.3. Myocardial Inflammation and the Diversity of Heart Failure
Dilated Cardiomyopathy (Idiopathic Non-Ischemic Cardiomyopathy)
1.4. Acute Myocardial Infarction with Ischemic Cardiomyopathy
1.5. Myocarditis
1.6. Hypertension and HFpEF
1.7. Infiltrative Heart Disease and Cardiac Amyloid
2. Discussion and Future Directions
Funding
Conflicts of Interest
References
- Abbas, A.K.L.; Andrew, H.; Pillai, S. Cellular and Molecular Immunology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2021; ISBN 978-0-323-75748-5. [Google Scholar]
- Libby, P.B.; Robert, O.; Mann, D.L.; Tomaselli, G.F.; Bhatt, D.L.; Soloman, S.D.; Braunwald, E. Braunwald’s Heart Disease, 12th ed.; Elsevier: Philadelphia, PA, USA, 2022; Volume 2, p. 2034. [Google Scholar]
- Mann, D.L.; Topkara, V.K.; Evans, S.; Barger, P.M. Innate immunity in the adult mammalian heart: For whom the cell tolls. Trans. Am. Clin. Clim. Assoc. 2010, 121, 34–51. [Google Scholar]
- Nguyen, L.S.; Cooper, L.T.; Kerneis, M.; Funck-Brentano, C.; Silvain, J.; Brechot, N.; Hekimian, G.; Ammirati, E.; Ben M’barek, B.; Redheuil, A.; et al. Systematic analysis of drug-associated myocarditis reported in the World Health Organization pharmacovigilance database. Nat. Commun. 2022, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Zhou, G.; Rokosh, G.; Hamid, T.; Prabhu, S.D. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation 2019, 139, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Khan, M.; Mohsin, S. The Regulatory Role of T Cell Responses in Cardiac Remodeling Following Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 5013. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, H.; Li, Z.; Dress, R.J.; Zhu, Y.; Zhang, S.; De Feo, D.; Kong, W.T.; Cai, P.; Shin, A.; et al. Dendritic cell type 3 arises from Ly6C+ monocyte-dendritic cell progenitors. Immunity 2023, 56, 1761–1777.e6. [Google Scholar] [CrossRef]
- Anderson, D.A.; Dutertre, C.-A.; Ginhoux, F.; Murphy, K.M. Genetic models of human and mouse dendritic cell development and function. Nat. Rev. Immunol. 2020, 21, 101–115. [Google Scholar] [CrossRef]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; da Costa, M.P.; e Sousa, C.R. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef]
- Naik, S.H.; Sathe, P.; Park, H.-Y.; Metcalf, D.; Proietto, A.I.; Dakic, A.; Carotta, S.; O’Keeffe, M.; Bahlo, M.; Papenfuss, A.; et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 2007, 8, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Onai, N.; Obata-Onai, A.; A Schmid, M.; Ohteki, T.; Jarrossay, D.; Manz, M.G. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat. Immunol. 2007, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Breton, G.; Zheng, S.; Valieris, R.; da Silva, I.T.; Satija, R.; Nussenzweig, M.C. Human dendritic cells (DCs) are derived from distinct circulating precursors that are precommitted to become CD1c+ or CD141+ DCs. J. Exp. Med. 2016, 213, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Dutertre, C.-A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Dress, R.J.; Dutertre, C.-A.; Giladi, A.; Schlitzer, A.; Low, I.; Shadan, N.B.; Tay, A.; Lum, J.; Kairi, M.F.B.M.; Hwang, Y.Y.; et al. Plasmacytoid dendritic cells develop from Ly6D+ lymphoid progenitors distinct from the myeloid lineage. Nat. Immunol. 2019, 20, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Chopin, M. Transcriptional Networks Driving Dendritic Cell Differentiation and Function. Immunity 2020, 52, 942–956. [Google Scholar] [CrossRef]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e8. [Google Scholar] [CrossRef] [PubMed]
- See, P.; Dutertre, C.-A.; Chen, J.; Günther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Händler, K.; Duan, K.; et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science 2017, 356, eaag3009. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Gillum, R.F. Idiopathic cardiomyopathy in the United States, 1970–1982. Am. Heart J. 1986, 111, 752–755. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Martino, T.A.; Liu, P.; Sole, M.J. Viral infection and the pathogenesis of dilated cardiomyopathy. Circ. Res. 1994, 74, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Durand, J.-B.; Bies, R.D.; Young, J.B.; Mann, D.L. Tumor Necrosis Factor-α and Tumor Necrosis Factor Receptors in the Failing Human Heart. Circulation 1996, 93, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Noutsias, M.; Pauschinger, M.; Schultheiss, H.; Hl, U.K. Phenotypic characterization of infiltrates in dilated cardiomyopathy—Diagnostic significance of T-lymphocytes and macrophages in inflammatory cardiomyopathy. Med. Sci. Monit. 2002, 8, 478–487. [Google Scholar]
- El-Hagrassy, M.M.; Banatvala, J.E.; Coltart, D.J. Coxsackie-B-Virus-Specific Igm Responses in Patients with Cardiac and Other Diseases. Lancet 1980, 316, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Muir, P.; Tilzey, A.; English, T.; Nicholson, F.; Signy, M.; Banatvala, J. Chronic Relapsing Pericarditis and Dilated Cardiomyopathy: Serological Evidence of Persistent Enterovirus Infection. Lancet 1989, 333, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.J.; Eglin, R.P. The Detection of Coxsackievirus RNA in Cardiac Tissue by in situ Hybridization. J. Gen. Virol. 1988, 6 Pt 2, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.M.; Miranda, Q.R.; Chason, J.L.; Lerner, A.M. Residual pathologic changes following murine coxsackie A and B myocarditis. Am. J. Pathol. 1969, 55, 253–265. [Google Scholar]
- Reyes, M.P.; Do, K.L.; Smith, F.; Lerner, A.M. A Mouse Model of Dilated-Type Cardiomyopathy Due to Coxsackievirus B3. J. Infect. Dis. 1981, 144, 232–236. [Google Scholar] [CrossRef]
- Eriksson, U.; Ricci, R.; Hunziker, L.; O Kurrer, M.; Oudit, G.Y.; Watts, T.H.; Sonderegger, I.; Bachmaier, K.; Kopf, M.; Penninger, J.M. Dendritic cell–induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 2003, 9, 1484–1490. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, E895–E1032. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef] [PubMed]
- Liehn, E.A.; Postea, O.; Curaj, A.; Marx, N. Repair after Myocardial Infarction, between Fantasy and Reality: The Role of Chemokines. J. Am. Coll. Cardiol. 2011, 58, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P. Importance of TIMI 3 flow. Circulation 2001, 104, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Kammler, J.; Kypta, A.; Hofmann, R.; Kerschner, K.; Grund, M.; Sihorsch, K.; Steinwender, C.; Lambert, T.; Helml, W.; Leisch, F. TIMI 3 flow after primary angioplasty is an important predictor for outcome in patients with acute myocardial infarction. Clin. Res. Cardiol. 2009, 98, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Prasada, S.; Rivera, A.; Nishtala, A.; Pawlowski, A.E.; Sinha, A.; Bundy, J.D.; Chadha, S.A.; Ahmad, F.S.; Khan, S.S.; Achenbach, C.; et al. Differential Associations of Chronic Inflammatory Diseases with Incident Heart Failure. JACC Heart Fail. 2020, 8, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S.; Sullivan, L.M.; Roubenoff, R.; Dinarello, C.A.; Harris, T.B.; Benjamin, E.J.; Sawyer, D.B.; Levy, D.; Wilson, P.W.; D’agostino, R.B.; et al. Inflammatory Markers and Risk of Heart Failure in Elderly Subjects without Prior Myocardial Infarction: The Framingham Heart Study. Circulation 2003, 107, 1486–1491. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the Mononuclear Phagocyte Network Underlies Chronic Inflammation and Disease Progression in Heart Failure: Critical importance of the cardiosplenic axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef]
- Baritussio, A.; Scatteia, A.; Bucciarelli-Ducci, C. Role of cardiovascular magnetic resonance in acute and chronic ischemic heart disease. Int. J. Cardiovasc. Imaging 2018, 34, 67–80. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef]
- Wernly, B.; Paar, V.; Aigner, A.; Pilz, P.M.; Podesser, B.K.; Förster, M.; Jung, C.; Hofbauer, J.P.; Tockner, B.; Wimmer, M.; et al. Anti-CD3 Antibody Treatment Reduces Scar Formation in a Rat Model of Myocardial Infarction. Cells 2020, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Santos-Zas, I.; Lemarié, J.; Zlatanova, I.; Cachanado, M.; Seghezzi, J.-C.; Benamer, H.; Goube, P.; Vandestienne, M.; Cohen, R.; Ezzo, M.; et al. Cytotoxic CD8+ T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 2021, 12, 1483. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T Lymphocytes Improves Wound Healing and Survival after Experimental Myocardial Infarction in Mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Noma, T.; Ishihara, Y.; Miyauchi, Y.; Takabatake, W.; Oomizu, S.; Yamaoka, G.; Ishizawa, M.; Namba, T.; Murakami, K.; et al. Prognostic Value of Circulating Regulatory T Cells for Worsening Heart Failure in Heart Failure Patients with Reduced Ejection Fraction. Int. Heart J. 2014, 55, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Blanton, R.M.; Carrillo-Salinas, F.J.; Alcaide, P. T-cell recruitment to the heart: Friendly guests or unwelcome visitors? Am. J. Physiol. Circ. Physiol. 2019, 317, H124–H140. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Colon, E.; Sampson, R.D.; Sattler, S.; Schneider, M.D.; Rosenthal, N.; Tonkin, J. Cardiac-Restricted IGF-1Ea Overexpression Reduces the Early Accumulation of Inflammatory Myeloid Cells and Mediates Expression of Extracellular Matrix Remodelling Genes after Myocardial Infarction. Mediat. Inflamm. 2015, 2015, 484357. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Panahi, M.; Baxan, N.; Ng, F.S.; Boyle, J.J.; Branca, J.; Bedard, O.; Hasham, M.G.; Benson, L.; Harding, S.E.; et al. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J. Cell. Mol. Med. 2020, 25, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) Cardiac Amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef]
- Sousa, M.M.; Du Yan, S.; Fernandes, R.; Guimarães, A.; Stern, D.; Saraiva, M.J. Familial Amyloid Polyneuropathy: Receptor for Advanced Glycation End Products-Dependent Triggering of Neuronal Inflammatory and Apoptotic Pathways. J. Neurosci. 2001, 21, 7576–7586. [Google Scholar] [CrossRef]
- Forte, E.; Perkins, B.; Sintou, A.; Kalkat, H.S.; Papanikolaou, A.; Jenkins, C.; Alsubaie, M.; Chowdhury, R.A.; Duffy, T.M.; Skelly, D.A.; et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology after Ischemic Tissue Damage in the Heart. Circulation 2021, 143, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Inui, H.; Nishida, M.; Ichii, M.; Nakaoka, H.; Asaji, M.; Ide, S.; Saito, S.; Saga, A.; Omatsu, T.; Tanaka, K.; et al. XCR1+ conventional dendritic cell-induced CD4+ T helper 1 cell activation exacerbates cardiac remodeling after ischemic myocardial injury. J. Mol. Cell. Cardiol. 2023, 176, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Van der Borght, K.; Scott, C.L.; Martens, L.; Sichien, D.; Van Isterdael, G.; Nindl, V.; Saeys, Y.; Boon, L.; Ludewig, B.; Gillebert, T.C.; et al. Myocarditis Elicits Dendritic Cell and Monocyte Infiltration in the Heart and Self-Antigen Presentation by Conventional Type 2 Dendritic Cells. Front. Immunol. 2018, 9, 2714. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.W. Myocarditis and dilated cardiomyopathy an inflammatory link. Cardiovasc. Res. 2003, 60, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, M.L.; Meijers, W.C.; Screever, E.M.; Qin, J.; Carroll, M.G.; Sun, X.; Tannous, E.; Zhang, Y.; Sugiura, A.; Taylor, B.C.; et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature 2022, 611, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Pistulli, R.; Andreas, E.; König, S.; Drobnik, S.; Kretzschmar, D.; Rohm, I.; Lichtenauer, M.; Heidecker, B.; Franz, M.; Mall, G.; et al. Characterization of dendritic cells in human and experimental myocarditis. ESC Heart Fail. 2020, 7, 2305–2317. [Google Scholar] [CrossRef]
- Neu, N.; Rose, N.R.; Beisel, K.W.; Herskowitz, A.; Gurri-Glass, G.; Craig, S.W. Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 1987, 139, 3630–3636. [Google Scholar] [CrossRef]
- Kodama, M.; Matsumoto, Y.; Fujiwara, M. In vivo lymphocyte-mediated myocardial injuries demonstrated by adoptive transfer of experimental autoimmune myocarditis. Circulation 1992, 85, 1918–1926. [Google Scholar] [CrossRef]
- Smith, S.C.; Allen, P.M. Myosin-induced acute myocarditis is a T cell-mediated disease. J. Immunol. 1991, 147, 2141–2147. [Google Scholar] [CrossRef]
- Pummerer, C.; Berger, P.; Frühwirth, M.; Ofner, C.; Neu, N. Cellular infiltrate, major histocompatibility antigen expression and immunopathogenic mechanisms in cardiac myosin-induced myocarditis. Lab. Investig. 1991, 65, 538–547. [Google Scholar] [PubMed]
- Anzai, A.; Mindur, J.E.; Halle, L.; Sano, S.; Choi, J.L.; He, S.; McAlpine, C.S.; Chan, C.T.; Kahles, F.; Valet, C.; et al. Self-reactive CD4+ IL-3+ T cells amplify autoimmune inflammation in myocarditis by inciting monocyte chemotaxis. J. Exp. Med. 2019, 216, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Heuser, J.S.; Kosanke, S.D.; Hemric, M.; Cunningham, M.W. Protection against Experimental Autoimmune Myocarditis Is Mediated by Interleukin-10-Producing T Cells that Are Controlled by Dendritic Cells. Am. J. Pathol. 2005, 167, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Drazner, M.H. The Progression of Hypertensive Heart Disease. Circulation 2011, 123, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Díez, J.; Butler, J. Growing Heart Failure Burden of Hypertensive Heart Disease: A Call to Action. Hypertension 2023, 80, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Galarraga, B.; Khan, F.; Kumar, P.; Pullar, T.; Belch, J.J.F. Etanercept improves inflammation-associated arterial stiffness in rheumatoid arthritis. Rheumatology 2009, 48, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Selzer, F.; Sutton-Tyrrell, K.; Fitzgerald, S.; Tracy, R.; Kuller, L.; Manzi, S. Vascular Stiffness in Women With Systemic Lupus Erythematosus. Hypertension 2001, 37, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Saleh, M.A.; Kirabo, A.; Itani, H.A.; Montaniel, K.R.C.; Xiao, L.; Chen, W.; Mernaugh, R.L.; Cai, H.; Bernstein, K.E.; et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J. Clin. Investig. 2016, 126, 1607. [Google Scholar] [CrossRef]
- Itani, H.A.; Xiao, L.; Saleh, M.A.; Wu, J.; Pilkinton, M.A.; Dale, B.L.; Barbaro, N.R.; Foss, J.D.; Kirabo, A.; Montaniel, K.R.; et al. CD70 Exacerbates Blood Pressure Elevation and Renal Damage in Response to Repeated Hypertensive Stimuli. Circ. Res. 2016, 118, 1233–1243. [Google Scholar] [CrossRef]
- Kirabo, A.; Fontana, V.; de Faria, A.P.; Loperena, R.; Galindo, C.L.; Wu, J.; Bikineyeva, A.T.; Dikalov, S.; Xiao, L.; Chen, W.; et al. DC isoketal-modified proteins activate T cells and promote hypertension. J. Clin. Investig. 2014, 124, 4642–4656. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II–induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Vinh, A.; Chen, W.; Blinder, Y.; Weiss, D.; Taylor, W.R.; Goronzy, J.J.; Weyand, C.M.; Harrison, D.G.; Guzik, T.J. Inhibition and Genetic Ablation of the B7/CD28 T-Cell Costimulation Axis Prevents Experimental Hypertension. Circulation 2010, 122, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Capcha, J.M.C.; Irion, C.I.; Seo, G.; Lambert, G.; Kamiar, A.; Yousefi, K.; Kanashiro-Takeuchi, R.; Takeuchi, L.; Saad, A.G.; et al. Mouse Model of Heart Failure with Preserved Ejection Fraction Driven by Hyperlipidemia and Enhanced Cardiac Low-Density Lipoprotein Receptor Expression. J. Am. Heart Assoc. 2022, 11, e027216. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, N.P.; Vieira, P.; Saraiva, M.J. Interleukin-1 signaling pathway as a therapeutic target in transthyretin amyloidosis. Amyloid 2014, 21, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.D.; Fernandes, R.; Saraiva, M.J. The heat shock response modulates transthyretin deposition in the peripheral and autonomic nervous systems. Neurobiol. Aging 2010, 31, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Tagoe, C.; Gallo, G.; Walker, J.R.; Kurian, S.; Salomon, D.R. Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 2012, 26, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Siegismund, C.S.; Escher, F.; Lassner, D.; Kühl, U.; Gross, U.; Fruhwald, F.; Wenzel, P.; Münzel, T.; Frey, N.; Linke, R.P.; et al. Intramyocardial inflammation predicts adverse outcome in patients with cardiac AL amyloidosis. Eur. J. Heart Fail. 2018, 20, 751–757. [Google Scholar] [CrossRef]
- Banypersad, S.M. The Evolving Role of Cardiovascular Magnetic Resonance Imaging in the Evaluation of Systemic Amyloidosis. Magn. Reson. Insights 2019, 12, 3519. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, D.; Jones, R.T.L.; Schroth, S.L.; Thorp, E.B.; Feinstein, M.J. Emerging Roles for Dendritic Cells in Heart Failure. Biomolecules 2023, 13, 1535. https://doi.org/10.3390/biom13101535
Saleh D, Jones RTL, Schroth SL, Thorp EB, Feinstein MJ. Emerging Roles for Dendritic Cells in Heart Failure. Biomolecules. 2023; 13(10):1535. https://doi.org/10.3390/biom13101535
Chicago/Turabian StyleSaleh, Danish, Rebecca T. L. Jones, Samantha L. Schroth, Edward B. Thorp, and Matthew J. Feinstein. 2023. "Emerging Roles for Dendritic Cells in Heart Failure" Biomolecules 13, no. 10: 1535. https://doi.org/10.3390/biom13101535
APA StyleSaleh, D., Jones, R. T. L., Schroth, S. L., Thorp, E. B., & Feinstein, M. J. (2023). Emerging Roles for Dendritic Cells in Heart Failure. Biomolecules, 13(10), 1535. https://doi.org/10.3390/biom13101535