
COVID-19 Pathology Sheds Further Light on Balance between Neutrophil Proteases and Their Inhibitors



{kind=link}
{kind=link}
Abstract
1. Introduction

2. Neutrophil Serine Protease
3. Human Alpha-1 Antitrypsin
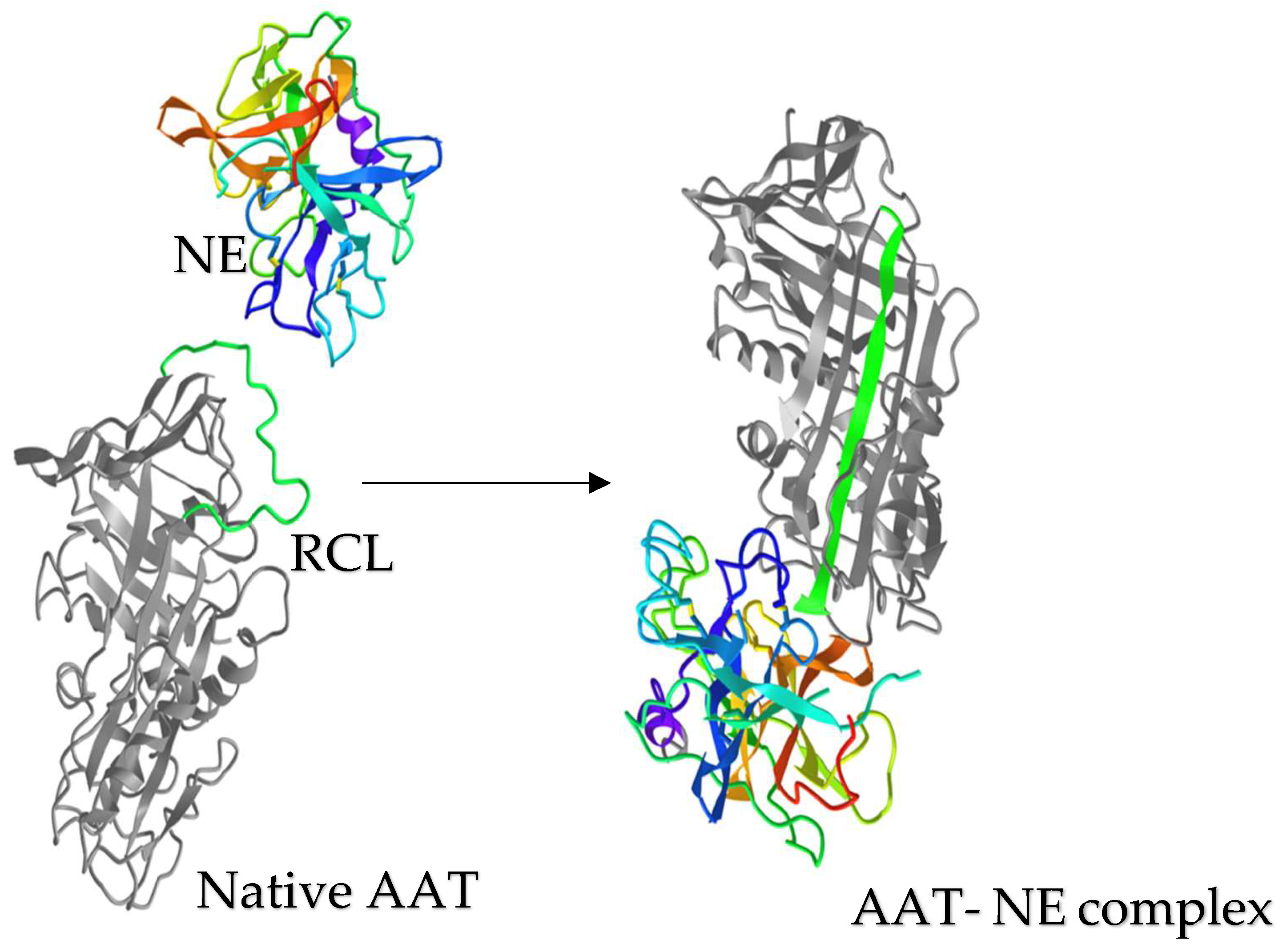
4. Alpha-1 Antitrypsin Structure and Inhibitory Mechanism
5. Alpha-1 Antitrypsin Deficiency
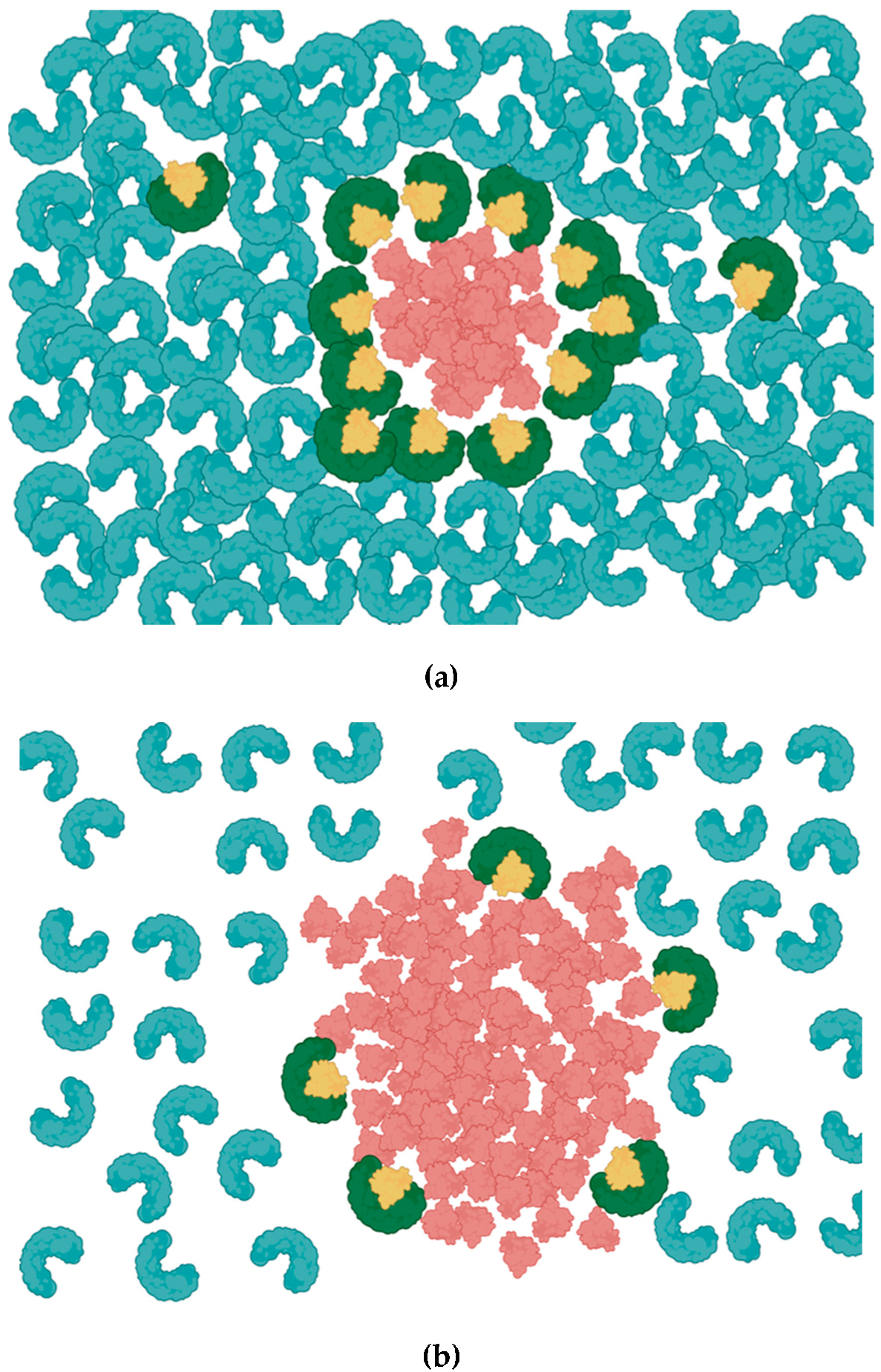
6. Effects of NETosis in Respiratory Diseases and COVID-19
7. AAT Supplementation to Reduce NET-Mediated Pathogenesis
8. Role of Neutrophil-Derived AAT
9. AAT Protection against COVID-19
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on Neutrophil Function in Severe Inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Allen, L.H. Phagocytosis and neutrophil extracellular traps. Fac. Rev. 2020, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kodama, T.; Yukioka, H.; Kato, T.; Kato, N.; Hato, F.; Kitagawa, S. Neutrophil elastase as a predicting factor for development of acute lung injury. Intern. Med. 2007, 46, 699–704. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef]
- Mócsai, A.; Walzog, B.; Lowell, C.A. Intracellular signalling during neutrophil recruitment. Cardiovasc. Res. 2015, 107, 373–385. [Google Scholar] [CrossRef]
- Gramegna, A.; Amati, F.; Terranova, L.; Sotgiu, G.; Tarsia, P.; Miglietta, D.; Calderazzo, M.A.; Aliberti, S.; Blasi, F. Neutrophil elastase in bronchiectasis. Respir. Res. 2017, 18, 211. [Google Scholar] [CrossRef]
- Strich, J.R.; Ramos-Benitez, M.J.; Randazzo, D.; Stein, S.R.; Babyak, A.; Davey, R.T.; Suffredini, A.F.; Childs, R.W.; Chertow, D.S. Fostamatinib Inhibits Neutrophils Extracellular Traps Induced by COVID-19 Patient Plasma: A Potential Therapeutic. J. Infect. Dis. 2021, 223, 981–984. [Google Scholar] [CrossRef]
- Park, J.A.; He, F.; Martin, L.D.; Li, Y.; Chorley, B.N.; Adler, K.B. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase C{delta}-mediated mechanism. Am. J. Pathol. 2005, 167, 651–661. [Google Scholar] [CrossRef]
- Von Bredow, C.; Wiesener, A.; Griese, M. Proteolysis of surfactant protein D by cystic fibrosis relevant proteases. Lung 2003, 181, 79–88. [Google Scholar] [CrossRef]
- Kao, S.S.; Ramezanpour, M.; Bassiouni, A.; Wormald, P.J.; Psaltis, A.J.; Vreugde, S. The effect of neutrophil serine proteases on human nasal epithelial cell barrier function. Int. Forum. Allergy Rhinol. 2019, 9, 1220–1226. [Google Scholar] [CrossRef]
- Demkow, U.; van Overveld, F.J. Role of elastases in the pathogenesis of chronic obstructive pulmonary disease: Implications for treatment. Eur. J. Med. Res. 2010, 15 (Suppl. S2), 27–35. [Google Scholar] [CrossRef]
- Mollica, V.; Rizzo, A.; Massari, F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020, 16, 2029–2033. [Google Scholar] [CrossRef]
- Mustafa, Z.; Zhanapiya, A.; Kalbacher, H.; Burster, T. Neutrophil Elastase and Proteinase 3 Cleavage Sites Are Adjacent to the Polybasic Sequence within the Proteolytic Sensitive Activation Loop of the SARS-CoV-2 Spike Protein. ACS Omega 2021, 6, 7181–7185. [Google Scholar] [CrossRef]
- McElvaney, O.F.; Asakura, T.; Meinig, S.L.; Torres-Castillo, J.L.; Hagan, R.S.; Gabillard-Lefort, C.; Murphy, M.P.; Thorne, L.B.; Borczuk, A.; Reeves, E.P.; et al. Protease-anti-protease compartmentalization in SARS-CoV-2 ARDS: Therapeutic implications. EBioMedicine 2022, 77, 103894. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Demkow, U. Neutrophil Extracellular Traps (NETs) in Severe SARS-CoV-2 Lung Disease. Int. J. Mol. Sci. 2021, 22, 8854. [Google Scholar] [CrossRef]
- Di Cera, E. Serine proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; Li, L.; Sahu, S.K.; Petersen, M.; Liu, X.; Melkonian, M.; Zhang, G.; Liu, H. Molecular evidence for origin, diversification and ancient gene duplication of plant subtilases (SBTs). Sci. Rep. 2019, 9, 12485. [Google Scholar] [CrossRef] [PubMed]
- Drescher, B.; Bai, F. Neutrophil in viral infections, friend or foe? Virus. Res. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- AhYoung, A.P.; Eckard, S.C.; Gogineni, A.; Xi, H.; Lin, S.J.; Gerhardy, S.; Cox, C.; Phung, Q.T.; Hackney, J.A.; Katakam, A.K.; et al. Neutrophil serine protease 4 is required for mast cell-dependent vascular leakage. Commun. Biol. 2020, 3, 687. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Pompili, S.; Latella, G.; Gaudio, E.; Sferra, R.; Vetuschi, A. The Charming World of the Extracellular Matrix: A Dynamic and Protective Network of the Intestinal Wall. Front. Med. 2021, 8, 610189. [Google Scholar] [CrossRef]
- Pham, C.T. Neutrophil serine proteases fine-tune the inflammatory response. Int. J. Biochem. Cell Biol. 2008, 40, 1317–1333. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Aliter, K.F.; Kar, S.; Mottamal, M. Sulfonated Nonsaccharide Heparin Mimetics Are Potent and Noncompetitive Inhibitors of Human Neutrophil Elastase. ACS Omega 2021, 6, 12699–12710. [Google Scholar] [CrossRef]
- Paone, G.; Conti, V.; Vestri, A.; Leone, A.; Puglisi, G.; Benassi, F.; Brunetti, G.; Schmid, G.; Cammarella, I.; Terzano, C. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis. Markers 2011, 31, 91–100. [Google Scholar] [CrossRef]
- Domon, H.; Nagai, K.; Maekawa, T.; Oda, M.; Yonezawa, D.; Takeda, W.; Hiyoshi, T.; Tamura, H.; Yamaguchi, M.; Kawabata, S.; et al. Neutrophil Elastase Subverts the Immune Response by Cleaving Toll-Like Receptors and Cytokines in Pneumococcal Pneumonia. Front. Immunol. 2018, 9, 732. [Google Scholar] [CrossRef]
- Aikawa, N.; Kawasaki, Y. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther. Clin. Risk Manag. 2014, 10, 621–629. [Google Scholar] [CrossRef]
- Sahoo, M.; Del Barrio, L.; Miller, M.A.; Re, F. Neutrophil elastase causes tissue damage that decreases host tolerance to lung infection with burkholderia species. PLoS Pathog. 2014, 10, e1004327. [Google Scholar] [CrossRef]
- Voynow, J.A.; Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef]
- Sanrattana, W.; Maas, C.; de Maat, S. SERPINs-From Trap to Treatment. Front. Med. 2019, 6, 25. [Google Scholar] [CrossRef]
- Sallenave, J.M. The role of secretory leukocyte proteinase inhibitor and elafin (elastase-specific inhibitor/skin-derived antileukoprotease) as alarm antiproteinases in inflammatory lung disease. Respir. Res. 2000, 1, 87–92. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Wrenger, S.; Immenschuh, S.; Olejnicka, B.; Greulich, T.; Welte, T.; Chorostowska-Wynimko, J. The Multifaceted Effects of Alpha1-Antitrypsin on Neutrophil Functions. Front. Pharmacol. 2018, 9, 341. [Google Scholar] [CrossRef]
- Engelmaier, A.; Weber, A. Sensitive and specific measurement of alpha(1)-antitrypsin activity with an elastase complex formation immunosorbent assay (ECFISA). J. Pharm. BioMed. Anal. 2022, 209, 114476. [Google Scholar] [CrossRef]
- Benarafa, C. Regulation of Neutrophil Serine Proteases by Intracellular Serpins. Serpin Fam. 2015, 59, 76. [Google Scholar]
- De Serres, F.; Blanco, I. Role of alpha-1 antitrypsin in human health and disease. J. Intern. Med. 2014, 276, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Torres-Durán, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Nita, I.M.; Serapinas, D.; Janciauskiene, S.M. Alpha1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int. J. Biochem. Cell Biol. 2007, 39, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Bergin, D.A.; Reeves, E.P.; Hurley, K.; Wolfe, R.; Jameel, R.; Fitzgerald, S.; McElvaney, N.G. The circulating proteinase inhibitor α-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci. Transl. Med. 2014, 6, 217ra211. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; Larsson, S.; Larsson, P.; Virtala, R.; Jansson, L.; Stevens, T. Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by alpha1-antitrypsin. Biochem. Biophys. Res. Commun. 2004, 321, 592–600. [Google Scholar] [CrossRef]
- Shapiro, L.; Pott, G.B.; Ralston, A.H. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001, 15, 115–122. [Google Scholar] [CrossRef]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Prelli Bozzo, C.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef]
- Fleixo-Lima, G.; Ventura, H.; Medini, M.; Bar, L.; Strauss, P.; Lewis, E.C. Mechanistic evidence in support of alpha1-antitrypsin as a therapeutic approach for type 1 diabetes. J. Diabetes Sci. Technol. 2014, 8, 1193–1203. [Google Scholar] [CrossRef]
- Ochayon, D.E.; Mizrahi, M.; Shahaf, G.; Baranovski, B.M.; Lewis, E.C. Human α1-Antitrypsin Binds to Heat-Shock Protein gp96 and Protects from Endogenous gp96-Mediated Injury In vivo. Front. Immunol. 2013, 4, 320. [Google Scholar] [CrossRef]
- Zhou, A.; Carrell, R.W.; Huntington, J.A. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J. Biol. Chem. 2001, 276, 27541–27547. [Google Scholar] [CrossRef]
- Jonigk, D.; Al-Omari, M.; Maegel, L.; Müller, M.; Izykowski, N.; Hong, J.; Hong, K.; Kim, S.H.; Dorsch, M.; Mahadeva, R.; et al. Anti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastase. Proc. Natl. Acad. Sci. USA 2013, 110, 15007–15012. [Google Scholar] [CrossRef]
- Renoux, C.; Odou, M.F.; Tosato, G.; Teoli, J.; Abbou, N.; Lombard, C.; Zerimech, F.; Porchet, N.; Chapuis Cellier, C.; Balduyck, M.; et al. Description of 22 new alpha-1 antitrypsin genetic variants. Orphanet J. Rare Dis. 2018, 13, 161. [Google Scholar] [CrossRef]
- Greulich, T. Alpha-1-Antitrypsin Deficiency: Disease Management and Learning from Studies. COPD 2017, 14, S8–S11. [Google Scholar] [CrossRef]
- Hutchison, D.C. Alpha 1-antitrypsin deficiency in Europe: Geographical distribution of Pi types S and Z. Respir. Med. 1998, 92, 367–377. [Google Scholar] [CrossRef]
- Jagger, A.M.; Waudby, C.A.; Irving, J.A.; Christodoulou, J.; Lomas, D.A. High-resolution ex vivo NMR spectroscopy of human Z α(1)-antitrypsin. Nat. Commun. 2020, 11, 6371. [Google Scholar] [CrossRef]
- McCarthy, C.; Saldova, R.; Wormald, M.R.; Rudd, P.M.; McElvaney, N.G.; Reeves, E.P. The role and importance of glycosylation of acute phase proteins with focus on alpha-1 antitrypsin in acute and chronic inflammatory conditions. J. Proteome Res. 2014, 13, 3131–3143. [Google Scholar] [CrossRef]
- Silverman, G.A.; Pak, S.C.; Perlmutter, D.H. Disorders of protein misfolding: Alpha-1-antitrypsin deficiency as prototype. J. Pediatr. 2013, 163, 320–326. [Google Scholar] [CrossRef]
- Scott, B.M.; Sheffield, W.P. Engineering the serpin α(1)-antitrypsin: A diversity of goals and techniques. Protein. Sci. 2020, 29, 856–871. [Google Scholar] [CrossRef]
- Hubbard, R.C.; Fells, G.; Gadek, J.; Pacholok, S.; Humes, J.; Crystal, R.G. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J. Clin. Investig. 1991, 88, 891–897. [Google Scholar] [CrossRef]
- O’Dwyer, C.A.; O’Brien, M.E.; Wormald, M.R.; White, M.M.; Banville, N.; Hurley, K.; McCarthy, C.; McElvaney, N.G.; Reeves, E.P. The BLT1 Inhibitory Function of α-1 Antitrypsin Augmentation Therapy Disrupts Leukotriene B4 Neutrophil Signaling. J. Immunol. 2015, 195, 3628–3641. [Google Scholar] [CrossRef]
- Bergin, D.A.; Reeves, E.P.; Meleady, P.; Henry, M.; McElvaney, O.J.; Carroll, T.P.; Condron, C.; Chotirmall, S.H.; Clynes, M.; O’Neill, S.J.; et al. α-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J. Clin. Investig. 2010, 120, 4236–4250. [Google Scholar] [CrossRef] [PubMed]
- Mansuy-Aubert, V.; Zhou, Q.L.; Xie, X.; Gong, Z.; Huang, J.Y.; Khan, A.R.; Aubert, G.; Candelaria, K.; Thomas, S.; Shin, D.J.; et al. Imbalance between neutrophil elastase and its inhibitor α1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013, 17, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Damas, C.; Azevedo, F.; Mota, A. Cutaneous Manifestation of Alpha-1 Antitrypsin Deficiency: A Case of Panniculitis. Indian J. Dermatol. 2018, 63, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, A.N.; Ralph, J.; O’Farrell, N.J.; Buckley, C.; Gulmann, C.; O’Kane, M.; Carroll, T.P.; McElvaney, N.G. Alpha-1 antitrypsin deficiency-associated panniculitis. J. Am. Acad. Dermatol. 2022, 87, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Neutrophils, from cradle to grave and beyond. Immunol. Rev. 2016, 273, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Taylor, E.B. Casting a wide NET: An update on uncontrolled NETosis in response to COVID-19 infection. Clin. Sci. 2022, 136, 1047–1052. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Xu, S.; Yin, Y.; Ageno, W.; De Stefano, V.; Zhao, Q.; Qi, X. Clinical significance of neutrophil extracellular traps biomarkers in thrombosis. Thromb. J. 2022, 20, 63. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Giaglis, S.; Hahn, S.; Blum, C.A.; Baumgartner, C.; Kutz, A.; van Breda, S.V.; Mueller, B.; Schuetz, P.; Christ-Crain, M.; et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: Secondary analysis of a randomised controlled trial. Eur. Respir. J. 2018, 51, 1701389. [Google Scholar] [CrossRef]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G.; et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef]
- Trivedi, A.; Khan, M.A.; Bade, G.; Talwar, A. Orchestration of Neutrophil Extracellular Traps (Nets), a Unique Innate Immune Function during Chronic Obstructive Pulmonary Disease (COPD) Development. Biomedicines 2021, 9, 53. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Silva, C.M.S.; Wanderley, C.W.S.; Veras, F.P.; Gonçalves, A.V.; Lima, M.H.F.; Toller-Kawahisa, J.E.; Gomes, G.F.; Nascimento, D.C.; Monteiro, V.V.S.; Paiva, I.M.; et al. Gasdermin-D activation by SARS-CoV-2 triggers NET and mediate COVID-19 immunopathology. Crit. Care 2022, 26, 206. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NETs of neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13, 838011. [Google Scholar] [CrossRef]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; Dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decoté-Ricardo, D.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J. Infect 2020, 81, e6–e12. [Google Scholar] [CrossRef]
- Kaur, S.; Tripathi, D.M.; Yadav, A. The Enigma of Endothelium in COVID-19. Front. Physiol. 2020, 11, 989. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Behzadifard, M.; Soleimani, M. NETosis and SARS-CoV-2 infection related thrombosis: A narrative review. Thromb. J. 2022, 20, 13. [Google Scholar] [CrossRef]
- Chen, W.; Pan, J.Y. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol. Proced. Online 2021, 23, 4. [Google Scholar] [CrossRef]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef]
- Staats, L.A.N.; Pfeiffer, H.; Knopf, J.; Lindemann, A.; Fürst, J.; Kremer, A.E.; Hackstein, H.; Neurath, M.F.; Muñoz, L.E.; Achenbach, S.; et al. IgA2 Antibodies against SARS-CoV-2 Correlate with NET Formation and Fatal Outcome in Severely Diseased COVID-19 Patients. Cells 2020, 9, 2676. [Google Scholar] [CrossRef]
- Steffen, U.; Koeleman, C.A.; Sokolova, M.V.; Bang, H.; Kleyer, A.; Rech, J.; Unterweger, H.; Schicht, M.; Garreis, F.; Hahn, J.; et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 2020, 11, 120. [Google Scholar] [CrossRef]
- Gößwein, S.; Lindemann, A.; Mahajan, A.; Maueröder, C.; Martini, E.; Patankar, J.; Schett, G.; Becker, C.; Wirtz, S.; Naumann-Bartsch, N.; et al. Citrullination Licenses Calpain to Decondense Nuclei in Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 2481. [Google Scholar] [CrossRef]
- Farley, K.; Stolley, J.M.; Zhao, P.; Cooley, J.; Remold-O’Donnell, E. A serpinB1 regulatory mechanism is essential for restricting neutrophil extracellular trap generation. J. Immunol. 2012, 189, 4574–4581. [Google Scholar] [CrossRef]
- Frenzel, E.; Korenbaum, E.; Hegermann, J.; Ochs, M.; Koepke, J.; Koczulla, A.R.; Welte, T.; Köhnlein, T.; Janciauskiene, S. Does augmentation with alpha1-antitrypsin affect neutrophil extracellular traps formation? Int. J. Biol. Sci. 2012, 8, 1023–1025. [Google Scholar] [CrossRef][Green Version]
- Yost, C.C.; Schwertz, H.; Cody, M.J.; Wallace, J.A.; Campbell, R.A.; Vieira-de-Abreu, A.; Araujo, C.V.; Schubert, S.; Harris, E.S.; Rowley, J.W.; et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J. Clin. Investig. 2016, 126, 3783–3798. [Google Scholar] [CrossRef]
- Niemann, M.A.; Baggott, J.E.; Miller, E.J. Binding of SPAAT, the 44-residue C-terminal peptide of alpha 1-antitrypsin, to proteins of the extracellular matrix. J. Cell Biochem. 1997, 66, 346–357. [Google Scholar] [CrossRef]
- Clemmensen, S.N.; Udby, L.; Borregaard, N. Subcellular fractionation of human neutrophils and analysis of subcellular markers. Methods Mol. Biol. 2014, 1124, 53–76. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.Y.; Cramer, E.M.; Massé, J.M.; Crystal, R.; Bassot, J.M.; Breton-Gorius, J. Alpha 1-antitrypsin is present within the primary granules of human polymorphonuclear leukocytes. Am. J. Pathol. 1991, 139, 623–628. [Google Scholar] [PubMed]
- Clemmensen, S.N.; Jacobsen, L.C.; Rørvig, S.; Askaa, B.; Christenson, K.; Iversen, M.; Jørgensen, M.H.; Larsen, M.T.; van Deurs, B.; Ostergaard, O.; et al. Alpha-1-antitrypsin is produced by human neutrophil granulocytes and their precursors and liberated during granule exocytosis. Eur. J. Haematol. 2011, 86, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, H.; Tebbutt, S.J. Leave no one behind: Inclusion of alpha-1 antitrypsin deficiency patients in COVID-19 vaccine trials. Eur. J. Hum. Genet. 2022, 30, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Netzel-Arnett, S.; Hooper, J.D.; Szabo, R.; Madison, E.L.; Quigley, J.P.; Bugge, T.H.; Antalis, T.M. Membrane anchored serine proteases: A rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003, 22, 237–258. [Google Scholar] [CrossRef]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ní Choileáin, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Azouz, N.P.; Klingler, A.M.; Callahan, V.; Akhrymuk, I.V.; Elez, K.; Raich, L.; Henry, B.M.; Benoit, J.L.; Benoit, S.W.; Noé, F.; et al. Alpha 1 Antitrypsin is an Inhibitor of the SARS-CoV-2-Priming Protease TMPRSS2. Pathog. Immun. 2021, 6, 55–74. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef]
- De Loyola, M.B.; Dos Reis, T.T.A.; de Oliveira, G.; da Fonseca Palmeira, J.; Argañaraz, G.A.; Argañaraz, E.R. Alpha-1-antitrypsin: A possible host protective factor against COVID-19. Rev. Med. Virol. 2021, 31, e2157. [Google Scholar] [CrossRef]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Hubbard, R.C.; Sellers, S.; Czerski, D.; Stephens, L.; Crystal, R.G. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA 1988, 260, 1259–1264. [Google Scholar] [CrossRef]
- Campos, M.A.; Geraghty, P.; Holt, G.; Mendes, E.; Newby, P.R.; Ma, S.; Luna-Diaz, L.V.; Turino, G.M.; Stockley, R.A. The Biological Effects of Double-Dose Alpha-1 Antitrypsin Augmentation Therapy. A Pilot Clinical Trial. Am. J. Respir. Crit. Care Med. 2019, 200, 318–326. [Google Scholar] [CrossRef]
- Rosendal, E.; Mihai, I.S.; Becker, M.; Das, D.; Frängsmyr, L.; Persson, B.D.; Rankin, G.D.; Gröning, R.; Trygg, J.; Forsell, M.; et al. Serine Protease Inhibitors Restrict Host Susceptibility to SARS-CoV-2 Infections. mBio 2022, 13, e0089222. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, V.; Radic, M. COVID-19 Pathology Sheds Further Light on Balance between Neutrophil Proteases and Their Inhibitors. Biomolecules 2023, 13, 82. https://doi.org/10.3390/biom13010082
Silva V, Radic M. COVID-19 Pathology Sheds Further Light on Balance between Neutrophil Proteases and Their Inhibitors. Biomolecules. 2023; 13(1):82. https://doi.org/10.3390/biom13010082
Chicago/Turabian StyleSilva, Vasuki, and Marko Radic. 2023. "COVID-19 Pathology Sheds Further Light on Balance between Neutrophil Proteases and Their Inhibitors" Biomolecules 13, no. 1: 82. https://doi.org/10.3390/biom13010082
APA StyleSilva, V., & Radic, M. (2023). COVID-19 Pathology Sheds Further Light on Balance between Neutrophil Proteases and Their Inhibitors. Biomolecules, 13(1), 82. https://doi.org/10.3390/biom13010082