
Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease


Abstract
:1. Introduction
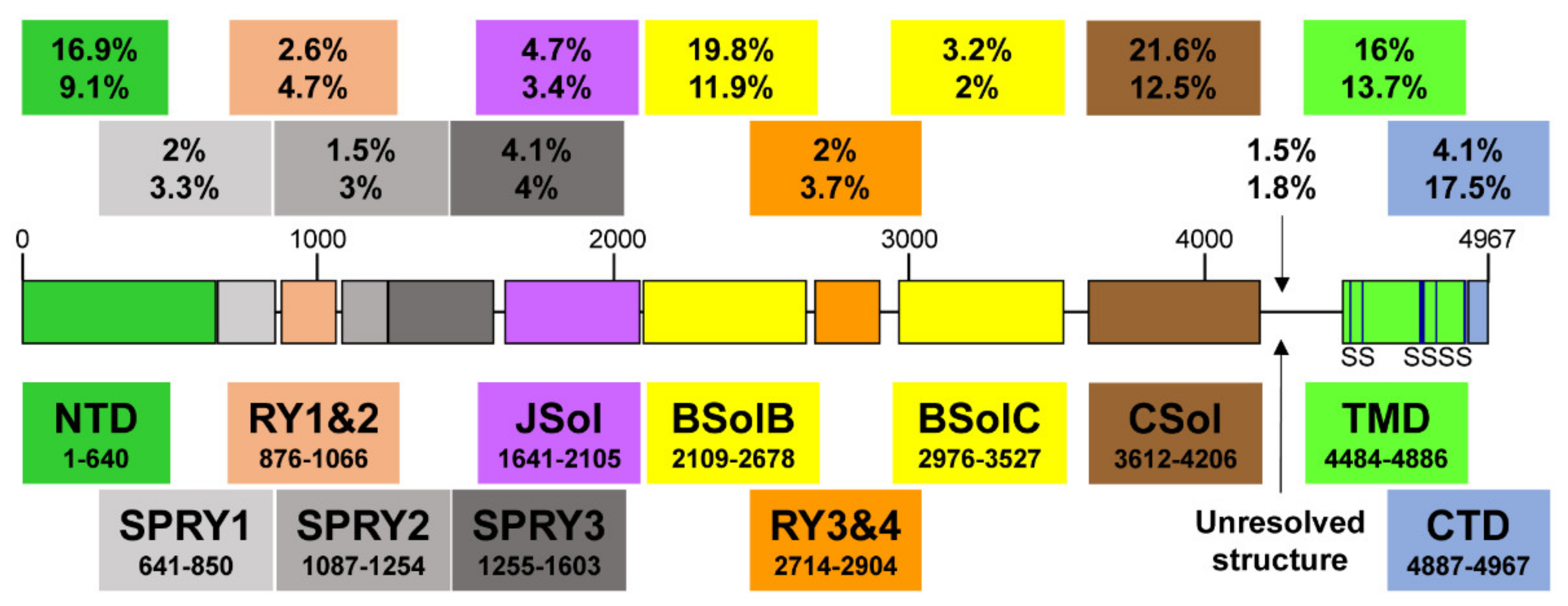
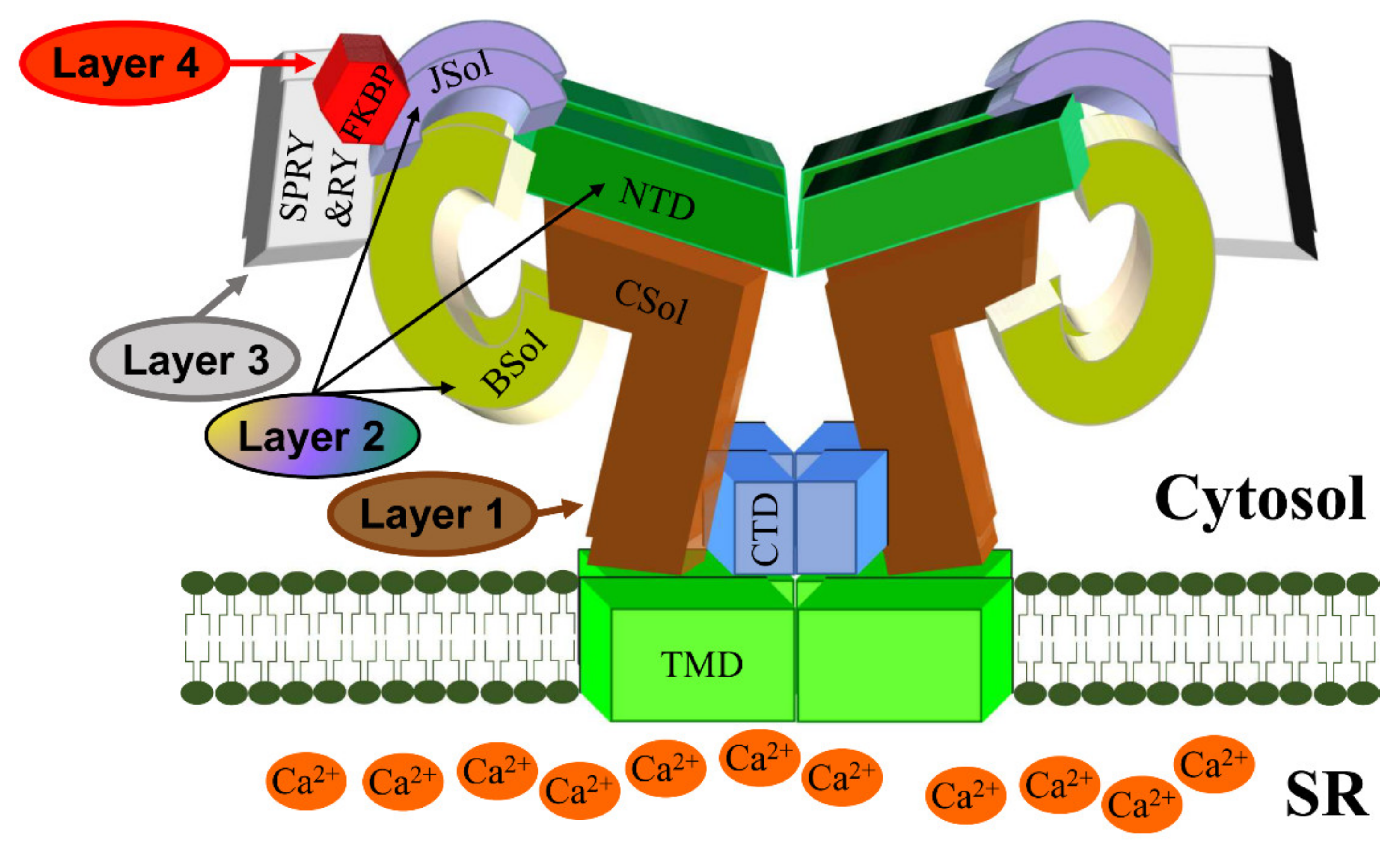
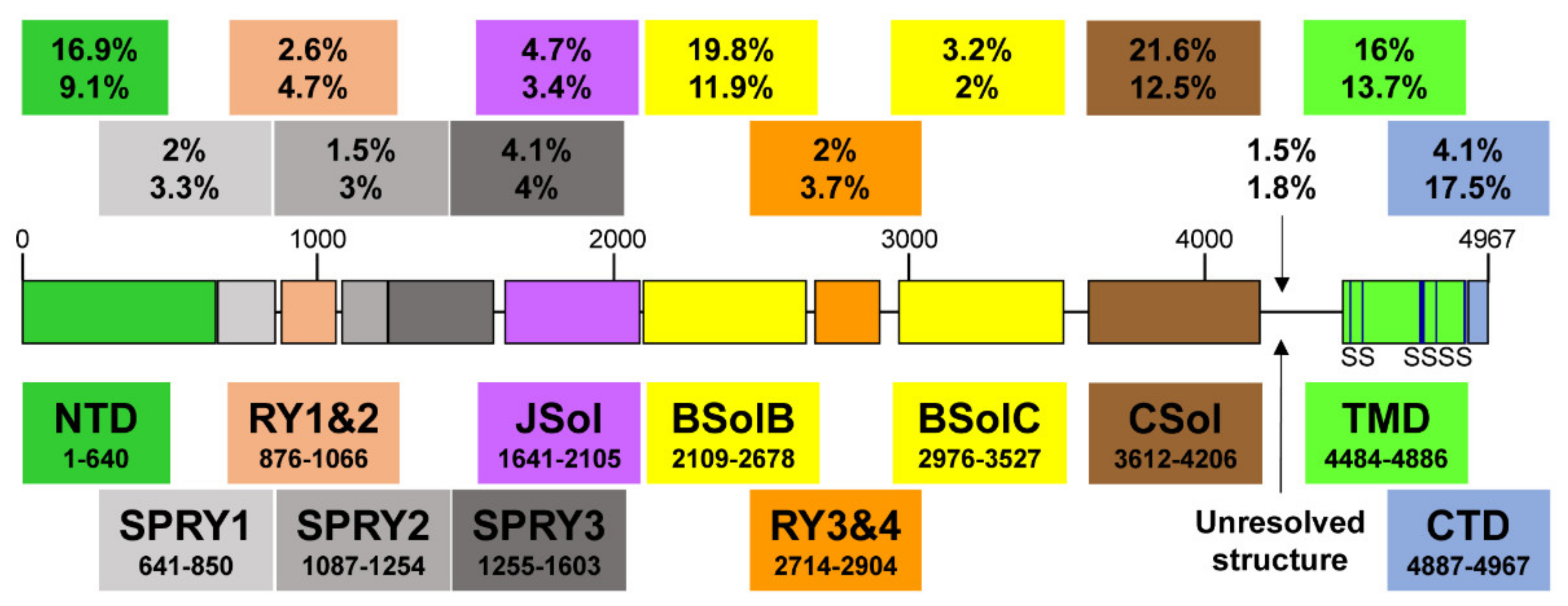
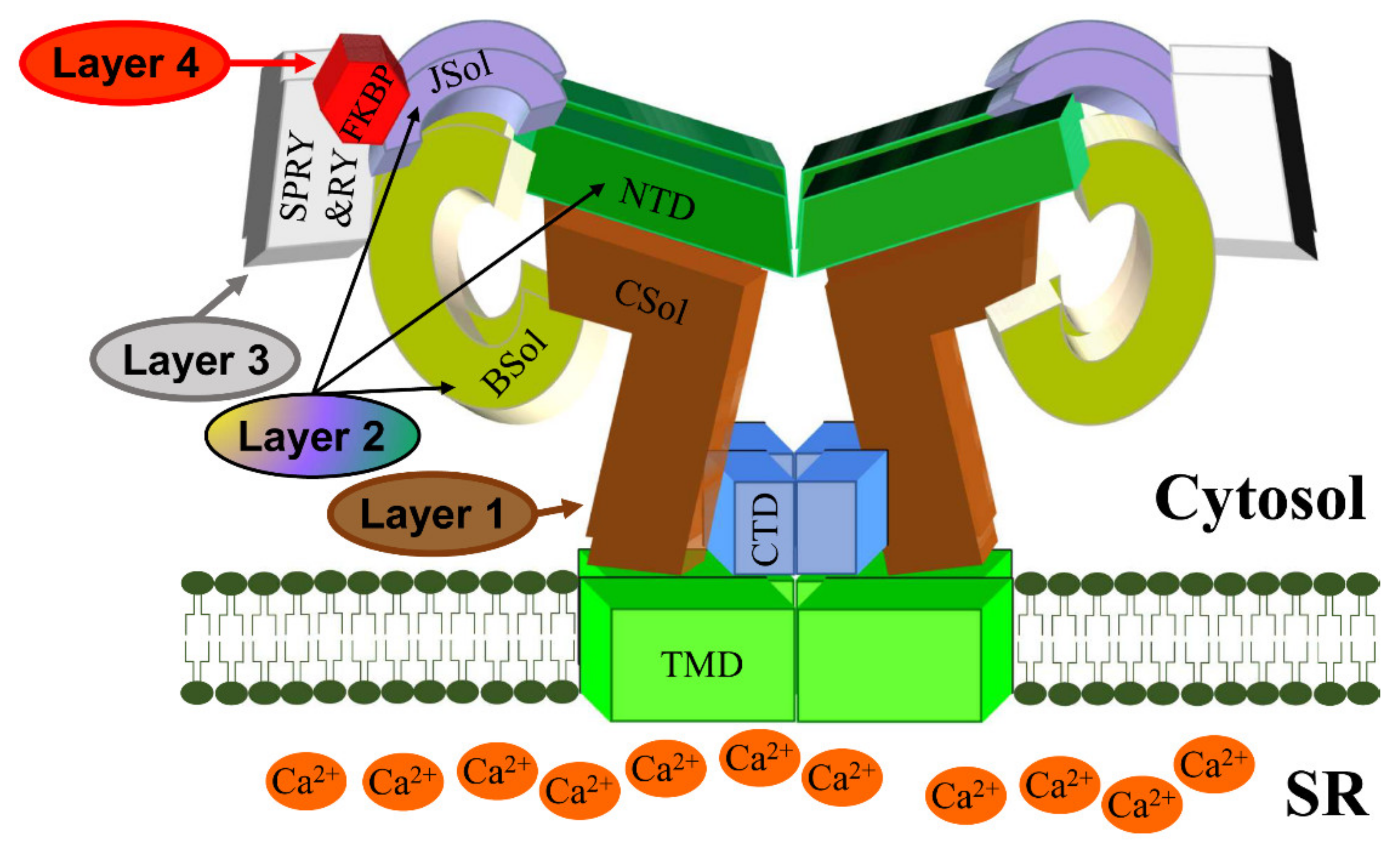
2. RyR2 Structure-–Function Relationships
RyR2 Channel Self-Assembly
3. Physiological RyR2 Regulators
3.1. Regulation of RyR2 by Accessory Proteins
3.1.1. FKBP12/12.6
3.1.2. Calmodulin (CaM)
3.1.3. Calsequestrin-2 (CSQ2)
4. Cardiac Disease Associated with RyR2 Mutations
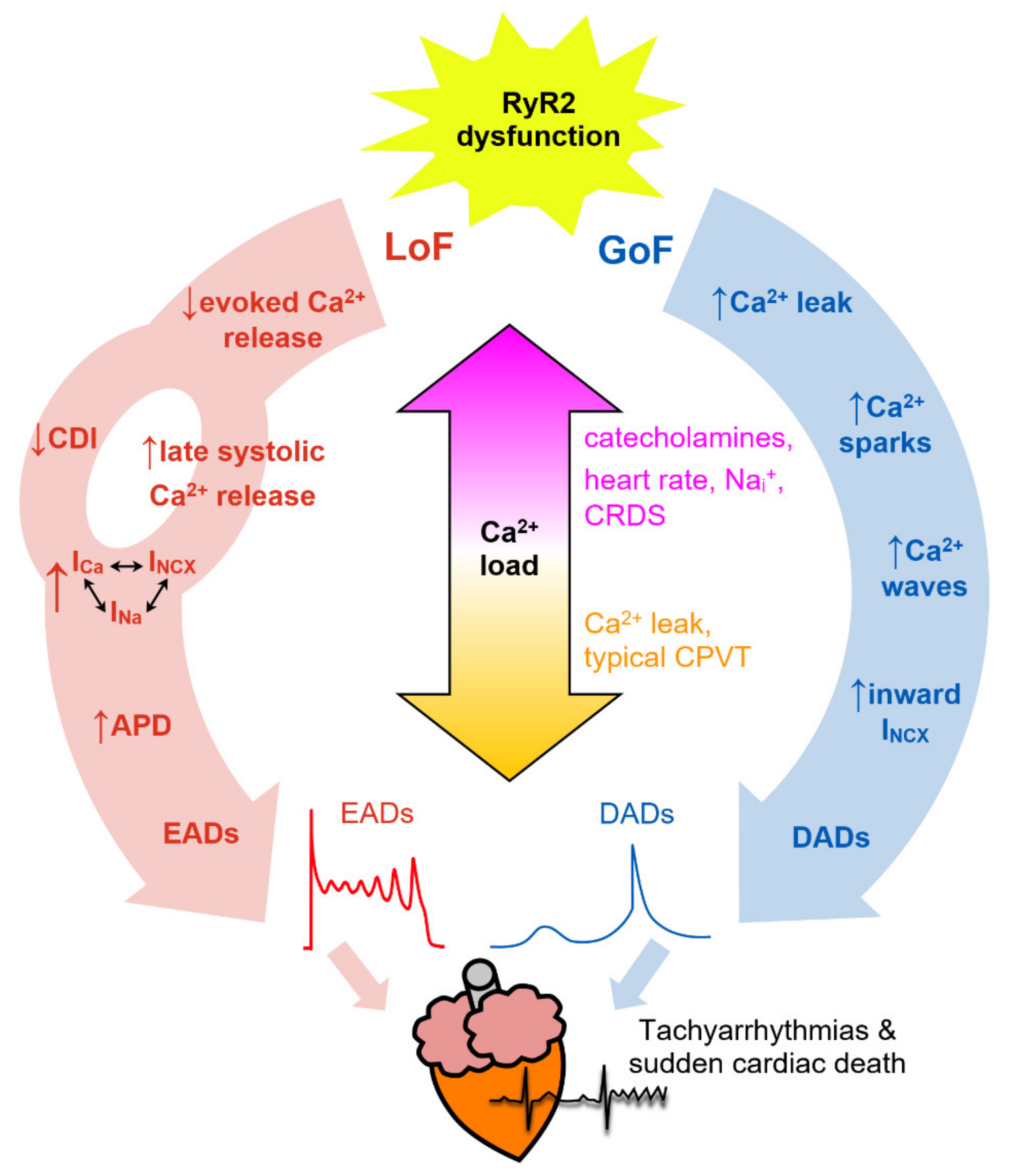
5. Molecular Mechanisms of Genetic RyR2 Disease
6. RyR2 Subcellular Geometry Influences Cardiac Ca2+ Release
6.1. RyR2 Clusters and Functional Ca2+ Release Units (CRU)
6.2. Pathological Fragmentation of CRU
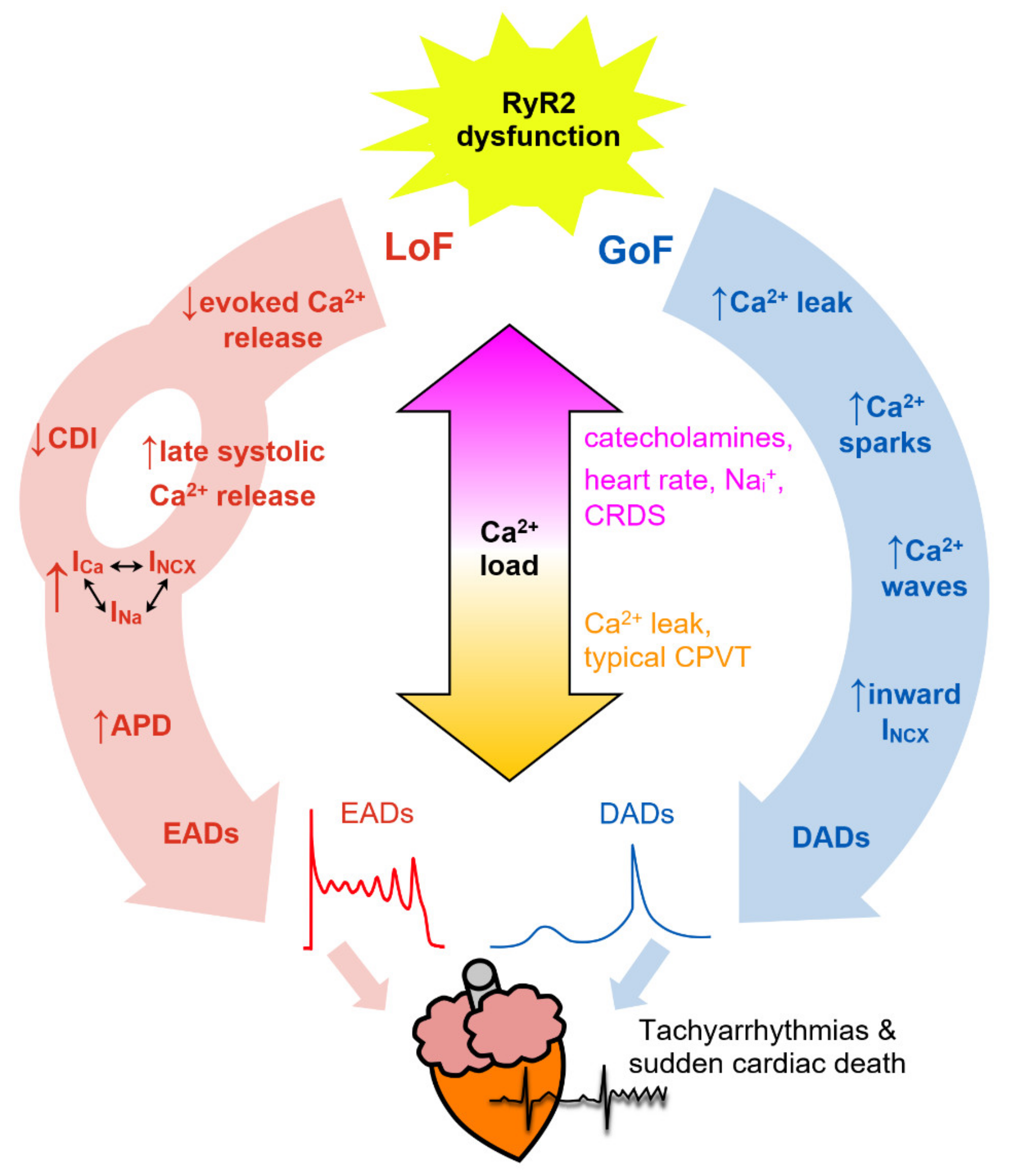
7. Arrhythmogenic Consequences of RyR2 Dysfunction
7.1. Ca2+ Sparks
7.2. Spontaneous Ca2+ Waves
7.3. Delayed Afterdepolarizations (DADs)
7.4. Early Afterdepolarizations (EADs)
8. Conclusions
Funding
Conflicts of Interest
References
- Bers, D. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Ackerman, M.J. Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annu. Rev. Med. 2009, 60, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef]
- Marx, S.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, P.; Brown, K.; Piippo, K.; Swan, H.; Devaney, J.; BrahmBhatt, B.; Donarum, E.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef]
- Priori, S.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Tiso, N.; Stephan, D.; Nava, A.; Bagattin, A.; Devaney, J.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e117. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef]
- Yano, M.; Yamamoto, T.; Ikeda, Y.; Matsuzaki, M. Mechanisms of Disease: Ryanodine receptor defects in heart failure and fatal arrhythmia. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 43–52. [Google Scholar] [CrossRef]
- Euden, J.; Mason, S.A.; Williams, A.J. Functional characterization of the cardiac ryanodine receptor pore-forming region. PLoS ONE 2013, 8, e66542. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Tripathy, A.; Lu, X.; Meissner, G. Evidence for a role of C-terminal amino acid residues in skeletal muscle Ca2+ release channel (ryanodine receptor) function. FEBS Lett. 1997, 412, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.; Zissimopoulos, S.; Lai, F. Oligomerization of the cardiac ryanodine receptor C-terminal tail. Biochem. J. 2003, 376, 795–799. [Google Scholar] [CrossRef]
- Bhat, M.; Zhao, J.; Takeshima, H.; Ma, J. Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys. J. 1997, 73, 1329–1336. [Google Scholar] [CrossRef] [Green Version]
- George, C.; Jundi, H.; Thomas, N.; Scoote, M.; Walters, N.; Williams, A.; Lai, F. Ryanodine receptor regulation by intramolecular interaction between cytoplasmic and transmembrane domains. Mol. Biol. Cell 2004, 15, 2627–2638. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Bhat, M.; Nishi, M.; Takeshima, H.; Ma, J. Molecular cloning of cDNA encoding a Drosophila ryanodine receptor and functional studies of the carboxyl-terminal calcium release channel. Biophys. J. 2000, 78, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Seidel, M.; de Meritens, C.R.; Johnson, L.; Parthimos, D.; Bannister, M.; Thomas, N.L.; Ozekhome-Mike, E.; Lai, F.A.; Zissimopoulos, S. Identification of an amino-terminus determinant critical for ryanodine receptor/Ca2+ release channel function. Cardiovasc. Res. 2021, 117, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Suetomi, T.; Yano, M.; Uchinoumi, H.; Fukuda, M.; Hino, A.; Ono, M.; Xu, X.; Tateishi, H.; Okuda, S.; Doi, M.; et al. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca2+ release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation 2011, 124, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Uchinoumi, H.; Yano, M.; Suetomi, T.; Ono, M.; Xu, X.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; Kobayashi, S.; et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ. Res. 2010, 106, 1413–1424. [Google Scholar] [CrossRef]
- Kimlicka, L.; Lau, K.; Tung, C.C.; Van Petegem, F. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat. Commun. 2013, 4, 1506. [Google Scholar] [CrossRef] [Green Version]
- Seidel, M.; Thomas, N.L.; Williams, A.J.; Lai, F.A.; Zissimopoulos, S. Dantrolene rescues aberrant N-terminus inter-subunit interactions in mutant pro-arrhythmic cardiac ryanodine receptors. Cardiovasc. Res. 2015, 105, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Zissimopoulos, S.; Viero, C.; Seidel, M.; Cumbes, B.; White, J.; Cheung, I.; Stewart, R.; Jeyakumar, L.H.; Fleischer, S.; Mukherjee, S.; et al. N-terminus oligomerization regulates the function of cardiac ryanodine receptors. J. Cell Sci. 2013, 126, 5042–5051. [Google Scholar] [CrossRef] [Green Version]
- Laver, D.R. Regulation of the RyR channel gating by Ca2+ and Mg2+. Biophys. Rev. 2018, 10, 1087–1095. [Google Scholar] [CrossRef]
- Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017, 149, 1065–1089. [Google Scholar] [CrossRef]
- Chi, X.; Gong, D.; Ren, K.; Zhou, G.; Huang, G.; Lei, J.; Zhou, Q.; Yan, N. Molecular basis for allosteric regulation of the type 2 ryanodine receptor channel gating by key modulators. Proc. Natl. Acad. Sci. USA 2019, 116, 25575–25582. [Google Scholar] [CrossRef] [Green Version]
- Murayama, T.; Ogawa, H.; Kurebayashi, N.; Ohno, S.; Horie, M.; Sakurai, T. A tryptophan residue in the caffeine-binding site of the ryanodine receptor regulates Ca2+ sensitivity. Commun. Biol. 2018, 1, 98. [Google Scholar] [CrossRef]
- Xu, L.; Chirasani, V.R.; Carter, J.S.; Pasek, D.A.; Dokholyan, N.V.; Yamaguchi, N.; Meissner, G. Ca2+-mediated activation of the skeletal-muscle ryanodine receptor ion channel. J. Biol. Chem. 2018, 293, 19501–19509. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, R.; Chen, B.; Zhong, X.; Kong, H.; Bai, Y.; Zhou, Q.; Xie, C.; Zhang, J.; Guo, A.; et al. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med. 2014, 20, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Zissimopoulos, S.; Lai, F. Ryanodine receptor structure, function and pathophysiology. In Calcium: A Matter of Life or Death; Elsevier: Amsterdam, The Netherlands, 2007; Volume 41, pp. 287–342. [Google Scholar]
- Zissimopoulos, S.; Seifan, S.; Maxwell, C.; Williams, A.J.; Lai, F.A. Disparities in the association of the ryanodine receptor and the FK506-binding proteins in mammalian heart. J. Cell Sci. 2012, 125, 1759–1769. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Cornea, R.L.; Huke, S.; Camors, E.; Yang, Y.; Picht, E.; Fruen, B.R.; Bers, D.M. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 2010, 106, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, L.; Ballester, L.; Cheng, D.; McIntyre, J.; Chang, P.; Olivey, H.; Rollins-Smith, L.; Barnett, J.; Murray, K.; Xin, H.-B.; et al. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem. Biophys. Res. Commun. 2001, 281, 979–986. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D. FK506 binding proteins: Cellular regulators of intracellular Ca2+ signalling. Eur. J. Pharmacol. 2013, 700, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Bai, X.C.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.C.; Li, X.; Scheres, S.H.; et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Balshaw, D.; Xu, L.; Yamaguchi, N.; Pasek, D.; Meissner, G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 2001, 276, 20144–20153. [Google Scholar] [CrossRef] [Green Version]
- Fruen, B.; Bardy, J.; Byrem, T.; Strasburg, G.; Louis, C. Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. 2000, 279, C724–C733. [Google Scholar] [CrossRef] [Green Version]
- Gong, D.; Chi, X.; Wei, J.; Zhou, G.; Huang, G.; Zhang, L.; Wang, R.; Lei, J.; Chen, S.R.W.; Yan, N. Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 2019, 572, 347–351. [Google Scholar] [CrossRef]
- Chazin, W.J.; Johnson, C.N. Calmodulin Mutations Associated with Heart Arrhythmia: A Status Report. Int. J. Mol. Sci. 2020, 21, 1418. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Mazzanti, A.; Santiago, D.J.; Kukavica, D.; Trancuccio, A.; Kovacic, J.C. Precision Medicine in Catecholaminergic Polymorphic Ventricular Tachycardia: JACC Focus Seminar 5/5. J. Am. Coll. Cardiol. 2021, 77, 2592–2612. [Google Scholar] [CrossRef]
- Handhle, A.; Ormonde, C.E.; Thomas, N.L.; Bralesford, C.; Williams, A.J.; Lai, F.A.; Zissimopoulos, S. Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci. 2016, 129, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Kelley, J.; Schmeisser, G.; Kobayashi, Y.; Jones, L. Complex formation between Junctin, Triadin, Calsequestrin, and the ryanodine receptor: Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997, 272, 23389–23397. [Google Scholar] [CrossRef] [Green Version]
- Sibbles, E.T.; Waddell, H.M.M.; Mereacre, V.; Jones, P.P.; Munro, M.L. The function and regulation of calsequestrin-2: Implications in calcium-mediated arrhythmias. Biophys. Rev. 2022, 14, 329–352. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Alcalai, R.; Arad, M.; Wolf, C.M.; Toka, O.; Conner, D.A.; Berul, C.I.; Eldar, M.; Seidman, C.E.; Seidman, J.G. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2007, 117, 1814–1823. [Google Scholar] [CrossRef]
- Gray, B.; Bagnall, R.D.; Lam, L.; Ingles, J.; Turner, C.; Haan, E.; Davis, A.; Yang, P.C.; Clancy, C.E.; Sy, R.W.; et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2016, 13, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Titus, E.W.; Lieve, K.V.; Roston, T.M.; Mazzanti, A.; Deiter, F.H.; Denjoy, I.; Ingles, J.; Till, J.; Robyns, T.; et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of CASQ2-Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2020, 142, 932–947. [Google Scholar] [CrossRef]
- Titus, E.W.; Deiter, F.H.; Shi, C.; Wojciak, J.; Scheinman, M.; Jura, N.; Deo, R.C. The structure of a Calsequestrin filament reveals mechanisms of familial arrhythmia. Nat. Struct. Mol. Biol. 2020, 27, 1142–1151. [Google Scholar] [CrossRef]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020, 598, 2817–2834. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [Green Version]
- Mazzanti, A.; Kukavica, D.; Trancuccio, A.; Memmi, M.; Bloise, R.; Gambelli, P.; Marino, M.; Ortiz-Genga, M.; Morini, M.; Monteforte, N.; et al. Outcomes of Patients with Catecholaminergic Polymorphic Ventricular Tachycardia Treated with beta-Blockers. JAMA Cardiol. 2022, 7, 504–512. [Google Scholar] [CrossRef]
- Kryshtal, D.O.; Blackwell, D.J.; Egly, C.L.; Smith, A.N.; Batiste, S.M.; Johnston, J.N.; Laver, D.R.; Knollmann, B.C. RYR2 Channel Inhibition Is the Principal Mechanism of Flecainide Action in CPVT. Circ. Res. 2021, 128, 321–331. [Google Scholar] [CrossRef]
- Bannister, M.L.; MacLeod, K.T.; George, C.H. Moving in the right direction: Elucidating the mechanisms of interaction between flecainide and the cardiac ryanodine receptor. Br. J. Pharmacol. 2022, 179, 2558–2563. [Google Scholar] [CrossRef]
- Benitah, J.P.; Gomez, A.M. Is the Debate on the Flecainide Action on the RYR2 in CPVT Closed? Circ. Res. 2021, 128, 332–334. [Google Scholar] [CrossRef]
- Roux-Buisson, N.; Gandjbakhch, E.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; Cosnay, P.; et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Results of a systematic screening. Heart Rhythm 2014, 11, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Shigemizu, D.; Aiba, T.; Nakagawa, H.; Ozaki, K.; Miya, F.; Satake, W.; Toda, T.; Miyamoto, Y.; Fujimoto, A.; Suzuki, Y.; et al. Exome Analyses of Long QT Syndrome Reveal Candidate Pathogenic Mutations in Calmodulin-Interacting Genes. PLoS ONE 2015, 10, e0130329. [Google Scholar] [CrossRef]
- Tester, D.J.; Kopplin, L.J.; Will, M.L.; Ackerman, M.J. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 1099–1105. [Google Scholar] [CrossRef]
- Fujii, Y.; Itoh, H.; Ohno, S.; Murayama, T.; Kurebayashi, N.; Aoki, H.; Blancard, M.; Nakagawa, Y.; Yamamoto, S.; Matsui, Y.; et al. A type 2 ryanodine receptor variant associated with reduced Ca2+ release and short-coupled torsades de pointes ventricular arrhythmia. Heart Rhythm 2017, 14, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Touat-Hamici, Z.; Blancard, M.; Ma, R.; Lin, L.; Iddir, Y.; Denjoy, I.; Leenhardt, A.; Yuchi, Z.; Guicheney, P. A SPRY1 domain cardiac ryanodine receptor variant associated with short-coupled torsade de pointes. Sci. Rep. 2021, 11, 5243. [Google Scholar] [CrossRef]
- Ohno, S.; Omura, M.; Kawamura, M.; Kimura, H.; Itoh, H.; Makiyama, T.; Ushinohama, H.; Makita, N.; Horie, M. Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction. Europace 2014, 16, 1646–1654. [Google Scholar] [CrossRef]
- Roston, T.M.; Guo, W.; Krahn, A.D.; Wang, R.; Van Petegem, F.; Sanatani, S.; Chen, S.R.; Lehman, A. A novel RYR2 loss-of-function mutation (I4855M) is associated with left ventricular non-compaction and atypical catecholaminergic polymorphic ventricular tachycardia. J. Electrocardiol. 2017, 50, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Medeiros-Domingo, A.; Gasperetti, A.; Breitenstein, A.; Steffel, J.; Guidetti, F.; Flammer, A.; Odening, K.; Ruschitzka, F.; Duru, F.; et al. Familial dilated cardiomyopathy associated with a novel heterozygous RYR2 early truncating variant. Cardiol. J. 2021, 28, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Muller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, F.J.; Bos, J.M.; Yuchi, Z.; Valdivia, C.R.; Hernandez, J.J.; Zhao, Y.T.; Henderlong, D.S.; Chen, Y.; Booher, T.R.; Marcou, C.A.; et al. Cardiac hypertrophy and arrhythmia in mice induced by a mutation in ryanodine receptor 2. JCI Insight 2019, 5, e126544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Yao, J.; Ni, M.; Wei, J.; Zhong, X.; Guo, W.; Zhang, L.; Wang, R.; Belke, D.; Chen, Y.X.; et al. Cardiac ryanodine receptor calcium release deficiency syndrome. Sci. Transl. Med. 2021, 13, eaba7287. [Google Scholar] [CrossRef]
- Tester, D.J.; Kim, C.S.J.; Hamrick, S.K.; Ye, D.; O’Hare, B.J.; Bombei, H.M.; Fitzgerald, K.K.; Haglund-Turnquist, C.M.; Atkins, D.L.; Nunez, L.A.O.; et al. Molecular characterization of the calcium release channel deficiency syndrome. JCI Insight 2020, 5, e135952. [Google Scholar] [CrossRef]
- Olubando, D.; Hopton, C.; Eden, J.; Caswell, R.; Lowri Thomas, N.; Roberts, S.A.; Morris-Rosendahl, D.; Venetucci, L.; Newman, W.G. Classification and correlation of RYR2 missense variants in individuals with catecholaminergic polymorphic ventricular tachycardia reveals phenotypic relationships. J. Hum. Genet. 2020, 65, 531–539. [Google Scholar] [CrossRef]
- Duvekot, J.C.; Baas, A.F.; Volker-Touw, C.M.L.; Bikker, H.; Schroer, C.; Breur, J. Early Lethal Noncompaction Cardiomyopathy in Siblings with Compound Heterozygous RYR2 Variant. Can. J. Cardiol. 2021, 37, 1864–1866. [Google Scholar] [CrossRef]
- Fernandez-Velasco, M.; Rueda, A.; Rizzi, N.; Benitah, J.P.; Colombi, B.; Napolitano, C.; Priori, S.G.; Richard, S.; Gomez, A.M. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2009, 104, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Guo, W.; Sun, B.; Hunt, D.J.; Wei, J.; Liu, Y.; Wang, Y.; Wang, R.; Jones, P.P.; Back, T.G.; et al. Enhanced Cytosolic Ca2+ Activation Underlies a Common Defect of Central Domain Cardiac Ryanodine Receptor Mutations Linked to Arrhythmias. J. Biol. Chem. 2016, 291, 24528–24537. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Xiao, B.; Yang, D.; Wang, R.; Choi, P.; Zhang, L.; Cheng, H.; Chen, S. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. USA 2004, 101, 13062–13067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrens, X.; Lenhart, S.; Huang, F.; Vest, J.; Reiken, S.; Mohler, P.; Sun, J.; Guatimosim, S.; Song, L.-S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Wang, R.; Xiao, B.; Kong, H.; Hunt, D.; Choi, P.; Zhang, L.; Chen, S. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 2005, 97, 1173–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zissimopoulos, S.; Thomas, N.L.; Jamaluddin, W.W.; Lai, F.A. FKBP12.6 binding of ryanodine receptors carrying mutations associated with arrhythmogenic cardiac disease. Biochem. J. 2009, 419, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Yamamoto, T.; Kobayashi, S.; Tamitani, M.; Hamada, Y.; Fukui, G.; Xu, X.; Nishimura, S.; Kato, T.; Uchinoumi, H.; et al. Ryanodine receptor-bound calmodulin is essential to protect against catecholaminergic polymorphic ventricular tachycardia. JCI Insight 2019, 4, e126112. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zahradnikova, A., Jr.; Rizzetto, R.; Boncompagni, S.; Rabesahala de Meritens, C.; Zhang, Y.; Joanne, P.; Marques-Sule, E.; Aguilar-Sanchez, Y.; Fernandez-Tenorio, M.; et al. Impaired Binding to Junctophilin-2 and Nanostructural Alteration in CPVT Mutation. Circ. Res. 2021, 129, e35–e52. [Google Scholar] [CrossRef]
- Jiang, D.; Chen, W.; Wang, R.; Zhang, L.; Chen, S. Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc. Natl. Acad. Sci. USA 2007, 104, 18309–18314. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wei, J.; Guo, W.; Sun, B.; Estillore, J.P.; Wang, R.; Yoruk, A.; Roston, T.M.; Sanatani, S.; Wilde, A.A.M.; et al. Human RyR2 (Ryanodine Receptor 2) Loss-of-Function Mutations: Clinical Phenotypes and In Vitro Characterization. Circ. Arrhythm. Electrophysiol. 2021, 14, e010013. [Google Scholar] [CrossRef]
- Roston, T.M.; Wei, J.; Guo, W.; Li, Y.; Zhong, X.; Wang, R.; Estillore, J.P.; Peltenburg, P.J.; Noguer, F.R.I.; Till, J.; et al. Clinical and Functional Characterization of Ryanodine Receptor 2 Variants Implicated in Calcium-Release Deficiency Syndrome. JAMA Cardiol. 2022, 7, 84–92. [Google Scholar] [CrossRef]
- Ormerod, J.O.M.; Ormondroyd, E.; Li, Y.; Taylor, J.; Wei, J.; Guo, W.; Wang, R.; Sarton, C.N.S.; McGuire, K.; Dreau, H.M.P.; et al. Provocation Testing and Therapeutic Response in a Newly Described Channelopathy: RyR2 Calcium Release Deficiency Syndrome. Circ. Genom. Precis. Med. 2022, 15, e003589. [Google Scholar] [CrossRef]
- Hirose, S.; Murayama, T.; Tetsuo, N.; Hoshiai, M.; Kise, H.; Yoshinaga, M.; Aoki, H.; Fukuyama, M.; Wuriyanghai, Y.; Wada, Y.; et al. Loss-of-function mutations in cardiac ryanodine receptor channel cause various types of arrhythmias including long QT syndrome. Europace 2022, 24, 497–510. [Google Scholar] [CrossRef]
- Zhong, X.; Guo, W.; Wei, J.; Tang, Y.; Liu, Y.; Zhang, J.Z.; Tan, V.H.; Zhang, L.; Wang, R.; Jones, P.P.; et al. Identification of loss-of-function RyR2 mutations associated with idiopathic ventricular fibrillation and sudden death. Biosci. Rep. 2021, 41, BSR20210209. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F. Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol. Rev. 1997, 77, 699–729. [Google Scholar] [CrossRef]
- Cabra, V.; Murayama, T.; Samsó, M. Ultrastructural Analysis of Self-Associated RyR2s. Biophys. J. 2016, 110, 2651–2662. [Google Scholar] [CrossRef] [Green Version]
- Baddeley, D.; Jayasinghe, I.D.; Lam, L.; Rossberger, S.; Cannell, M.B.; Soeller, C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 22275–22280. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Jayasinghe, I.; Crossman, D.J.; Baddeley, D.; Soeller, C. Nanoscale analysis of ryanodine receptor clusters in dyadic couplings of rat cardiac myocytes. J. Mol. Cell. Cardiol. 2015, 80, 45–55. [Google Scholar] [CrossRef]
- Shen, X.; den Brink, J.; Hou, Y.; Colli, D.; Le, C.; Kolstad, T.R.; MacQuaide, N.; Carlson, C.R.; Kekenes-Huskey, P.M.; Edwards, A.G.; et al. 3D dSTORM imaging reveals novel detail of ryanodine receptor localization in rat cardiac myocytes. J. Physiol. 2019, 597, 399–418. [Google Scholar] [CrossRef]
- Macquaide, N.; Tuan, H.-T.M.; Hotta, J.-I.; Sempels, W.; Lenaerts, I.; Holemans, P.; Hofkens, J.; Jafri, M.S.; Willems, R.; Sipido, K.R. Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc. Res. 2015, 108, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Munro, M.L.; van Hout, I.; Aitken-Buck, H.M.; Sugunesegran, R.; Bhagwat, K.; Davis, P.J.; Lamberts, R.R.; Coffey, S.; Soeller, C.; Jones, P.P. Human Atrial Fibrillation Is Not Associated with Remodeling of Ryanodine Receptor Clusters. Front. Cell Dev. Biol. 2021, 9, 633704. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Sobie, E.A.; Guatimosim, S.; Gómez-Viquez, L.; Song, L.-S.; Hartmann, H.; Saleet Jafri, M.; Lederer, W.J. The Ca2+ leak paradox and “rogue ryanodine receptors”: SR Ca2+ efflux theory and practice. Prog. Biophys. Mol. Biol. 2006, 90, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Galice, S.; Xie, Y.; Yang, Y.; Sato, D.; Bers, D.M. Size Matters: Ryanodine Receptor Cluster Size Affects Arrhythmogenic Sarcoplasmic Reticulum Calcium Release. J. Am. Heart Assoc. 2018, 7, e008724. [Google Scholar] [CrossRef] [Green Version]
- Laver, D.R.; Kong, C.H.T.; Imtiaz, M.S.; Cannell, M.B. Termination of calcium-induced calcium release by induction decay: An emergent property of stochastic channel gating and molecular scale architecture. J. Mol. Cell. Cardiol. 2013, 54, 98–100. [Google Scholar] [CrossRef]
- Sheard, T.M.D.; Hurley, M.E.; Colyer, J.; White, E.; Norman, R.; Pervolaraki, E.; Narayanasamy, K.K.; Hou, Y.; Kirton, H.M.; Yang, Z.; et al. Three-Dimensional and Chemical Mapping of Intracellular Signaling Nanodomains in Health and Disease with Enhanced Expansion Microscopy. ACS Nano 2019, 13, 2143–2157. [Google Scholar] [CrossRef]
- Munro, M.L.; Jayasinghe, I.D.; Wang, Q.; Quick, A.; Wang, W.; Baddeley, D.; Wehrens, X.H.T.; Soeller, C. Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. J. Cell Sci. 2016, 129, 4388–4398. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Porta, M.; Qin, J.; Ramos, J.; Nani, A.; Shannon, T.R.; Fill, M. Flux regulation of cardiac ryanodine receptor channels. J. Gen. Physiol. 2010, 135, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Zima, A.V.; Picht, E.; Bers, D.M.; Blatter, L.A. Termination of cardiac Ca2+ sparks: Role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008, 103, e105–e115. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, D.; Fill, M. Pernicious Attrition and Inter-RyR2 CICR Current Control in Cardiac Muscle. J. Mol. Cell. Cardiol. 2013, 58, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Eisner, D.A.; Trafford, A.W.; Díaz, M.E.; Overend, C.L.; O’Neill, S.C. The control of Ca release from the cardiac sarcoplasmic reticulum: Regulation versus autoregulation. Cardiovasc. Res. 1998, 38, 589–604. [Google Scholar] [CrossRef] [Green Version]
- Zima, A.V.; Bovo, E.; Bers, D.M.; Blatter, L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010, 588, 4743–4757. [Google Scholar] [CrossRef]
- Kashimura, T.; Briston, S.J.; Trafford, A.W.; Napolitano, C.; Priori, S.G.; Eisner, D.A.; Venetucci, L.A. In the RyR2R4496C Mouse Model of CPVT, β-Adrenergic Stimulation Induces Ca Waves by Increasing SR Ca Content and Not by Decreasing the Threshold for Ca Waves. Circ. Res. 2010, 107, 1483–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Sobie, E.A.; Song, L.-S.; Lederer, W.J. Local recovery of Ca2+ release in rat ventricular myocytes. J. Physiol. 2005, 565, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Janicek, R.; Fernandez-Tenorio, M.; Courtehoux, M.; Matas, L.; Gerbaud, P.; Gomez, A.M.; Egger, M.; Niggli, E. Uptake-leak balance of SR Ca2+ determines arrhythmogenic potential of RyR2R420Q+/− cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 170, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Janicek, R.; Agarwal, H.; Gómez, A.M.; Egger, M.; Ellis-Davies, G.C.R.; Niggli, E. Local recovery of cardiac calcium-induced calcium release interrogated by ultra-effective, two-photon uncaging of calcium. J. Physiol. 2021, 599, 3841–3852. [Google Scholar] [CrossRef] [PubMed]
- Brunello, L.; Slabaugh, J.L.; Radwanski, P.B.; Ho, H.-T.; Belevych, A.E.; Lou, Q.; Chen, H.; Napolitano, C.; Lodola, F.; Priori, S.G.; et al. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc. Natl. Acad. Sci. USA 2013, 110, 10312–10317. [Google Scholar] [CrossRef] [Green Version]
- Kryshtal, D.O.; Gryshchenko, O.; Gomez-Hurtado, N.; Knollmann, B.C. Impaired calcium-calmodulin-dependent inactivation of Cav1.2 contributes to loss of sarcoplasmic reticulum calcium release refractoriness in mice lacking calsequestrin 2. J. Mol. Cell. Cardiol. 2015, 82, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Nivala, M.; Ko, C.Y.; Nivala, M.; Weiss, J.N.; Qu, Z. Criticality in Intracellular Calcium Signaling in Cardiac Myocytes. Biophys. J. 2012, 102, 2433–2442. [Google Scholar] [CrossRef] [Green Version]
- Izu, L.T.; Means, S.A.; Shadid, J.N.; Chen-Izu, Y.; Balke, C.W. Interplay of ryanodine receptor distribution and calcium dynamics. Biophys. J. 2006, 91, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Chen-Izu, Y.; Ward, C.W.; Stark, W.; Banyasz, T.; Sumandea, M.P.; Balke, C.W.; Izu, L.T.; Wehrens, X.H.T. Phosphorylation of RyR2 and shortening of RyR2 cluster spacing in spontaneously hypertensive rat with heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H2409–H2417. [Google Scholar] [CrossRef] [Green Version]
- Fowler, E.D.; Benoist, D.; Drinkhill, M.J.; Stones, R.; Helmes, M.; Wüst, R.C.I.; Stienen, G.J.M.; Steele, D.S.; White, E. Decreased creatine kinase is linked to diastolic dysfunction in rats with right heart failure induced by pulmonary artery hypertension. J. Mol. Cell. Cardiol. 2015, 86, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, E.D.; Drinkhill, M.J.; Norman, R.; Pervolaraki, E.; Stones, R.; Steer, E.; Benoist, D.; Steele, D.S.; Calaghan, S.C.; White, E. Beta1-adrenoceptor antagonist, metoprolol attenuates cardiac myocyte Ca2+ handling dysfunction in rats with pulmonary artery hypertension. J. Mol. Cell. Cardiol. 2018, 120, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiess, F.; Vallmitjana, A.; Wang, R.; Cheng, H.; ter Keurs, H.E.D.J.; Chen, J.; Hove-Madsen, L.; Benitez, R.; Chen, S.R.W. Distribution and Function of Cardiac Ryanodine Receptor Clusters in Live Ventricular Myocytes. J. Biol. Chem. 2015, 290, 20477–20487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, D.J.; Curran, J.W.; Bers, D.M.; Lederer, W.J.; Stern, M.D.; Ríos, E.; Shannon, T.R. Ca Sparks Do Not Explain all Ryanodine Receptor-Mediated SR Ca Leak in Mouse Ventricular Myocytes. Biophys. J. 2010, 98, 2111–2120. [Google Scholar] [CrossRef] [Green Version]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e272–e391. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Pianca, N.; Borile, G.; Da Broi, F.; Richter, C.; Campione, M.; Lehnart, S.E.; Luther, S.; Corrado, D.; Miquerol, L.; et al. Optogenetic determination of the myocardial requirements for extrasystoles by cell type-specific targeting of ChannelRhodopsin-2. Proc. Natl. Acad. Sci. USA 2015, 112, E4495–E4504. [Google Scholar] [CrossRef] [Green Version]
- Borile, G.; Zaglia, T.; Lehnart, S.E.; Mongillo, M. Multiphoton Imaging of Ca2+ Instability in Acute Myocardial Slices from a RyR2R2474S Murine Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Clin. Med. 2021, 10, 2821. [Google Scholar] [CrossRef]
- Wu, J.; Wu, J.; Zipes, D.P. Early Afterdepolarizations, U Waves, and Torsades de Pointes. Circulation 2002, 105, 675–676. [Google Scholar] [CrossRef]
- Volders, P.G.; Vos, M.A.; Szabo, B.; Sipido, K.R.; de Groot, S.H.; Gorgels, A.P.; Wellens, H.J.; Lazzara, R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc. Res. 2000, 46, 376–392. [Google Scholar] [CrossRef] [Green Version]
- Kistamás, K.; Veress, R.; Horváth, B.; Bányász, T.; Nánási, P.P.; Eisner, D.A. Calcium Handling Defects and Cardiac Arrhythmia Syndromes. Front. Pharmacol. 2020, 11, 72. [Google Scholar] [CrossRef]
- Pott, C.; Eckardt, L.; Goldhaber, J.I. Triple Threat: The Na+/Ca2+ Exchanger in the Pathophysiology of Cardiac Arrhythmia, Ischemia and Heart Failure. Curr. Drug Targets 2011, 12, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Xie, L.-H.; Olcese, R.; Karagueuzian, H.S.; Chen, P.-S.; Garfinkel, A.; Weiss, J.N. Early afterdepolarizations in cardiac myocytes: Beyond reduced repolarization reserve. Cardiovasc. Res. 2013, 99, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Fowler, E.D.; Kong, C.H.T.; Hancox, J.C.; Cannell, M.B. Late Ca2+ Sparks and Ripples During the Systolic Ca2+ Transient in Heart Muscle Cells. Circ. Res. 2018, 122, 473–478. [Google Scholar] [CrossRef]
- Fowler, E.D.; Wang, N.; Hezzell, M.; Chanoit, G.; Hancox, J.C.; Cannell, M.B. Arrhythmogenic late Ca2+ sparks in failing heart cells and their control by action potential configuration. Proc. Natl. Acad. Sci. USA 2020, 117, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Litwin, S.E.; Zhang, D.; Bridge, J.H.B. Dyssynchronous Ca2+ Sparks in Myocytes from Infarcted Hearts. Circ. Res. 2000, 87, 1040–1047. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Rees, C.M.; Terentyev, D.; Choi, B.-R.; Koren, G.; Karma, A. NCX-Mediated Subcellular Ca2+ Dynamics Underlying Early Afterdepolarizations in LQT2 Cardiomyocytes. Biophys. J. 2018, 115, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.G.; Mørk, H.; Stokke, M.K.; Lipsett, D.B.; Sjaastad, I.; Richard, S.; Sejersted, O.M.; Louch, W.E. Sarcoplasmic Reticulum Calcium Release Is Required for Arrhythmogenesis in the Mouse. Front. Physiol. 2021, 12, 744730. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Li, Y.; Greensmith, D.J.; Eisner, D.A.; Venetucci, L. Biphasic decay of the Ca transient results from increased sarcoplasmic reticulum Ca leak. J. Physiol. 2016, 594, 611–623. [Google Scholar] [CrossRef]
- Benitah, J.-P.; Perrier, R.; Mercadier, J.-J.; Pereira, L.; Gómez, A.M. RyR2 and Calcium Release in Heart Failure. Front. Physiol. 2021, 12, 734210. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, T.; Ginsburg, K.S.; Mishra, S.; Brown, J.H.; Bers, D.M. CaMKIIδC Slows [Ca]i Decline in Cardiac Myocytes by Promoting Ca Sparks. Biophys. J. 2012, 102, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.; Bers, D. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003, 92, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Denegri, M.; Dun, W.; Boncompagni, S.; Lodola, F.; Protasi, F.; Napolitano, C.; Boyden, P.A.; Priori, S.G. Abnormal Propagation of Calcium Waves and Ultrastructural Remodeling in Recessive Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2013, 113, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.T.; Valdivia, C.R.; Gurrola, G.B.; Powers, P.P.; Willis, B.C.; Moss, R.L.; Jalife, J.; Valdivia, H.H. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc. Natl. Acad. Sci. USA 2015, 112, E1669–E1677. [Google Scholar] [CrossRef] [Green Version]
- Szentandrássy, N.; Magyar, Z.É.; Hevesi, J.; Bányász, T.; Nánási, P.P.; Almássy, J. Therapeutic Approaches of Ryanodine Receptor-Associated Heart Diseases. Int. J. Mol. Sci. 2022, 23, 4435. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Domain | Coordinates (Amino Acids) | Length (Amino Acids) | Number of Mutations | Mutation Frequency Overall (%) | Mutation Frequency within Domain (%) |
|---|---|---|---|---|---|
| NTD | 1–640 | 640 | 58 | 16.9 | 9.1 |
| SPRY1 | 641–850 | 209 | 7 | 2.0 | 3.3 |
| RY1&2 | 876–1066 | 190 | 9 | 2.6 | 4.7 |
| SPRY2 | 1087–1254 | 167 | 5 | 1.5 | 3.0 |
| SPRY3 | 1255–1603 | 348 | 14 | 4.1 | 4.0 |
| JSol | 1641–2105 | 464 | 16 | 4.7 | 3.4 |
| BSolB | 2109–2678 | 569 | 68 | 19.8 | 11.9 |
| RY3&4 | 2714–2904 | 190 | 7 | 2.0 | 3.7 |
| BSolC | 2976–3527 | 551 | 11 | 3.2 | 2.0 |
| CSol | 3612–4206 | 594 | 74 | 21.6 | 12.5 |
| Unresolved | 4207–4483 | 276 | 5 | 1.5 | 1.8 |
| TMD | 4484–4886 | 402 | 55 | 16.0 | 13.7 |
| CTD | 4887–4967 | 80 | 14 | 4.1 | 17.5 |
| Mutation | Domain | Disease | References |
|---|---|---|---|
| Duplication 1 | - | CRDS | [67] |
| L433P 2 | NTD | ARVD2 | [22] |
| G570D | NTD | CRDS | [79] |
| Q2275H | BSolB | CRDS | [80] |
| Q3774L | CSol | CRDS | [66] |
| Q3925E | CSol | CRDS | [79] |
| I3995V | CSol | CRDS | [66] |
| M4109R | CSol | CRDS | [79] |
| D4112N | CSol | CRDS | [66] |
| A4142T | CSol | CRDS | [81] |
| E4146D | CSol | long QT syndrome | [82] |
| E4146K | CSol | CRDS | [83] |
| R4147K | CSol | CRDS | [79] |
| S4168P | CSol | long QT syndrome | [82] |
| T4196I | CSol | CRDS | [66] |
| A4203V | CSol | CRDS | [79] |
| A4204V | CSol | CRDS | [79] |
| E4451del | Unresolved | CRDS | [80] |
| F4499C | TMD | CRDS | [80] |
| K4594Q | TMD | long QT syndrome | [82] |
| V4606E | TMD | CRDS | [80] |
| R4608Q | TMD | CRDS | [80] |
| R4608W | TMD | CRDS | [80] |
| D4646A | TMD | CRDS | [66] |
| I4855M | TMD | left ventricular noncompaction | [62] |
| A4860G 3 | TMD | CRDS | [66,78] |
| Q4879H | TMD | CRDS | [66] |
| G4935R | TMD | CRDS | [83] |
| S4938F | TMD | torsade de pointes | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fowler, E.D.; Zissimopoulos, S. Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease. Biomolecules 2022, 12, 1030. https://doi.org/10.3390/biom12081030
Fowler ED, Zissimopoulos S. Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease. Biomolecules. 2022; 12(8):1030. https://doi.org/10.3390/biom12081030
Chicago/Turabian StyleFowler, Ewan Douglas, and Spyros Zissimopoulos. 2022. "Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease" Biomolecules 12, no. 8: 1030. https://doi.org/10.3390/biom12081030
APA StyleFowler, E. D., & Zissimopoulos, S. (2022). Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease. Biomolecules, 12(8), 1030. https://doi.org/10.3390/biom12081030