
Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Plan
2.2. Inclusion and Exclusion Criteria
2.3. Data Extraction
3. Results
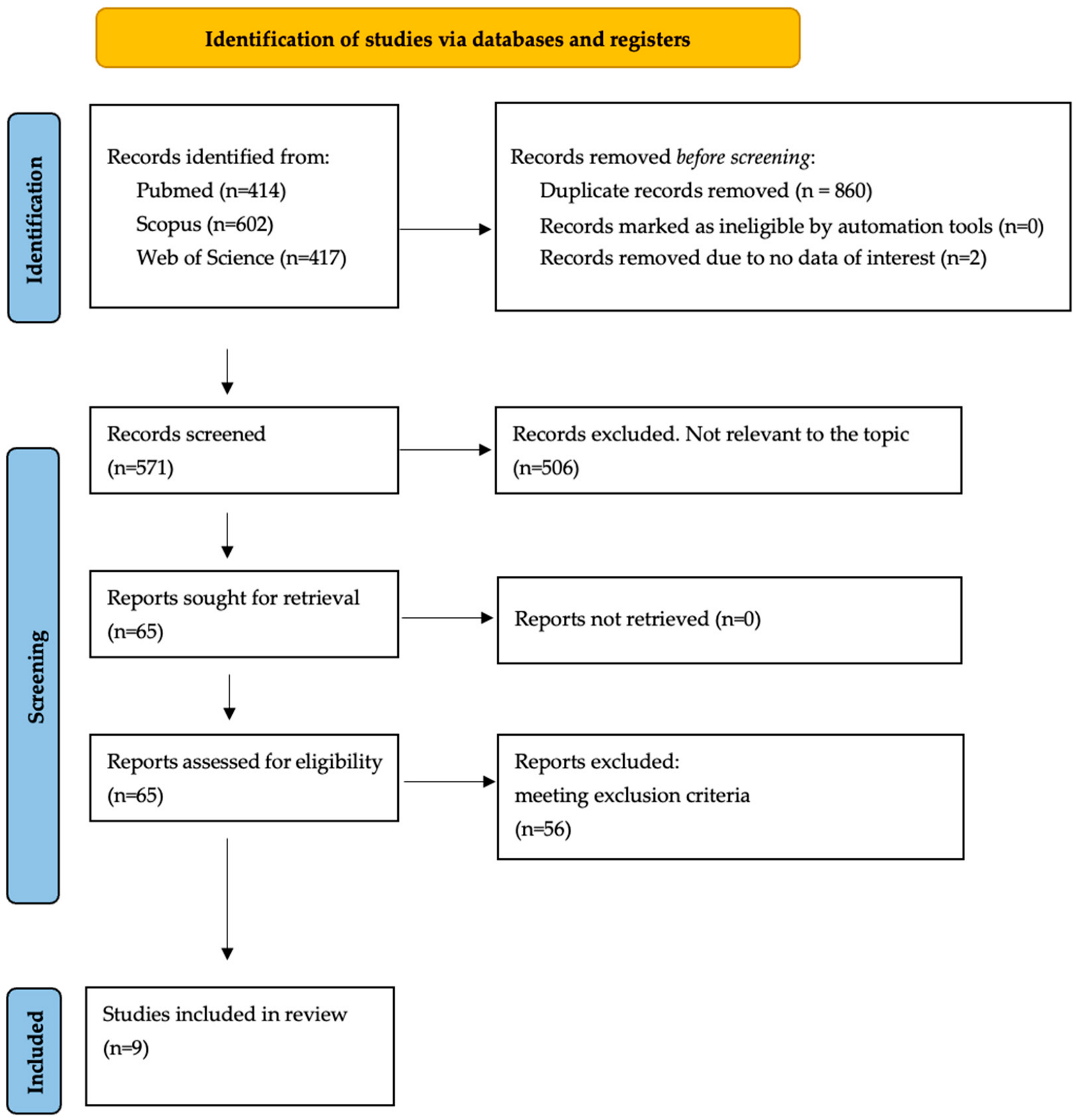
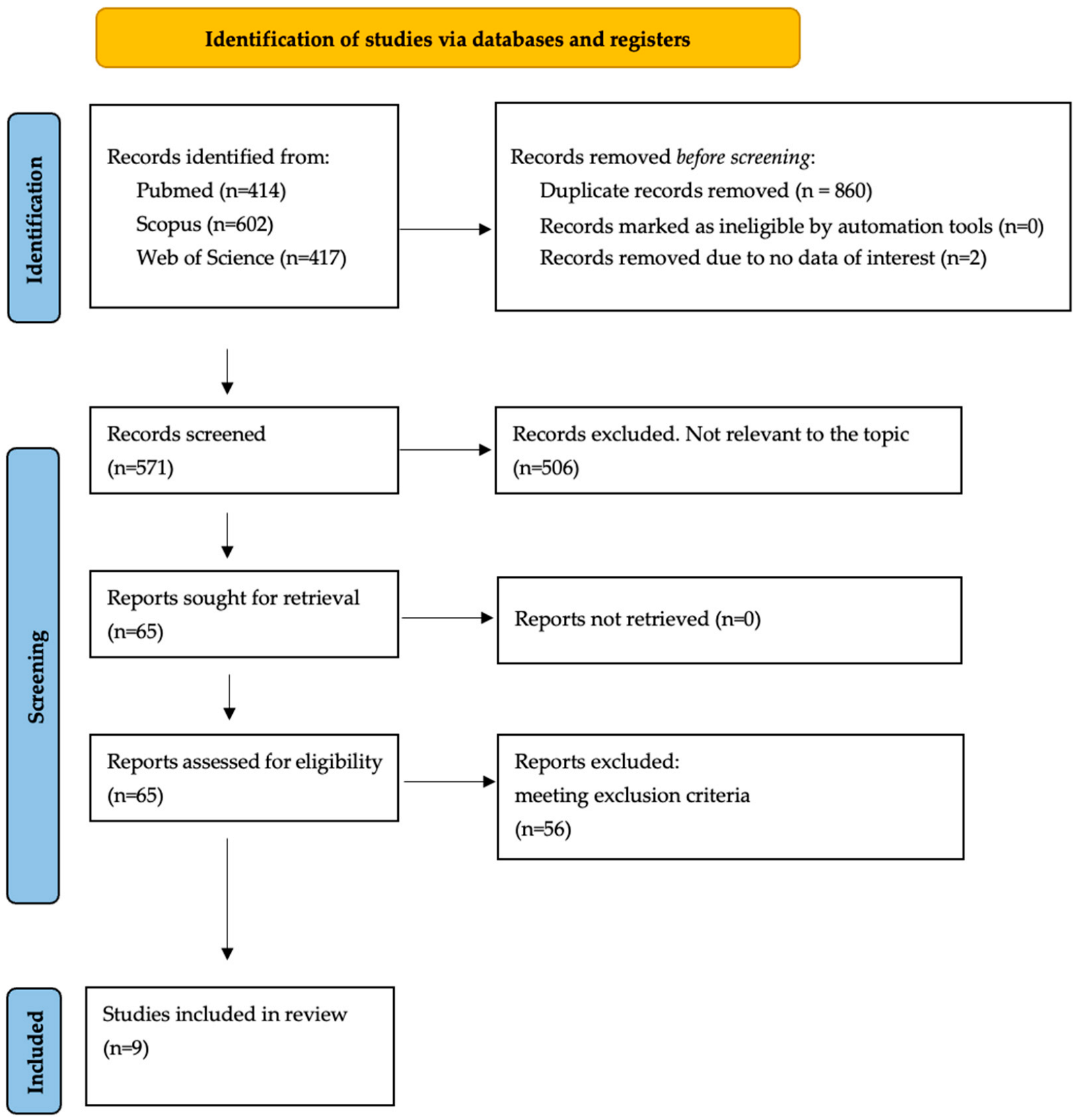
3.1. Study Retrieval
3.2. Clinical Feature of Patient with Hot-Phase Episodes
3.3. Diagnosis of a Cardiac Disease in Patients with Hot-Phase Episodes
3.4. Arrhythmic Symptoms and HF in Patients with Hot-Phase Episodes
3.5. Genetic Background in Patient with Hot-Phase Episodes
4. Discussion
4.1. Hot-Phase in ACM Patients: An Historical Perspective
4.2. Hot-Phase in ACM Patients: General Considerations on Available Clinical Studies
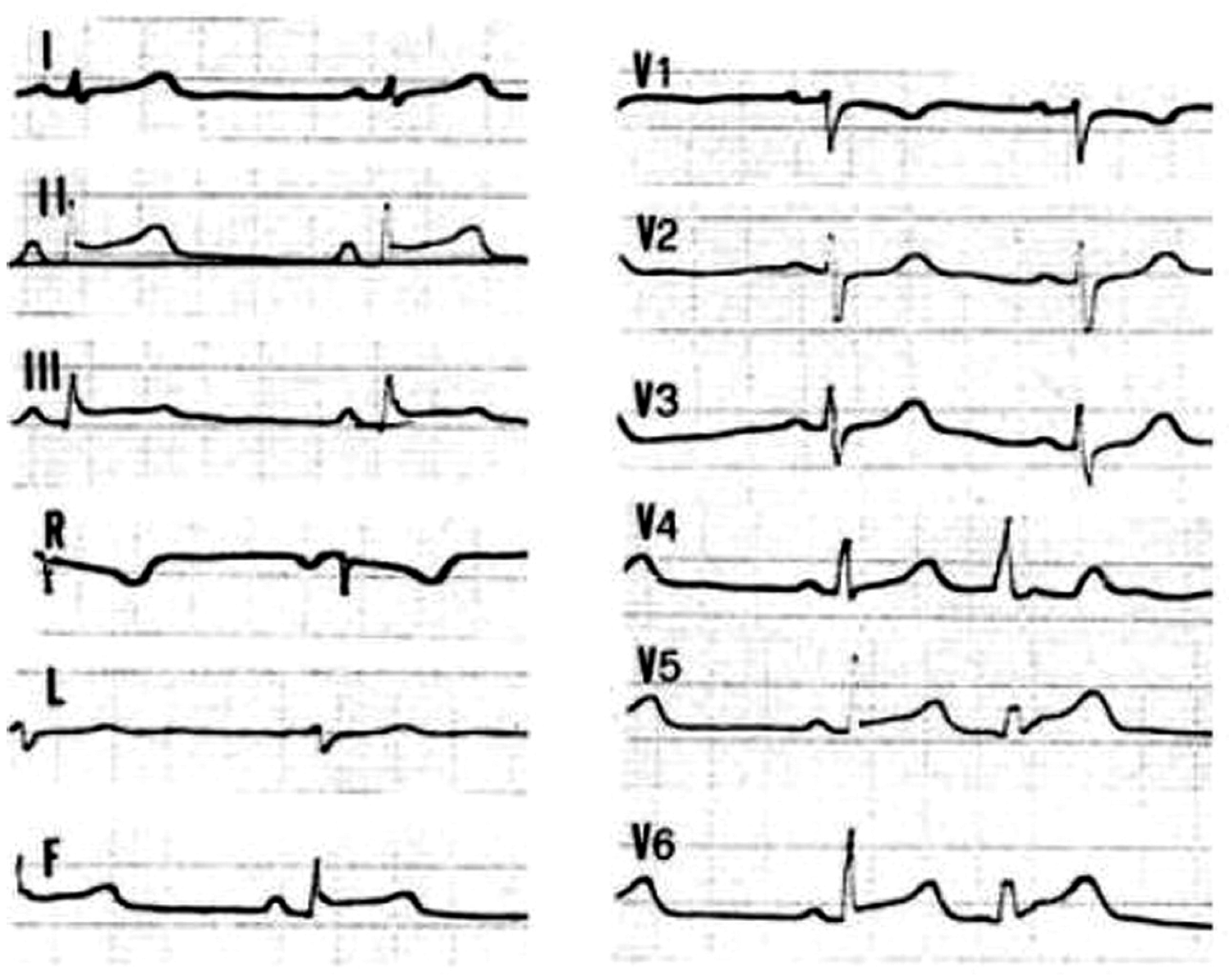
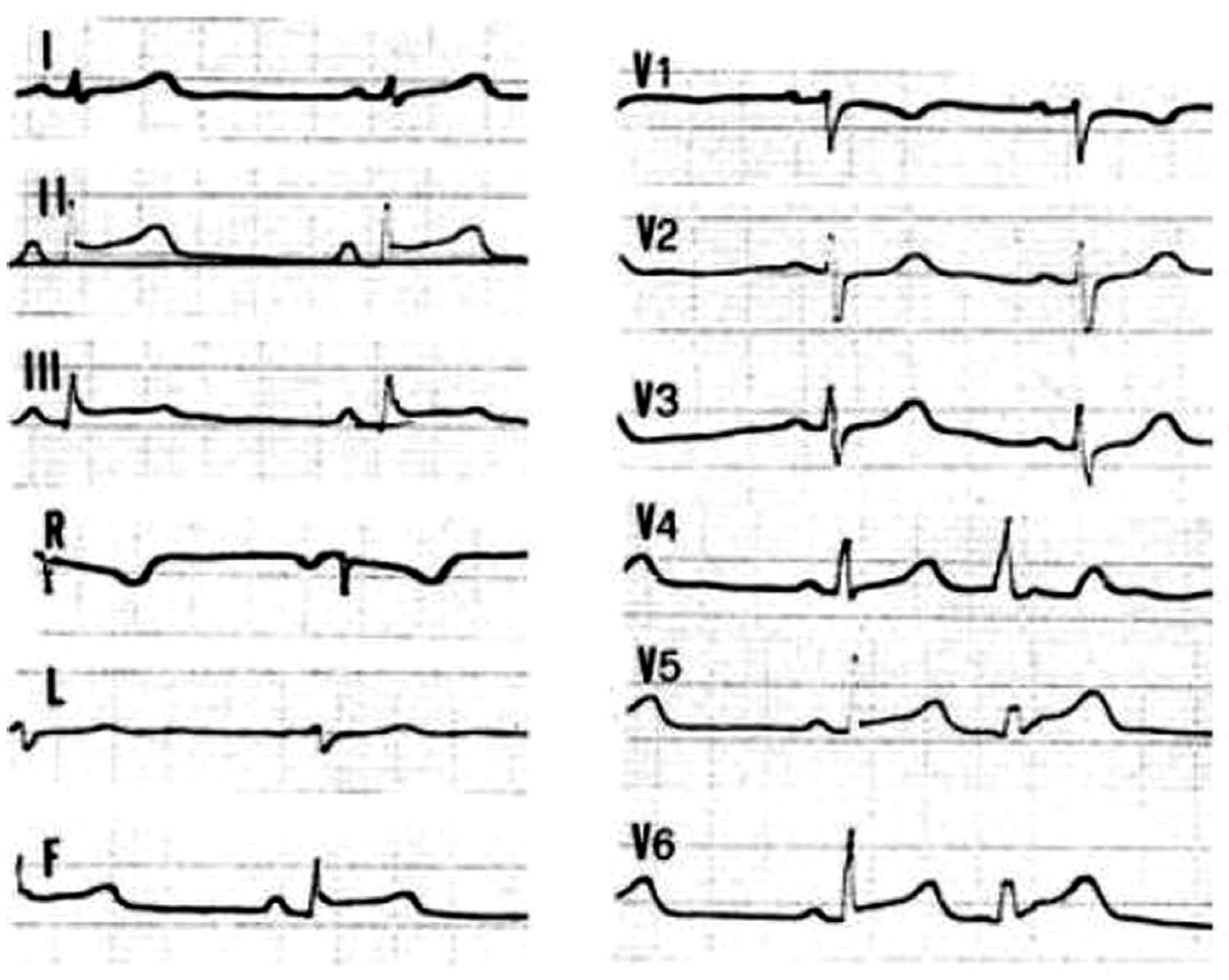
4.3. Hot-Phase Episodes in ACM Patients: Clinical Features
4.4. Hot-Phase and ALVC Forms
4.5. Hot-Phase in ACM Patients: Genetic Background
4.6. Arrhythmic Burden in ACM Patients with Hot-Phase of the Disease
4.7. Role of Inflammation in the Pathogenesis of ACM
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right Ventricular Dysplasia: A Report of 24 Adult Cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and Genetic Characterization of Families with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Provides Novel Insights into Patterns of Disease Expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-Dominant Arrhythmogenic Cardiomyopathy: An Under-Recognized Clinical Entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef]
- Bariani, R.; Cason, M.; Rigato, I.; Cipriani, A.; Celeghin, R.; De Gaspari, M.; Bueno Marinas, M.; Mattesi, G.; Pergola, V.; Rizzo, S.; et al. Clinical Profile and Long-Term Follow-up of a Cohort of Patients with Desmoplakin Cardiomyopathy. Heart Rhythm 2022, 19, 1315–1324. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.A.; Thiene, G.; et al. Clinical Profile of Four Families with Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Dominant Desmoplakin Mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- Bariani, R.; Cipriani, A.; Rizzo, S.; Celeghin, R.; Bueno Marinas, M.; Giorgi, B.; De Gaspari, M.; Rigato, I.; Leoni, L.; Zorzi, A.; et al. ‘Hot Phase’ Clinical Presentation in Arrhythmogenic Cardiomyopathy. EP Eur. 2021, 23, 907–917. [Google Scholar] [CrossRef]
- Tramer, M.R.; Reynolds, D.J.M.; Moore, R.A.; McQuay, H.J. Impact of Covert Duplicate Publication on Meta-Analysis: A Case Study. BMJ 1997, 315, 635–640. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of Myocarditis in Arrhythmogenic Right Ventricular Dysplasia. Heart Rhythm 2015, 12, 766–773. [Google Scholar] [CrossRef]
- Martins, D.; Ovaert, C.; Khraiche, D.; Boddaert, N.; Bonnet, D.; Raimondi, F. Myocardial Inflammation Detected by Cardiac MRI in Arrhythmogenic Right Ventricular Cardiomyopathy: A Paediatric Case Series. Int. J. Cardiol. 2018, 271, 81–86. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, E.S.; Chandler, S.F.; Hylind, R.J.; Beausejour Ladouceur, V.; Blume, E.D.; VanderPluym, C.; Powell, A.J.; Fynn-Thompson, F.; Roberts, A.E.; Sanders, S.P.; et al. Phenotypic Manifestations of Arrhythmogenic Cardiomyopathy in Children and Adolescents. J. Am. Coll. Cardiol. 2019, 74, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; Le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; Le Tourneau, T.; Conan, E.; et al. Familial Screening in Case of Acute Myocarditis Reveals Inherited Arrhythmogenic Left Ventricular Cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Wang, W.; Murray, B.; Tichnell, C.; Gilotra, N.A.; Zimmerman, S.L.; Gasperetti, A.; Scheel, P.; Tandri, H.; Calkins, H.; James, C.A. Clinical Characteristics and Risk Stratification of Desmoplakin Cardiomyopathy. EP Eur. 2022, 24, 268–277. [Google Scholar] [CrossRef]
- Graziosi, M.; Ditaranto, R.; Rapezzi, C.; Pasquale, F.; Lovato, L.; Leone, O.; Parisi, V.; Potena, L.; Ferrara, V.; Minnucci, M.; et al. Clinical Presentations Leading to Arrhythmogenic Left Ventricular Cardiomyopathy. Open Heart 2022, 9, e001914. [Google Scholar] [CrossRef]
- Scheel, P.J.; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134. [Google Scholar] [CrossRef]
- Hisaoka, T.; Kawai, S.; Ohi, H.; Ishijima, M.; Okada, R.; Hayashida, N.; Saiki, S.; Kobayashi, H.; Yoshimura, H. Two Cases of Chronic Myocarditis Mimicking Arrhythmogenic Right Ventricular Dysplasia. Heart Vessel. Suppl. 1990, 5, 51–54. [Google Scholar]
- Sabel, K.-G.; Blomström-Lundqvist, C.; Olsson, S.B.; Eneström, S. Arrhythmogenic Right Ventricular Dysplasia in Brother and Sister: Is It Related to Myocarditis? Pediatr. Cardiol. 1990, 11, 113–116. [Google Scholar] [CrossRef]
- Hofmann, R.; Trappe, H.-J.; Klein, H.; Kemnitz, J. Chronic (or Healed) Myocarditis Mimicking Arrhythmogenic Right Ventricular Dysplasia. Eur. Heart J. 1993, 14, 717–720. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic Right Ventricular Cardiomyopathy: Dysplasia, Dystrophy, or Myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.J. Role of Genetic Analysis in the Management of Patients With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef]
- Lin, Y.-N.; Ibrahim, A.; Marbán, E.; Cingolani, E. Pathogenesis of Arrhythmogenic Cardiomyopathy: Role of Inflammation. Basic Res. Cardiol. 2021, 116, 39. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular Mechanisms of Arrhythmogenic Cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Alcalde, M.; Campuzano, O.; Bellin, M. Inflammation in the Pathogenesis of Arrhythmogenic Cardiomyopathy: Secondary Event or Active Driver? Front. Cardiovasc. Med. 2021, 8, 784715. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C. Arrhythmogenic Left Ventricular Cardiomyopathy. Heart 2022, 108, 733. [Google Scholar] [CrossRef]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Perazzolo Marra, M.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic Cardiomyopathy. Orphanet J. Rare Dis. 2016, 11, 33. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.D.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Bariani, R.; Rigato, I.; Cason, M.; Bueno Marinas, M.; Celeghin, R.; Pilichou, K.; Bauce, B. Genetic Background and Clinical Features in Arrhythmogenic Left Ventricular Cardiomyopathy: A Systematic Review. J. Clin. Med. 2022, 11, 4313. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Alcalde, M.; Iglesias, A.; Barahona-Dussault, C.; Sarquella-Brugada, G.; Benito, B.; Arzamendi, D.; Flores, J.; Leung, T.K.; Talajic, M.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Severe Structural Alterations Are Associated with Inflammation. J. Clin. Pathol. 2012, 65, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with Myocardial Inflammation and Fibrotic Remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; Van Den Hoff, M.J.B.; De Bakker, J.M.T.; et al. Myocyte Necrosis Underlies Progressive Myocardial Dystrophy in Mouse Dsg2-Related Arrhythmogenic Right Ventricular Cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Lubos, N.; van der Gaag, S.; Gerçek, M.; Kant, S.; Leube, R.E.; Krusche, C.A. Inflammation Shapes Pathogenesis of Murine Arrhythmogenic Cardiomyopathy. Basic Res. Cardiol. 2020, 115, 42. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An Autoantibody Identifies Arrhythmogenic Right Ventricular Cardiomyopathy and Participates in Its Pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence from Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies with Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Barzaghi, F.; Cicalese, M.P.; Di Resta, C.; Slavich, M.; Benedetti, S.; Giangiobbe, S.; Rizzo, S.; Palmisano, A.; Esposito, A.; et al. Immunosuppressive therapy in childhood-onset arrhythmogenic inflammatory cardiomyopathy. Pacing Clin. Electrophysiol. 2021, 44, 552–556. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Reference | Aim and Design of the Study | Study Population | Main Results | Patients with Hot-Phase | Genetic Variants Detected in Patients with Hot-Phase | Conclusions |
|---|---|---|---|---|---|---|
| Sen-Chowdhry, JACC 2008 [5] | Aim: to investigate the clinical-genetic profile of ALVC patients | 42 patients (22 M) with ALVC | - Patients showed arrhythmia or chest pain, but not HF - Desmosomal mutations identified in 45% of patients - In 50% previously diagnosed with viral myocarditis or DCM | 11 patients | DSP, PKP2, DSG2 | -ACM is distinguished from DCM by a propensity towards arrhythmias exceeding the degree of ventricular dysfunction. |
| Lopez Ayala, Hearth Rhythm 2015 [10] | Aim: to evaluate the genetic basis of myocarditis in ACM and investigate the association with a poorer prognosis and a higher risk of ventricular arrhythmias | 131 patients - 84 ACM (62% M) - 47 ALVC (47% M) | - Hot-phase as first clinical presentation in 6 out of 7 cases - In 2 patients, hot-phase episode preceded a worsening of LV systolic function, and in an additional 2 patients an episode of ventricular tachycardia | 7 patients (3 M, 4 F), mean age at symptom 30 years (min 14-max 45 years) 3 ARVC, 2 ALVC, 2 NR | DSP, LIM domain binding protein | -Acute myocarditis reflects an active phase of inflammation in ACM, leading to changes in the phenotype and abrupt complication of the disease. -An active phase of inflammation should be suspected in the presence of myocarditis associated with a family history of ACM. |
| Martins, Int J Cardiol 2018 [11] | Aim: to study the relationship between myocardial inflammation detected at CMR and ACM in a pediatric population | ACM patients < 18 years with clinical suspicion of myocarditis who had genetic testing for inherited cardiomyopathies | Six ACM patients experiencing myocarditis-like episodes with chest pain and troponin elevation. - Hot-phase episodes were likely exercise-induced in 50% of cases | 6 patients (5 M, 1 F), mean age at symptoms 9 years (min 2, max 15 years) 1 ARVC, 3 BIV, 2 ALVC | DSP, PKP2, DSG2 | - ACM can present as recurrent myocarditis-like episodes with CMR evidence of myocardial inflammation despite absent infectious trigger in children. |
| De Witt, JACC 2020 [12] | Aim: to describe the diverse phenotype, genotype, and outcomes in pediatric and adolescent patients affected with ACM | ACM patients < 21 years, divided into three groups (ARVC, ALVC, BIV forms) | - 32 patients (mean age 15.1 ± 3,8 years), 22 probands (16 ARVC, 9 BIV, 7 ALVC) | 6 patients, min age 6 max 21 years. 1 ARVC, 3 BIV, 2 ALVC | DSP, DES, PKP2 | -ACM in the young has highly varied phenotypic expression incorporating life-threatening arrhythmias, HF and hot-phases. |
| Piriou, ESC Heart Failure 2020 [13] | Aim: to assess the risk of patients with acute myocarditis of carrying an associated genetic variant involved in familial cardiomyopathies | Families with at least one individual with a documented episode of acute myocarditis and at least one individual affected with a cardiomyopathy or with a history of SD | - Six families (33 subjects) were identified - In the 5 families with a DSP variant, genetic testing was triggered by the association of an acute myocarditis with a single case of apparent DCM or SCD - Among 28 DSP variant carriers 39% had ALVC - Family history of SCD was frequent | 6 patients (3 M, 3 F), mean age at symptoms 20 years (min 9, max 41 years) 1 BIV, 5 ALVC | DSP, DSG2 | -Comprehensive familial screening, including genetic testing in the case of acute myocarditis associated with a family history of cardiomyopathy or SCD, revealed unknown misdiagnosed ALVC patients. -Genetic testing should be advised in patients who experience acute myocarditis and have a family history of cardiomyopathy or SCD. |
| Smith, Circulation 2020 [14] | Aim: to systematically analyze the clinical spectrum of DSP cardiomyopathy | 107 patients with DSP mutations and 81 with PKP2 mutations identified at 6 tertiary referral centers of DCM and ACM | - ALVC forms were exclusively present among patients with DSP mutation carriers -LV EF <55% strongly associated with severe ventricular arrhythmias in DSP patients -RVEF < 45% associated with severe arrhythmias in PKP2 group | 16 out of 107 DSP mutation carriers (15%) with troponin release 1 BIV, 15 ALVC | DSP | -DSP cardiomyopathy is a distinct form of ACM cardiomyopathy, characterized by episodic myocardial injury, left ventricular fibrosis and a high incidence of ventricular arrhythmias. |
| Bariani, Europace 2021 [8] | -Aim: to evaluate the clinical features of patients affected by ACM presenting with chest pain and myocardial enzyme release in the setting of normal coronary arteries (‘hot-phase’) | -ACM patients presenting with chest pain and/or myocardial necrosis markers elevation in the setting of normal coronary arteries | -Among 530 patients fulfilling ARVC TFC, 23 (5%) experienced hot-phase episodes - Genetic testing was positive in 77% of cases and pathogenic -DSP was the most frequent involved gene - No patient complained of sustained ventricular arrhythmia or died suddenly during the hot-phase | 23 patients (12 M, mean age at symptoms 24 years, min 10–max 71 years) 5 ARVC, 9 BIV, 6 ALVC | DSP, DSG2, PKP2, DES | -Hot-phase represents an uncommon clinical presentation of ACM, which often occurs in pediatric patients and carriers of DSP gene mutations. Tissue characterization, family history, and genetic test represent fundamental diagnostic tools for differential diagnosis. |
| Wang, Europace 2022 [15] | -Aim: to characterize the diagnosis, natural history, and risk for ventricular arrhythmia andheart failure in DSP cardiomyopathy | 91 patients (49% probands), enrolled in the Johns Hopkins ARVC registry who carry pathogenic or likely pathogenic DSP variants | -ALVC forms were common (28%) -Hot-phase episodes in 22% of patients -In univariate regression, myocardial injury was associated with sustained ventricular arrhythmia and HF - LVEF <35% and RV dysfunction were prognostic for sustained ventricular arrhythmia | 20 patients (mean age 27.5 years) 14 ALVC | DSP | -DSP cardiomyopathy affects both ventricles and carries high risk for ventricular arrhythmia and heart failure. -Myocardial injury is associated with worse disease outcomes. |
| Graziosi, Open Heart 2022 [16] | -Aim: to describe a cohort of patients with ALVC, focusing on the spectrum of the clinical presentations | 52 patients (63% M) diagnosed with ALVC retrospectively evaluated | -21 patients (41%) had normal echocardiogram, 13 (25%) a HNDC and 17 (33%) a DCM. -29 (62%) carried a pathogenic/likely pathogenic variant -30 patients (57%) had a previous different diagnosis with a diagnostic delay of 6 years | 8 patients (4 M), mean age at symptoms 36 years (min 27, max 60 years) | DSP | -ALVC is hidden in different clinical scenarios, with a phenotypic spectrum ranging from normal LV to HNDC and DCM. -Ventricular arrhythmias, chest pain, heart failure and SCD are the main clinical presentations. -Familial screening is essential for the affected relatives’ identification. |
| Reference | Definition |
|---|---|
| Sen-Chowdhry et al., JACC 2008 [5] | Chest pain and enzyme rise with unobstructed coronary arteries |
| Lopez Ayala et al., Hearth Rhythm 2015 [10] | Chest pain and enzyme rise with unobstructed coronary arteries. In one patient post-mortem evaluation identified myocardial inflammation |
| Martins et al., Int J Cardiol 2018 [11] | CMR inflammation criteria |
| De Witt et al., JACC 2020 [12] | Myocardial inflammation was diagnosed in presence of chest pain with elevated serum troponin with or without ST-segment changes on ECG, in the absence of fever or of infective diseases |
| Piriou et al., ESC Heart Failure 2020 [13] | Diagnosis of myocardial inflammation was based on the Lake Louise Criteria that were applicable before the end of 2018 |
| Smith et al., Circulation 2020 [14] | Episodic chest pain as a primary symptom independent of arrhythmias, and significant troponin elevation (greater than upper limit of normal as per specific laboratory reference ranges) in the absence of obstructive coronary disease on coronary angiography |
| Bariani et al., Europace 2021 [8] | Chest pain and myocardial enzyme release in the setting of normal coronary arteries. Eleven patients underwent EMB patients which showed that myocarditis-like features, i.e., Foci of inflammatory infiltration associated with oedema and necrosis of the cardiomyocytes, were found in seven patients |
| Wang et al., Europace 2022 [15] | Myocardial injury was defined as chest pain, serum cardiac troponin elevation greater than the upper limit of normal, as per local laboratory reference ranges, and the absence of obstructive coronary disease on coronary angiogram |
| Graziosi et al., Open Heart 2022 [16] | Chest pain: patients requiring hospital admission or outpatient evaluation because of acute or chronic chest pain, respectively |
| Study | N. Subjects | Gene | c.DNA | Amioacid Change | ACMG |
|---|---|---|---|---|---|
| Sen Chowdhry et al., JACC 2008 [5] | 1 | DSP | c.3045del | p.Arg1015Serfs*3 | Pathogenic |
| Sen Chowdhry et al., JACC 2008 [5] | 1 | DSP | c.1325C>T | p.Ser442Phe | Likely pathogenic |
| Lopez-Ajala et al., Heart Rhythm 2015 [10] | 1 | DSP | c.5318del | p.Leu1773Tyrfs*8 | Pathogenic |
| Lopez-Ajala et al., Heart Rhythm 2015 [10] | 4 | DSP | c.1339C>T | p.Gln447* | Pathogenic |
| Lopez-Ajala et al., Heart Rhythm 2015 [10]. | 2 | LDB3 | c.1051A>G | p.Thr351Ala | Likely benign |
| Martins et al., Int J Cardiol 2018 [11] | 1 | DSG2 | c.2410del | p.Thr804Leufs*4 | Likely pathogenic |
| Martins et al., Int J Cardiol 2018 [11] | 1 | PKP2 | c.2062T>C | p.Ser688Pro | Likely pathogenic |
| Martins et al., Int J Cardiol 2018 [11] | 1 | DSP | c.8392_8393del | p.Thr2798Trpfs*53 | Likely pathogenic |
| Martins et al., Int J Cardiol 2018 [11] | 1 | DSP | c.1691C>T | p.Thr564Ile | Likely pathogenic |
| Martins et al., Int J Cardiol 2018 [11] | 1 | PKP2 | c.2014-1G>C | Pathogenic | |
| Martins et al., Int J Cardiol 2018 [11] | 1 | DSP | c.4372C>T | p.Arg1458* | Pathogenic |
| De Witt et al., JACC 2020 [12] | 1 | DES | c.347A>G | p.Asn116Ser | Pathogenic |
| De Witt et al., JACC 2020 [12] | 1 | DSP | c.1873C>T | p.Gln625* | Pathogenic |
| DSP | c.6442G>A | p.Ala2148Thr | VUS | ||
| De Witt et al., JACC 2020 [12] | 1 | PKP2 | c.1162C>T | p.Arg388Trp | Likely pathogenic |
| PKP2 | c.2301del | p.Glu769Lysfs*31 | Likely pathogenic | ||
| De Witt et al., JACC 2020 [12] | 1 | PKP2 | c.2509del | p.Ser837Valfs*94 | Pathogenic |
| De Witt et al., JACC 2020 [12] | 1 | DSP | c.3526del | p.Val1176Phefs*20 | Likely pathogenic |
| DSG2 | c.1003A>G | p.Thr335Ala | VUS | ||
| De Witt et al., JACC 2020 [12] | 1 | DSP | c.2920del | p.Thr974Leufs*3 | Pathogenic |
| Piriou et al., ESC Heart Failure 2020 [13] | 1 | DSP | c.3925del | p.His1309Thrfs*40 | Likely pathogenic |
| Piriou et al., ESC Heart Failure 2020 [13] | 1 | DSP | c.1856del | p.Tyr619Serfs*17 | Likely pathogenic |
| Piriou et al., ESC Heart Failure 2020 [13] | 1 | DSP | c.1396C>T | p.Leu466Phe | VUS |
| MYBPC3 | c.1153G>A | p.Val385Met | VUS | ||
| Piriou et al., ESC Heart Failure 2020 [13] | 1 | DSP | c.2610del | p.Ile870Metfs*19 | Likely pathogenic |
| Piriou et al., ESC Heart Failure 2020 [13] | 1 | DSP | c.3211C>T | p.Gln1071* | Likely pathogenic |
| Piriou et al., ESC Heart Failure 2020 [13] | 1 | DSG2 | c.146G>A | p.Arg49His | Pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | PKP2 | c.2447_2448del | p.Thr816Argfs*10 | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.7461_7464del | p.Asp2489Metfs*17 | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSG2 | c.2032del | p.Gly679Alafs*3 | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | PKP2 | c.84del | p.Ser29Alafs*10 | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.3889C>T | p.Gln1297* | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 2 | DSP | c.897C>G | p.Ser299Arg | Pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DES | c.346A>G | p.Asn116Asp | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.2821C>T | p.Arg941* | Pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.3475G>T | p.Glu1159* | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.944G>C | p.Arg315Pro | VUS |
| Bariani et al., Europace 2021 [8] | 2 | DSG2 | c.1672C>T | p.Gln558* | Pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.6323C>A | p.Ser2108* | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 2 | PKP2 | c.175C>T | p.Gln59* | Pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | PKP2 | c.1027C>T | p.Gln343* | Likely pathogenic |
| Bariani et al., Europace 2021 [8] | 1 | DSP | c.3889C>T | p.Gln1297* | Likely pathogenic |
| Graziosi et al., Open Heart 2022 [16] | 1 | DSP | c.6496C>T | p.Arg2166* | Pathogenic |
| Graziosi et al., Open Heart 2022 [16] | 1 | DSP | c.2611_2614del | p.Asp871Asnfs*17 | Pathogenic |
| Graziosi et al., Open Heart 2022 [16] | 1 | DSP | c.448C>T | p.Arg150* | Pathogenic |
| Graziosi et al., Open Heart 2022 [16] | 1 | DSP | c.6850C>T | p.Arg2284* | Pathogenic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bariani, R.; Rigato, I.; Cipriani, A.; Bueno Marinas, M.; Celeghin, R.; Basso, C.; Corrado, D.; Pilichou, K.; Bauce, B. Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease. Biomolecules 2022, 12, 1324. https://doi.org/10.3390/biom12091324
Bariani R, Rigato I, Cipriani A, Bueno Marinas M, Celeghin R, Basso C, Corrado D, Pilichou K, Bauce B. Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease. Biomolecules. 2022; 12(9):1324. https://doi.org/10.3390/biom12091324
Chicago/Turabian StyleBariani, Riccardo, Ilaria Rigato, Alberto Cipriani, Maria Bueno Marinas, Rudy Celeghin, Cristina Basso, Domenico Corrado, Kalliopi Pilichou, and Barbara Bauce. 2022. "Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease" Biomolecules 12, no. 9: 1324. https://doi.org/10.3390/biom12091324
APA StyleBariani, R., Rigato, I., Cipriani, A., Bueno Marinas, M., Celeghin, R., Basso, C., Corrado, D., Pilichou, K., & Bauce, B. (2022). Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease. Biomolecules, 12(9), 1324. https://doi.org/10.3390/biom12091324