
New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia





Abstract
:
1. Introduction
2. Materials and Methods
2.1. FTD Mouse Model: CaMKII-TDP-43
2.2. ALS Mouse Model
2.3. Animal Management, Sacrifice, and Sampling
2.4. Human Samples
2.5. Analysis of mRNA Levels by Quantitative Real-Time PCR
2.6. RNAscope
2.7. Immunoblotting
2.8. Measurement of Thiobarbituric Acid Reactive Substances (TBARs)
2.9. Statistical Analyses
3. Results
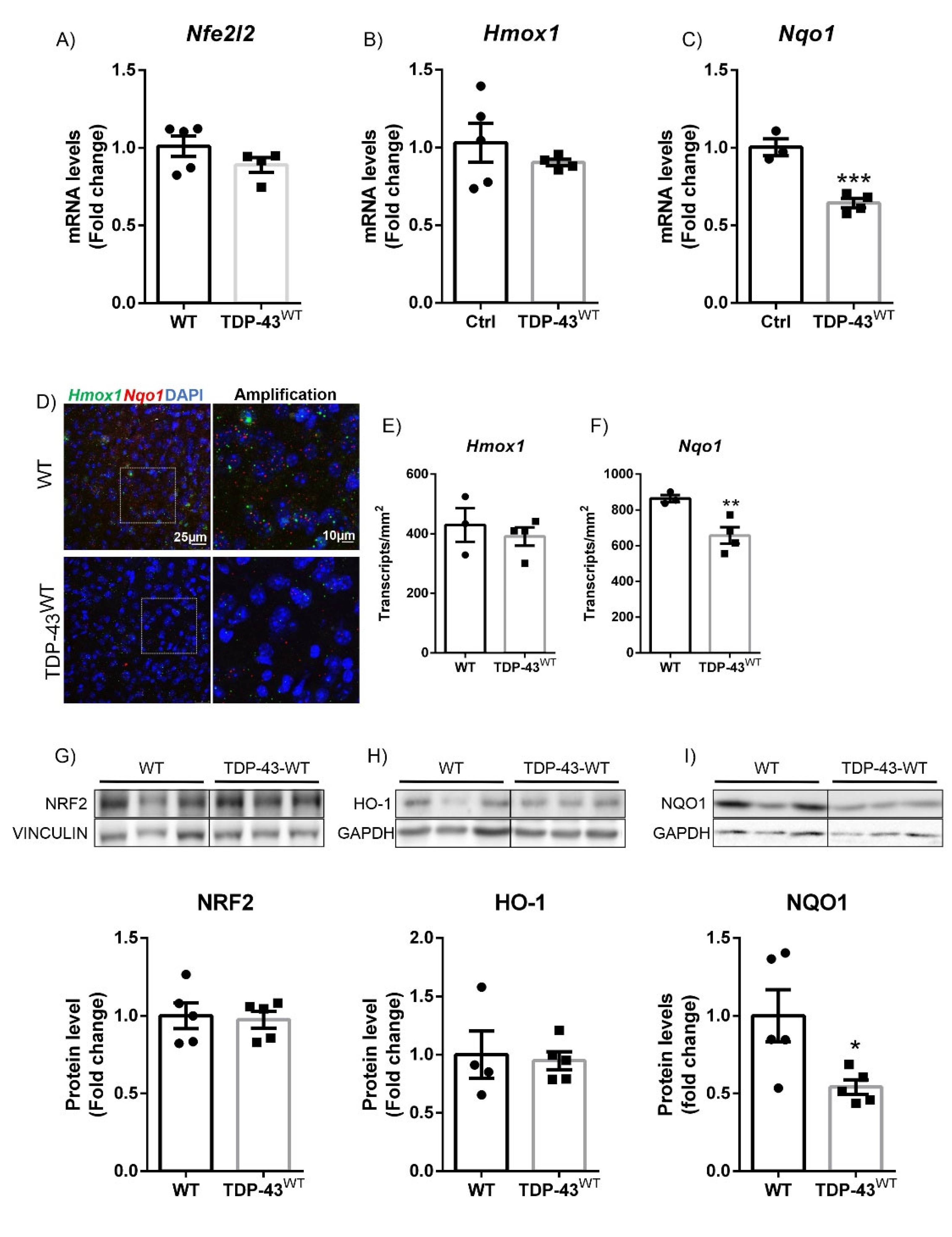
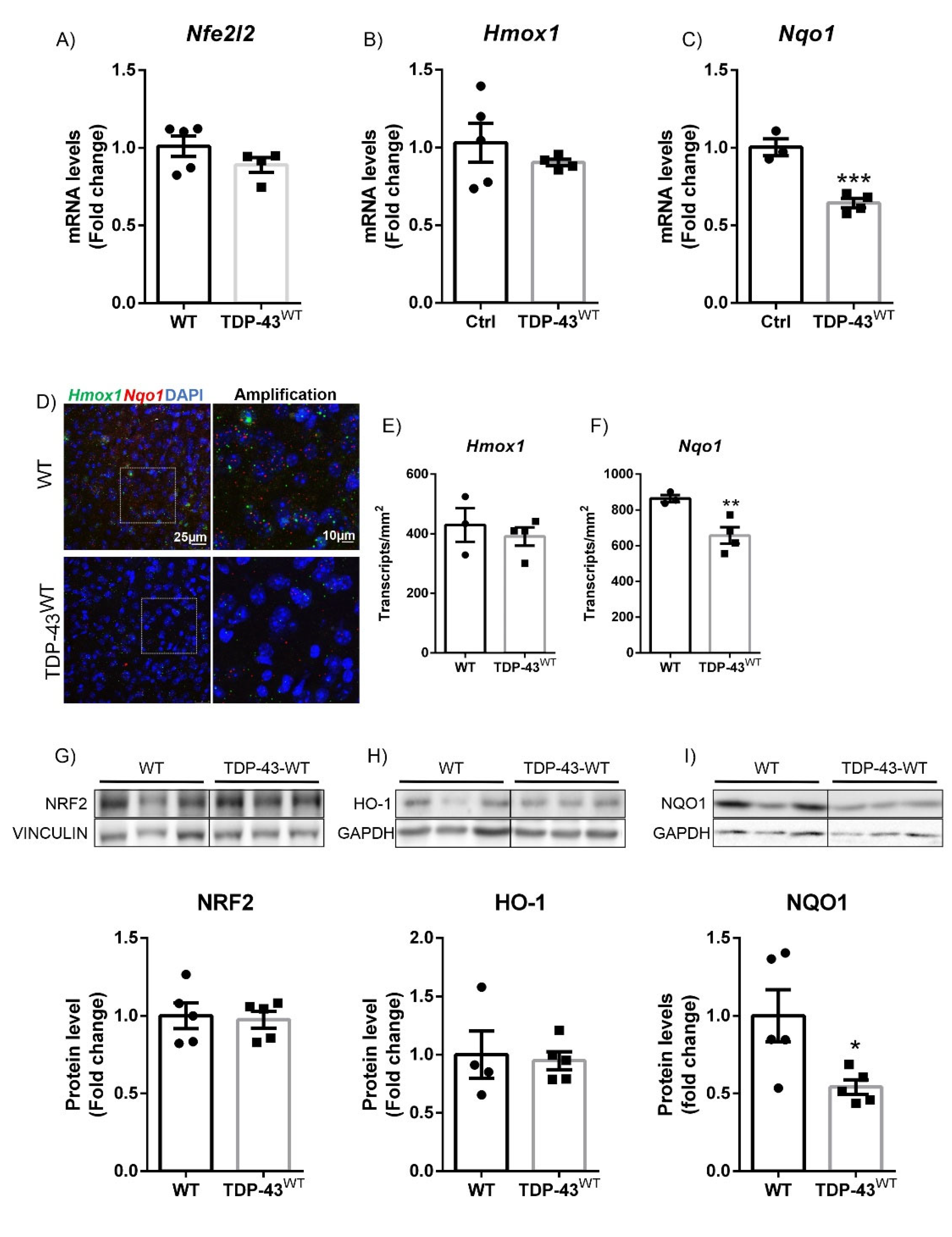
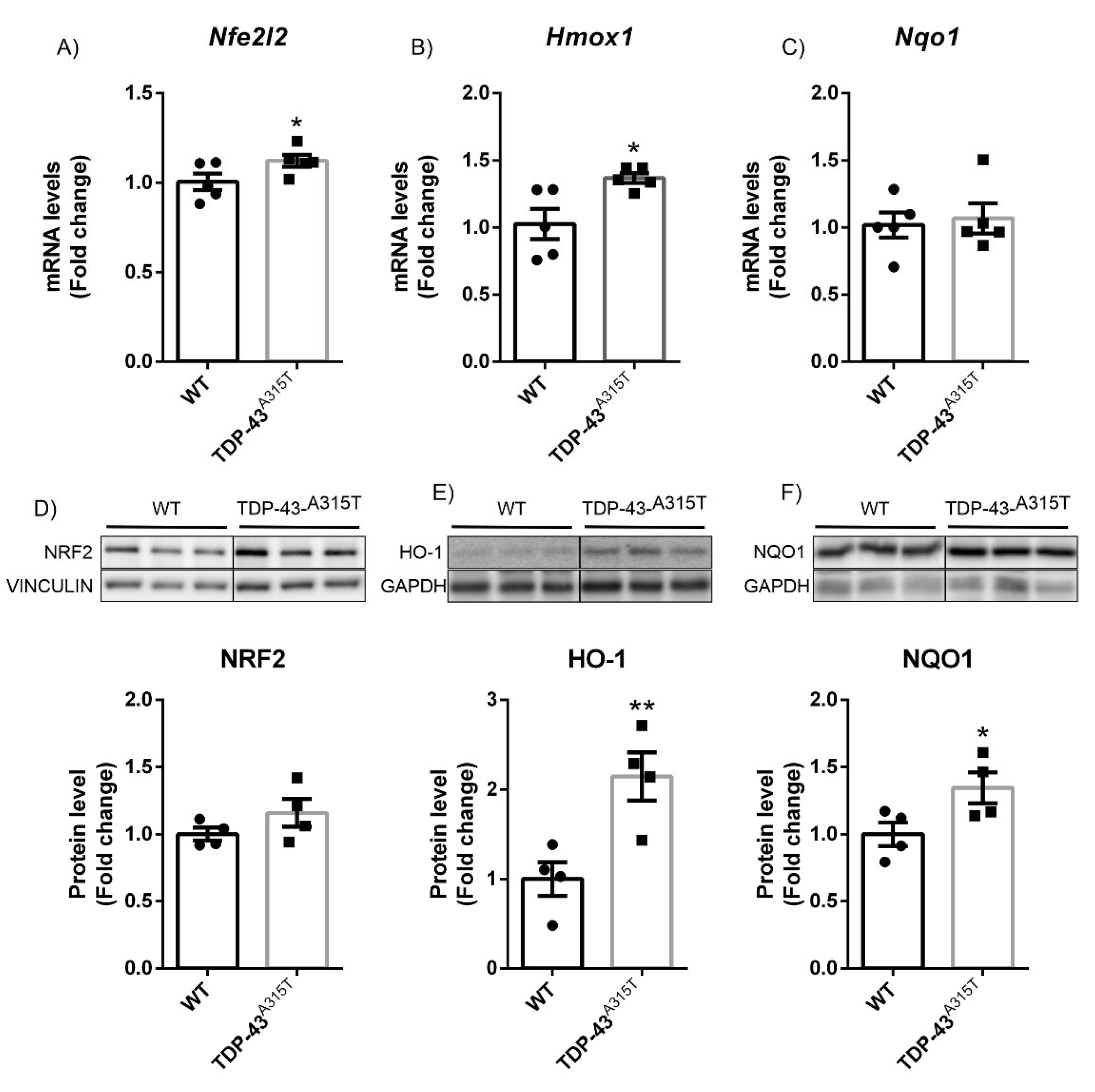
3.1. TDP-43 Overexpression Decreased NQO1 Expression in a Model of FTD
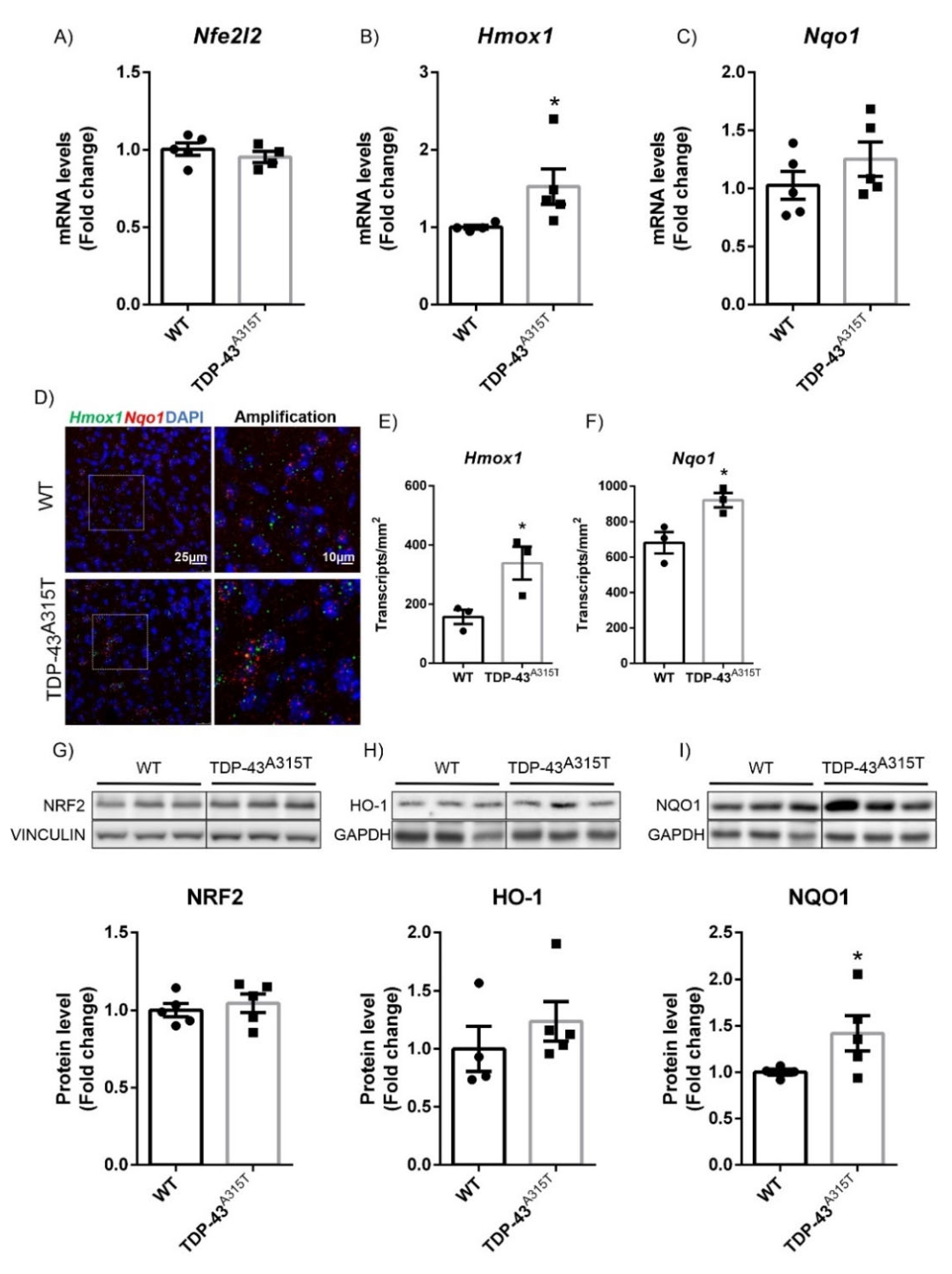
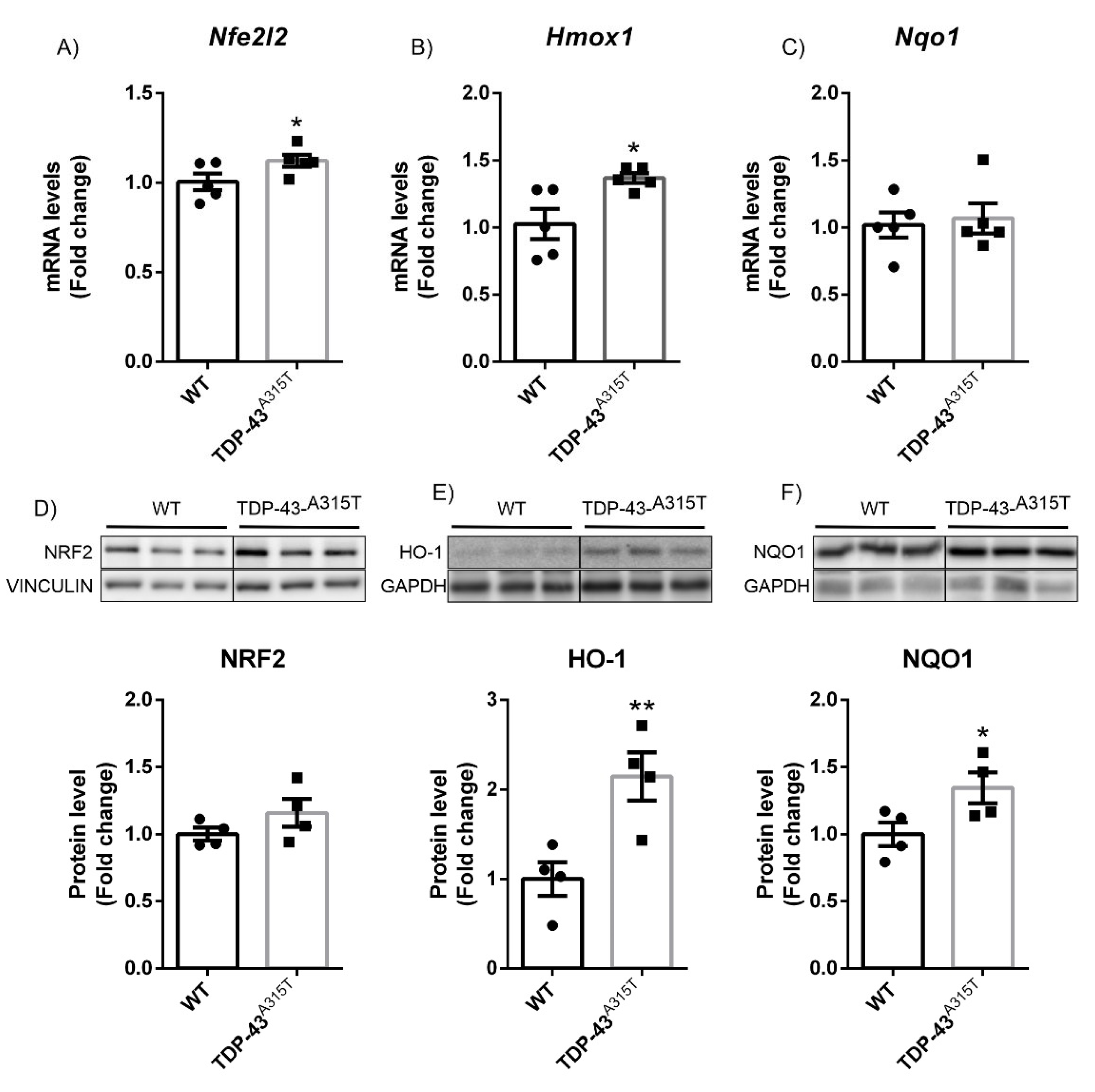
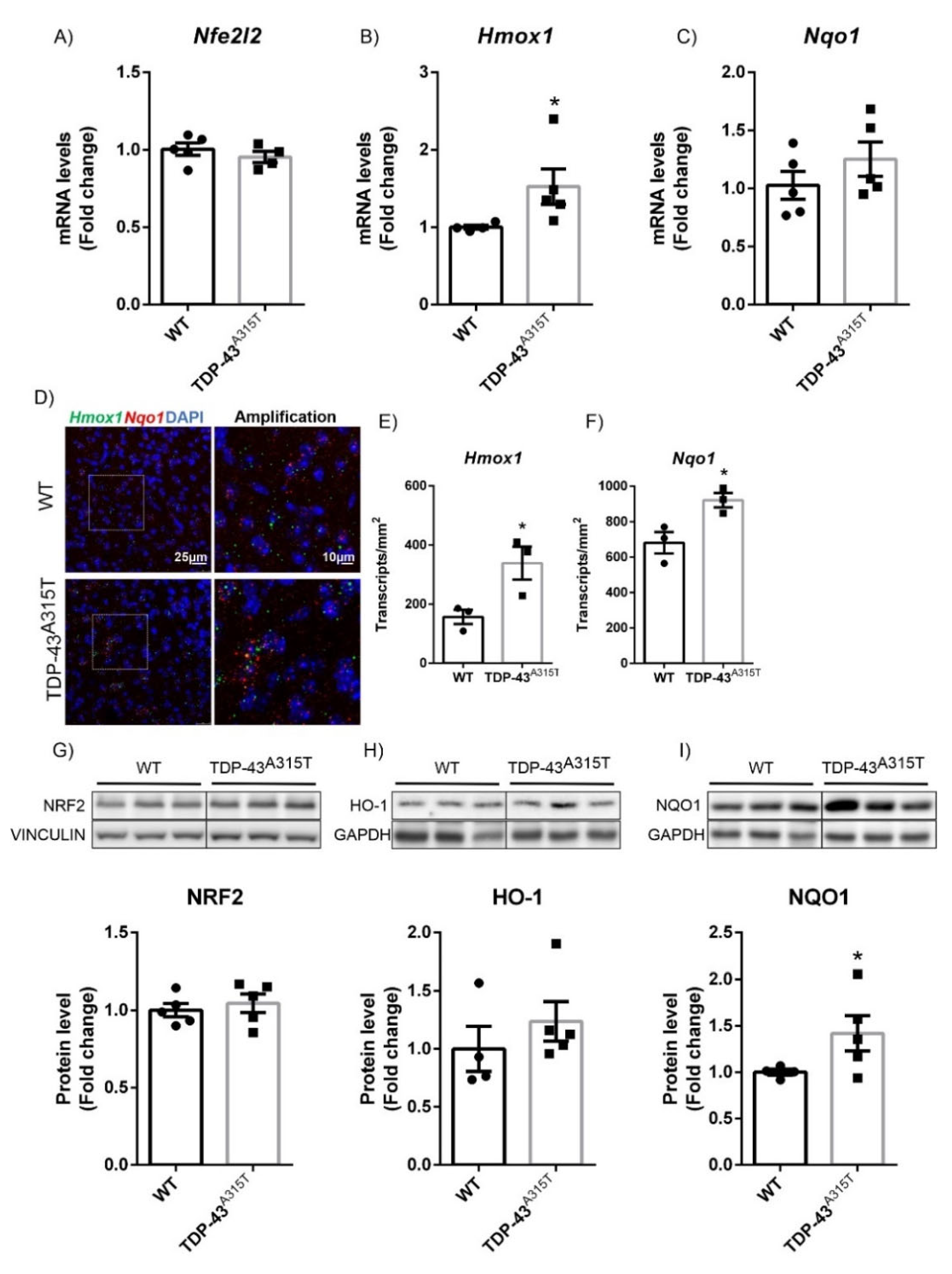
3.2. A315T Mutation in TDP-43-Induced NRF2 Signaling in a Model of Amyotrophic Lateral Sclerosis (ALS)
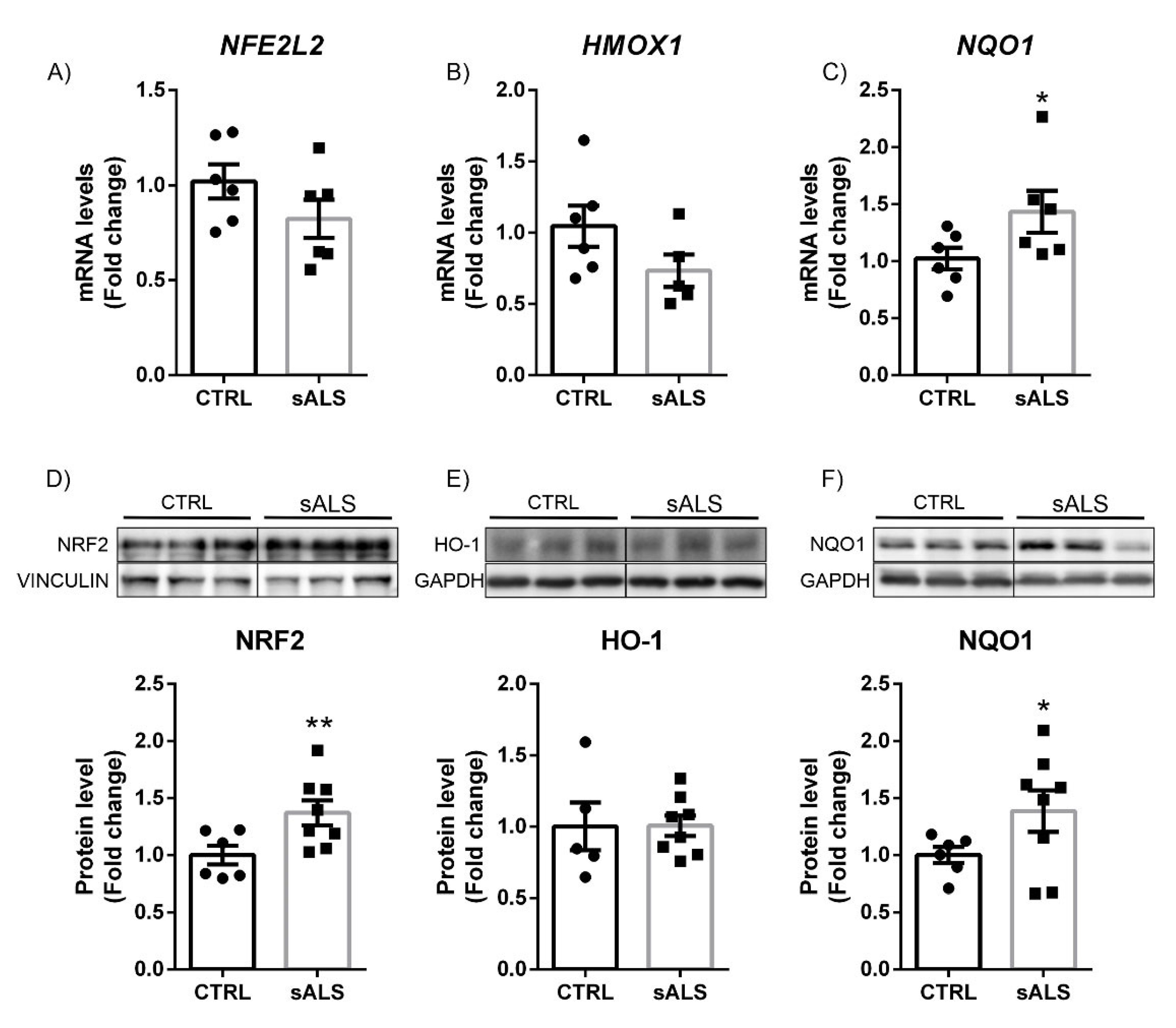
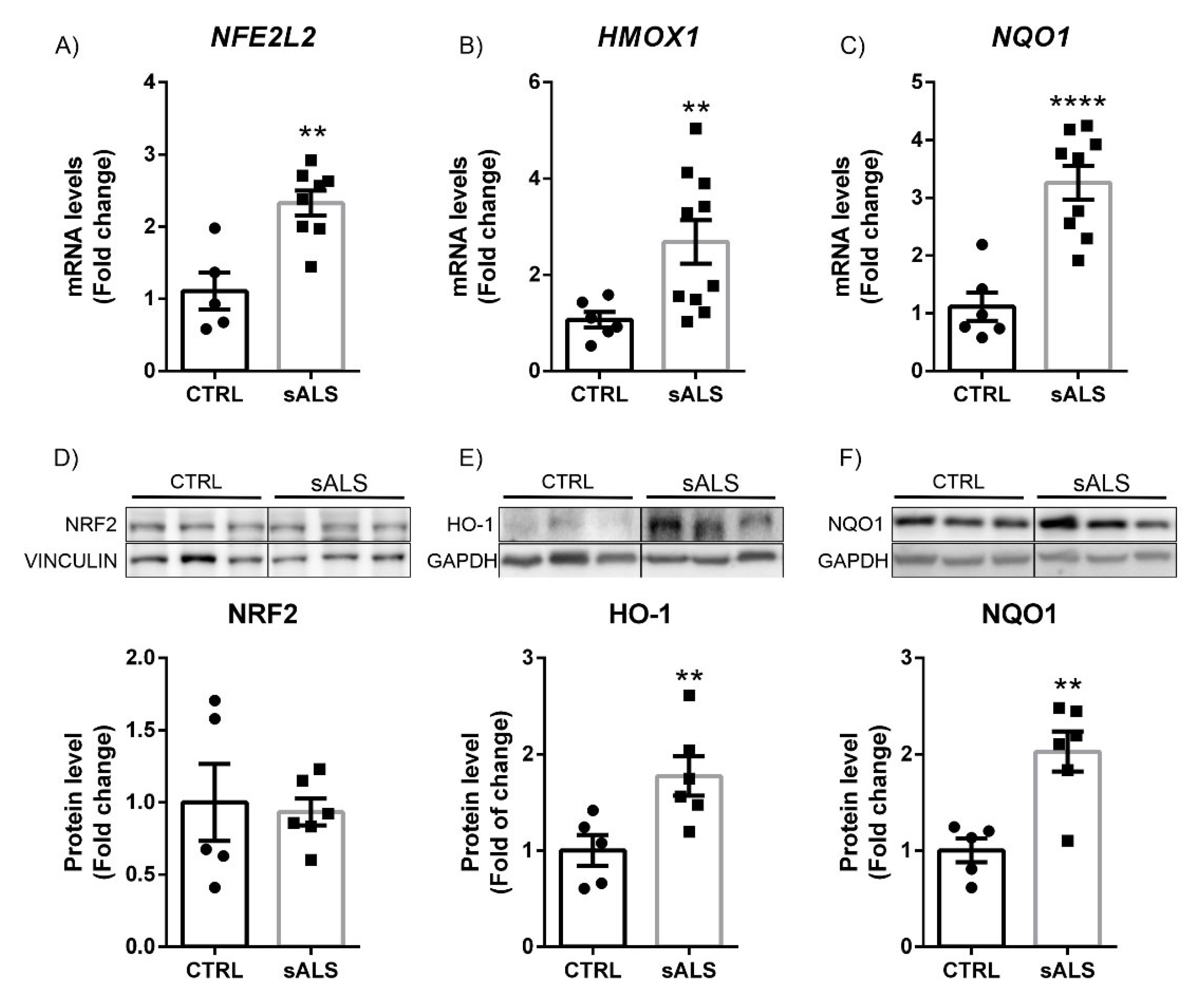
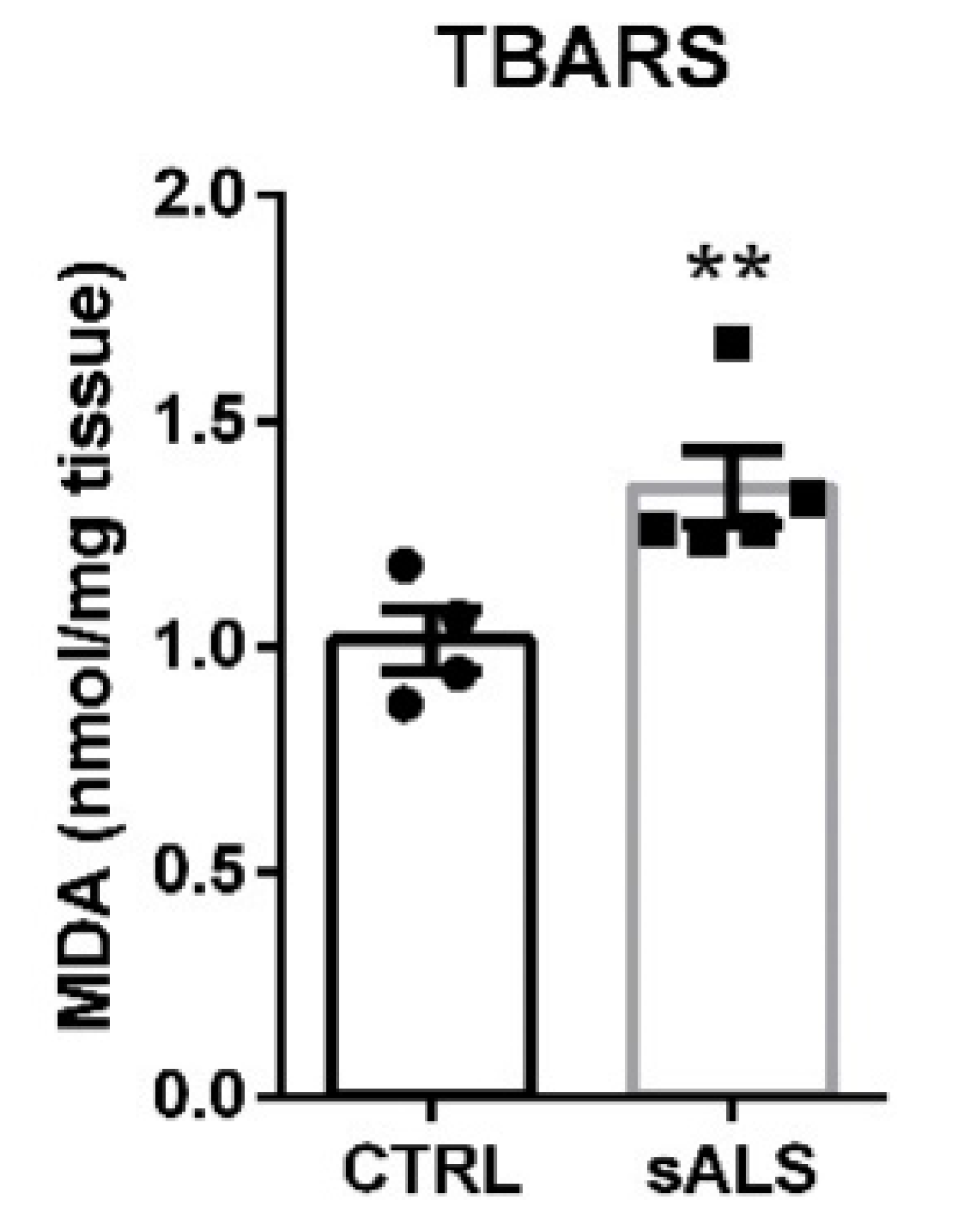
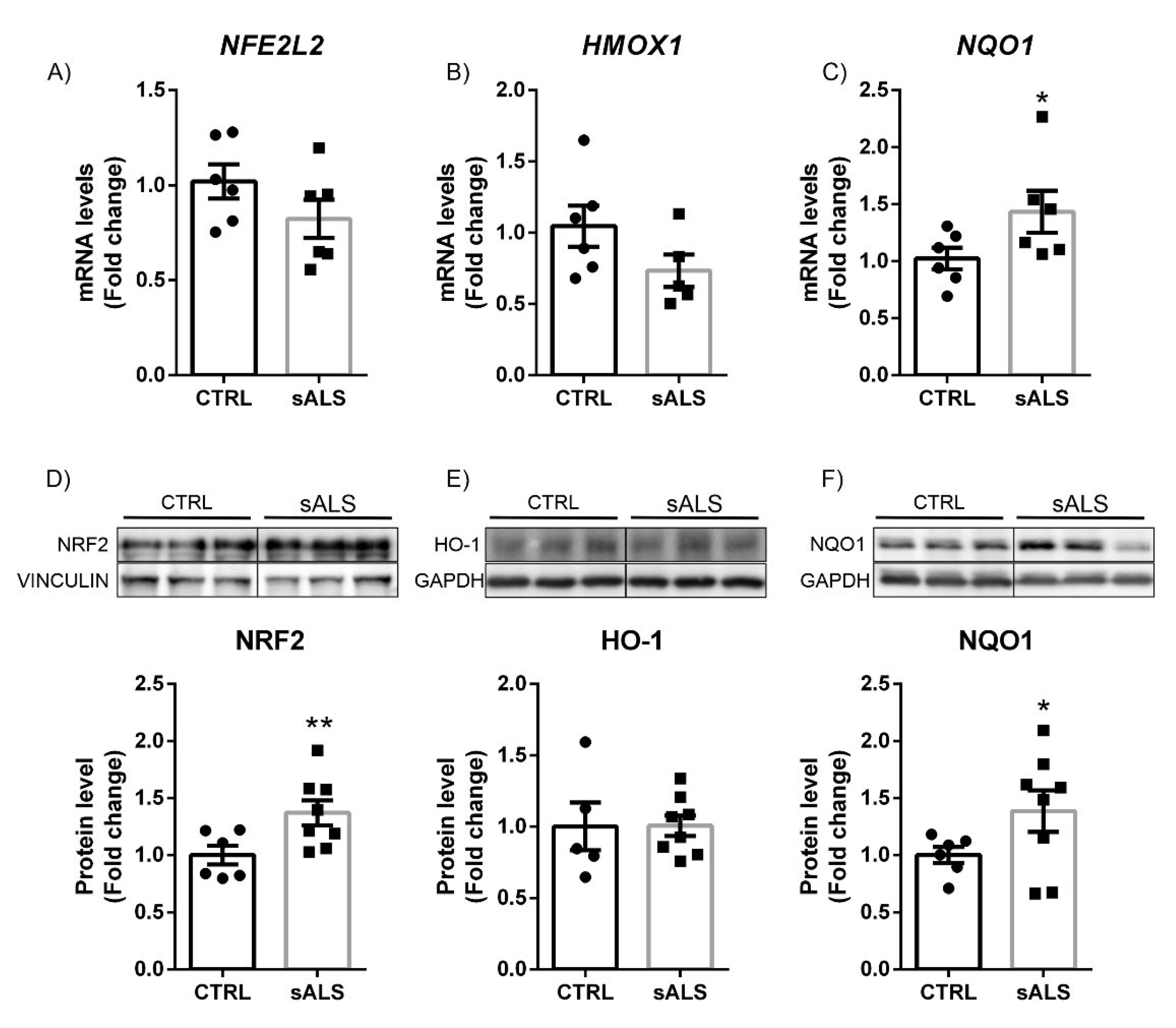
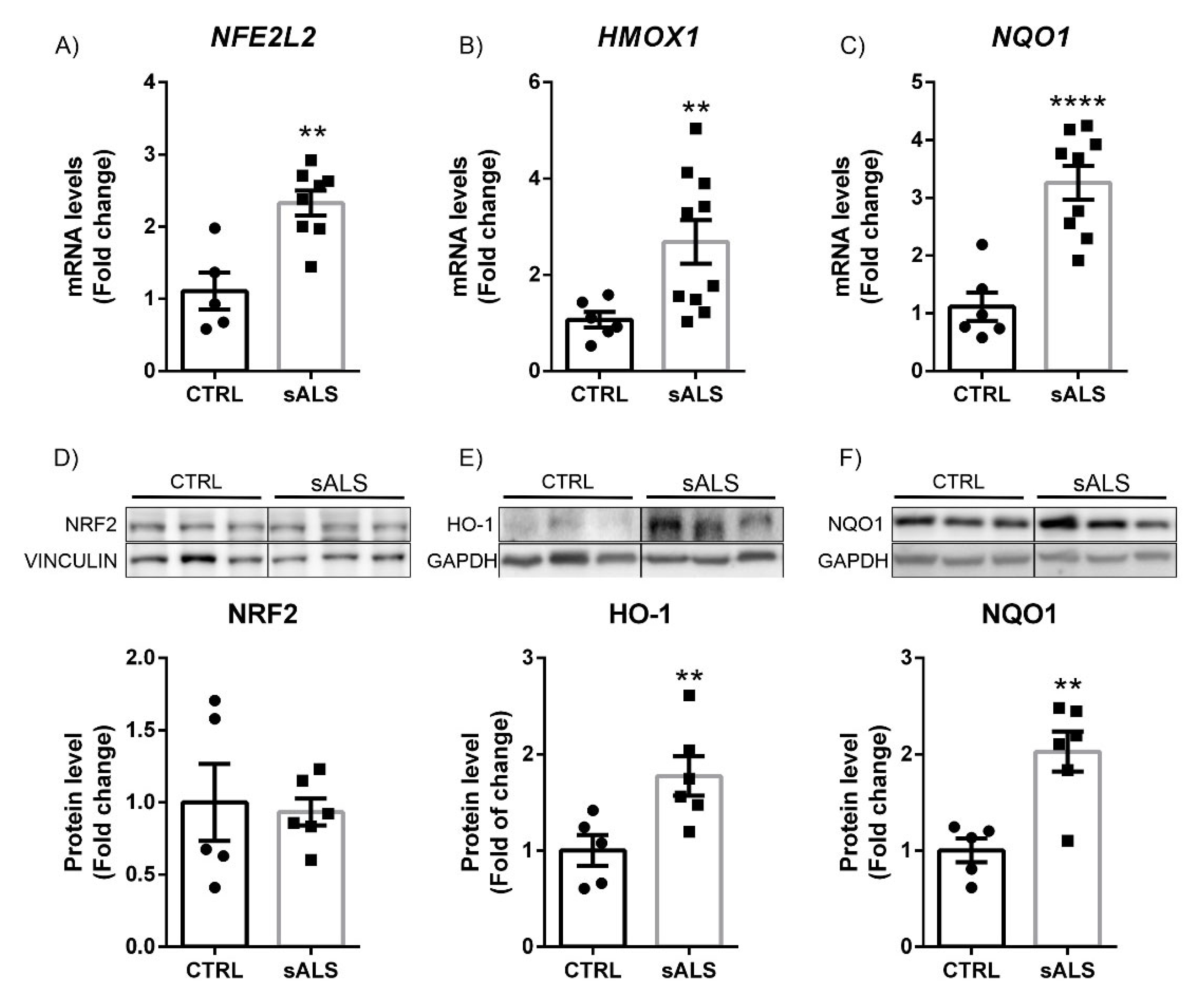
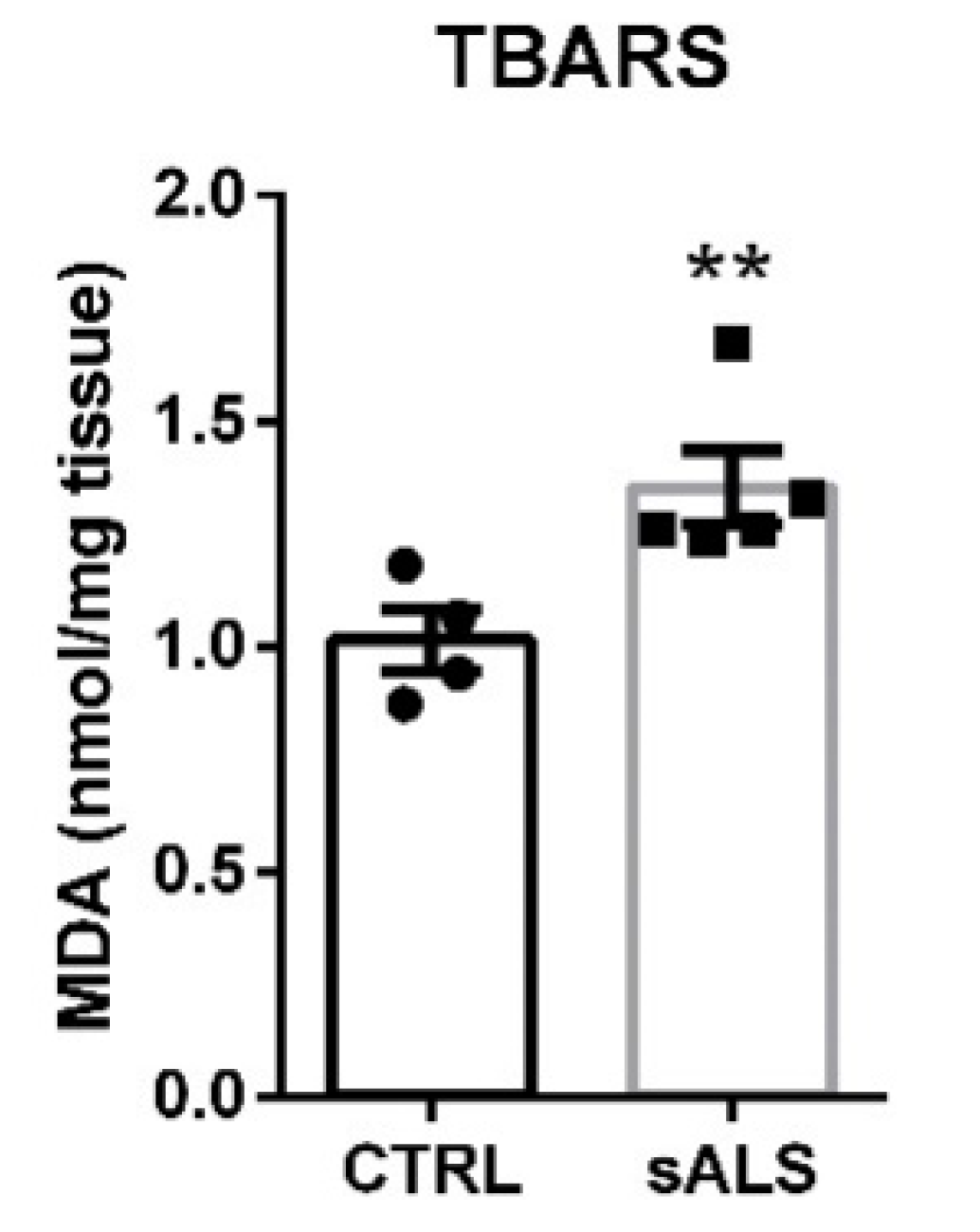
3.3. Altered NRF2 Signaling Pathway in ALS Human Samples
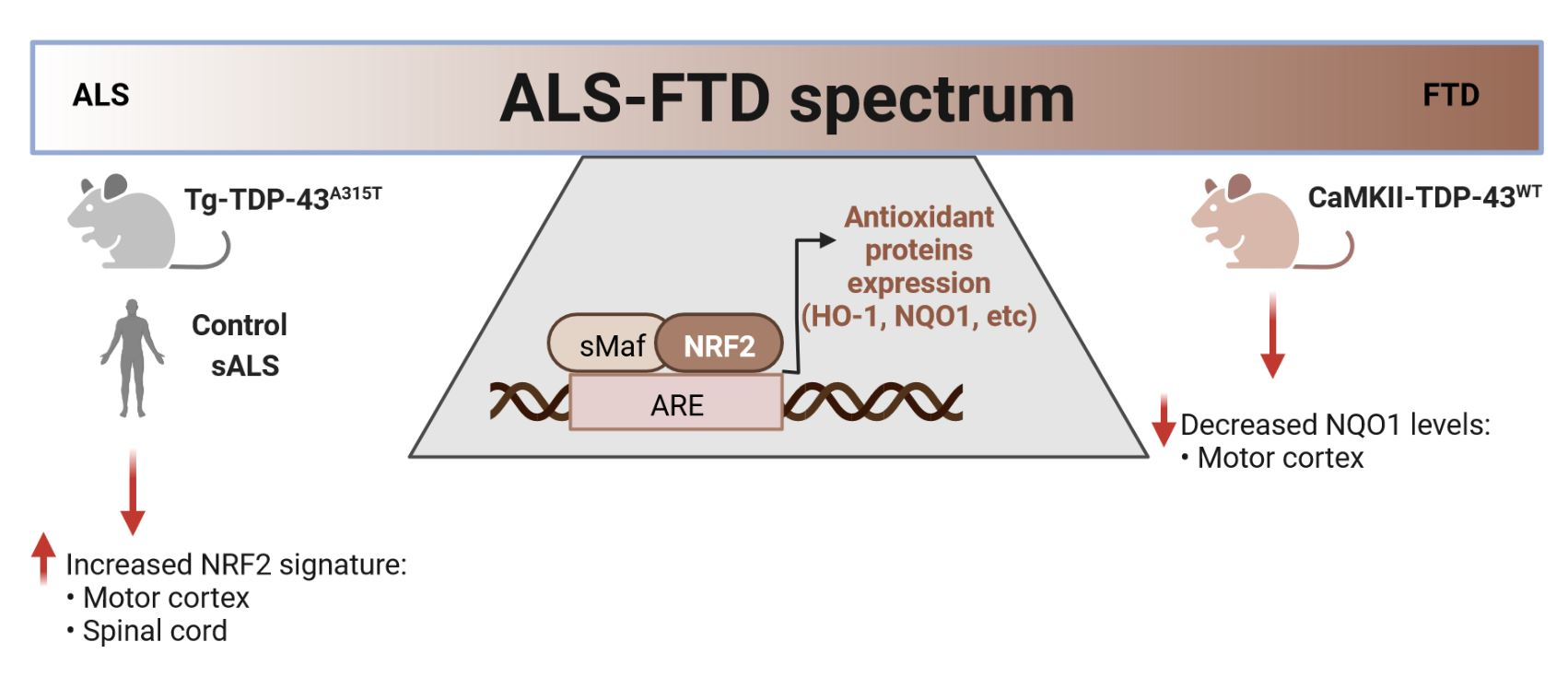
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, F.B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Marin, B. ALS and frontotemporal dementia belong to a common disease spectrum. Rev. Neurol. 2017, 173, 273–279. [Google Scholar] [CrossRef]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Liscic, R.M.; Grinberg, L.T.; Zidar, J.; Gitcho, M.A.; Cairns, N.J. ALS and FTLD: Two faces of TDP-43 proteinopathy. Eur. J. Neurol. 2008, 15, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurother. J. Am. Soc. Exp. NeuroTher. 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Koren, S.A.; Galvis-Escobar, S.; Abisambra, J.F. Tau-mediated dysregulation of RNA: Evidence for a common molecular mechanism of toxicity in frontotemporal dementia and other tauopathies. Neurobiol. Dis. 2020, 141, 104939. [Google Scholar] [CrossRef]
- Dumanchin, C.; Camuzat, A.; Campion, D.; Verpillat, P.; Hannequin, D.; Dubois, B.; Saugier-Veber, P.; Martin, C.; Penet, C.; Charbonnier, F.; et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum. Mol. Genet. 1998, 7, 1825–1829. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A. Brain-Protective Mechanisms of Transcription Factor NRF2: Toward a Common Strategy for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Minj, E.; Yadav, R.K.; Mehan, S. Targeting Abnormal Nrf2/HO-1 Signaling in Amyotrophic Lateral Sclerosis: Current Insights on Drug Targets and Influences on Neurological Disorders. Curr. Mol. Med. 2021, 21, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Bono, S.; Feligioni, M.; Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The NRF2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Arslanbaeva, L.; Bisaglia, M. Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules 2022, 27, 1471. [Google Scholar] [CrossRef]
- Suzen, S.; Tucci, P.; Profumo, E.; Buttari, B.; Saso, L. A Pivotal Role of Nrf2 in Neurodegenerative Disorders: A New Way for Therapeutic Strategies. Pharmaceuticals 2022, 15, 692. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Kugler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rabano, A.; Kugler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.J.; Yang, C.H.; Fang, Y.H.; Cho, K.H.; Chien, W.L.; Wang, W.T.; Wu, T.W.; Lin, C.P.; Fu, W.M.; Shen, C.K. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med. 2010, 207, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [PubMed]
- Galán-Ganga, M.; Rodríguez-Cueto, C.; Merchán-Rubira, J.; Hernández, F.; Ávila, J.; Posada-Ayala, M.; Lanciego, J.L.; Luengo, E.; Lopez, M.G.; Rábano, A.; et al. Cannabinoid receptor CB2 ablation protects against TAU induced neurodegeneration. Acta Neuropathol. Commun. 2021, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Erben, L.; Buonanno, A. Detection and Quantification of Multiple RNA Sequences Using Emerging Ultrasensitive Fluorescent In Situ Hybridization Techniques. Curr. Protoc. Neurosci. 2019, 87, e63. [Google Scholar] [CrossRef]
- Garcia, E.; Limon, D.; Perez-De La Cruz, V.; Giordano, M.; Diaz-Muñoz, M.; Maldonado, P.D.; Herrera-Mundo, M.N.; Pedraza-Chaverri, J.; Santamaria, A. Lipid peroxidation, mitochondrial dysfunction and neurochemical and behavioural deficits in different neurotoxic models: Protective role of S-allylcysteine. Free Radic. Res. 2008, 42, 892–902. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Porras, G.; Arribas-Blázquez, M.; Maestro, I.; Borrego-Hernández, D.; Boya, P.; Cerdán, S.; García-Redondo, A.; Martínez, A.; Martin-Requero, Á. Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 3007. [Google Scholar] [CrossRef]
- Cohen, T.J.; Lee, V.M.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L., 3rd; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008, 63, 535–538. [Google Scholar] [CrossRef]
- Rodríguez-Cueto, C.; Gómez-Almería, M.; García Toscano, L.; Romero, J.; Hillard, C.J.; de Lago, E.; Fernández-Ruiz, J. Inactivation of the CB(2) receptor accelerated the neuropathological deterioration in TDP-43 transgenic mice, a model of amyotrophic lateral sclerosis. Brain Pathol. 2021, 31, e12972. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Gunay, A.; Chandel, N.S.; Ozdinler, P.H. Mitochondrial dysregulation occurs early in ALS motor cortex with TDP-43 pathology and suggests maintaining NAD+ balance as a therapeutic strategy. Sci. Rep. 2022, 12, 4287. [Google Scholar] [CrossRef] [PubMed]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Martínez-González, L.; Rodríguez-Cueto, C.; Cabezudo, D.; Bartolomé, F.; Andrés-Benito, P.; Ferrer, I.; Gil, C.; Martín-Requero, Á.; Fernández-Ruiz, J.; Martínez, A.; et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1δ kinase inhibitor treatment. Sci. Rep. 2020, 10, 4449. [Google Scholar] [CrossRef]
- Martín-Cámara, O.; Arribas, M.; Wells, G.; Morales-Tenorio, M.; Martín-Requero, Á.; Porras, G.; Martínez, A.; Giorgi, G.; López-Alvarado, P.; Lastres-Becker, I.; et al. Multitarget Hybrid Fasudil Derivatives as a New Approach to the Potential Treatment of Amyotrophic Lateral Sclerosis. J. Med. Chem. 2022, 65, 1867–1882. [Google Scholar] [CrossRef]
- Bergström, P.; von Otter, M.; Nilsson, S.; Nilsson, A.C.; Nilsson, M.; Andersen, P.M.; Hammarsten, O.; Zetterberg, H. Association of NFE2L2 and KEAP1 haplotypes with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 130–137. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Gutierrez, C.; Bonora, N.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Bates, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Abrogating mitochondrial ROS in neurons or astrocytes reveals cell-specific impact on mouse behaviour. Redox Biol. 2021, 41, 101917. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef]
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 Activation in Astrocytes Protects against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Burton, N.C.; Kutzke, J.; Gan, L.; Johnson, D.A.; Schäfer, M.; Werner, S.; Johnson, J.A. Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PLoS ONE 2013, 8, e56625. [Google Scholar] [CrossRef]
- Lau, A.; Tian, W.; Whitman, S.A.; Zhang, D.D. The predicted molecular weight of Nrf2: It is what it is not. Antioxid. Redox Signal. 2013, 18, 91–93. [Google Scholar] [CrossRef]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef]
- Mimoto, T.; Miyazaki, K.; Morimoto, N.; Kurata, T.; Satoh, K.; Ikeda, Y.; Abe, K. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res. 2012, 1446, 109–118. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Henry, R.G.; Langmore, S.; Kramer, J.H.; Miller, B.L.; Lomen-Hoerth, C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch. Neurol. 2007, 64, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Espejo-Porras, F.; García-Toscano, L.; Rodríguez-Cueto, C.; Santos-García, I.; de Lago, E.; Fernandez-Ruiz, J. Targeting glial cannabinoid CB(2) receptors to delay the progression of the pathological phenotype in TDP-43 (A315T) transgenic mice, a model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2019, 176, 1585–1600. [Google Scholar] [CrossRef]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Clinic Presentation | Age | Gender | Samples |
|---|---|---|---|---|
| BCPA00870 | ALS | 71 | M | FROZEN MOTOR CX |
| BCPA00746 | ALS | 67 | F | FROZEN MOTOR CX |
| BCPA00694 | ALS | 77 | M | FROZEN MOTOR CX |
| BCPA00658 | ALS | 77 | M | FROZEN MOTOR CX |
| BCPA00493 | ALS | 79 | F | FROZEN MOTOR CX |
| BCPA00405 | ALS | 80 | F | FROZEN MOTOR CX |
| BCPA00347 | ALS | 67 | F | FROZEN MOTOR CX |
| BCPA00481 | ALS | 71 | F | FROZEN MOTOR CX |
| BCPA00364 | Control | 43 | M | FROZEN MOTOR CX |
| BCPA00537 | Control | 74 | F | FROZEN MOTOR CX |
| BCPA00587 | Control | 83 | F | FROZEN MOTOR CX |
| BCPA00662 | Control | 58 | F | FROZEN MOTOR CX |
| BCPA00849 | Control | 61 | F | FROZEN MOTOR CX |
| BCPA00972 | Control | 83 | M | FROZEN MOTOR CX |
| 381 | ALS | 72 | M | FROZEN SPINAL CORD |
| 1216 | ALS | 78 | M | FROZEN SPINAL CORD |
| 1344 | ALS | 60 | F | FROZEN SPINAL CORD |
| 1407 | ALS | 65 | F | FROZEN SPINAL CORD |
| 1872 | ALS | 66 | M | FROZEN SPINAL CORD |
| 1971 | ALS | 71 | F | FROZEN SPINAL CORD |
| 1694 | Control | 58 | M | FROZEN SPINAL CORD |
| 1697 | Control | 78 | M | FROZEN SPINAL CORD |
| 1818 | Control | 78 | M | FROZEN SPINAL CORD |
| 1858 | Control | 83 | F | FROZEN SPINAL CORD |
| 1888 | Control | 93 | F | FROZEN SPINAL CORD |
| 1937 | Control | 83 | F | FROZEN SPINAL CORD |
| 1949 | Control | 86 | M | FROZEN SPINAL CORD |
| Gene Product | Forward Primer | Reverse Primer |
|---|---|---|
| β-ACTIN | 5′ TCCTTCCTGGGCATGGAG 3′ | 5′ AGGAGGAGCAATGATCTTGATCTT 3′ |
| HMOX1 | 5′ TGCTCAACATCCAGCTCTTTGA 3′ | 5′ GCAGAATCTTGCACTTTGTTGCT 3′ |
| Hmox1 | 5′ CACAGATGGCGTCACTTCGTC 3′ | 5′ GTGAGGACCCACTGGAGGAG 3′ |
| NFE2L2 | 5′CCCGAAGCACGCTGAAGGCA 3′ | 5′ CCAGGCGGTGGGTCTCCGTA 3′ |
| NQO1 | 5′ GTTCATAGGAGAGTTTGCTT 3′ | 5′ TAGAACCTCAACTGACACTT 3′ |
| Nfe2l2 | 5′ CCCGAAGCACGCTGAAGGCA 3′ | 5′ CCAGGCGGTGGGTCTCCGTA 3′ |
| Nqo1 | 5′ GGTAGCGGCTCCATGTACTC 3′ | 5′ CATCCTTCCAGGATCTGCAT 3′ |
| TBP | 5′ TGCACAGGAGCCAAGAGTGAA 3′ | 5′ CACATCACAGCTCCCCACCA 3′ |
| Antibody | Source | Catalog Number | Dilution |
|---|---|---|---|
| GAPDH (6C5) | Santa Cruz Biotechnologies () | sc-32233 | 1:1000 |
| HO-1 | Millipore | AB1284 | 1:1000 |
| IBA1 | Wako | 019-19741 | 1:500 |
| LAMIN B | Santa Cruz Biotechnologies | sc-6217 | 1:1000 |
| NRF2 | Abyntek | AJ1555a | 1:2000 |
| NQO1 | Abcam | ab2346 | 1:2000 |
| VINCULIN (N3C1) | Gentex | GTX109749 | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lastres-Becker, I.; de Lago, E.; Martínez, A.; Fernández-Ruiz, J. New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Biomolecules 2022, 12, 1200. https://doi.org/10.3390/biom12091200
Lastres-Becker I, de Lago E, Martínez A, Fernández-Ruiz J. New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Biomolecules. 2022; 12(9):1200. https://doi.org/10.3390/biom12091200
Chicago/Turabian StyleLastres-Becker, Isabel, Eva de Lago, Ana Martínez, and Javier Fernández-Ruiz. 2022. "New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia" Biomolecules 12, no. 9: 1200. https://doi.org/10.3390/biom12091200
APA StyleLastres-Becker, I., de Lago, E., Martínez, A., & Fernández-Ruiz, J. (2022). New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Biomolecules, 12(9), 1200. https://doi.org/10.3390/biom12091200