
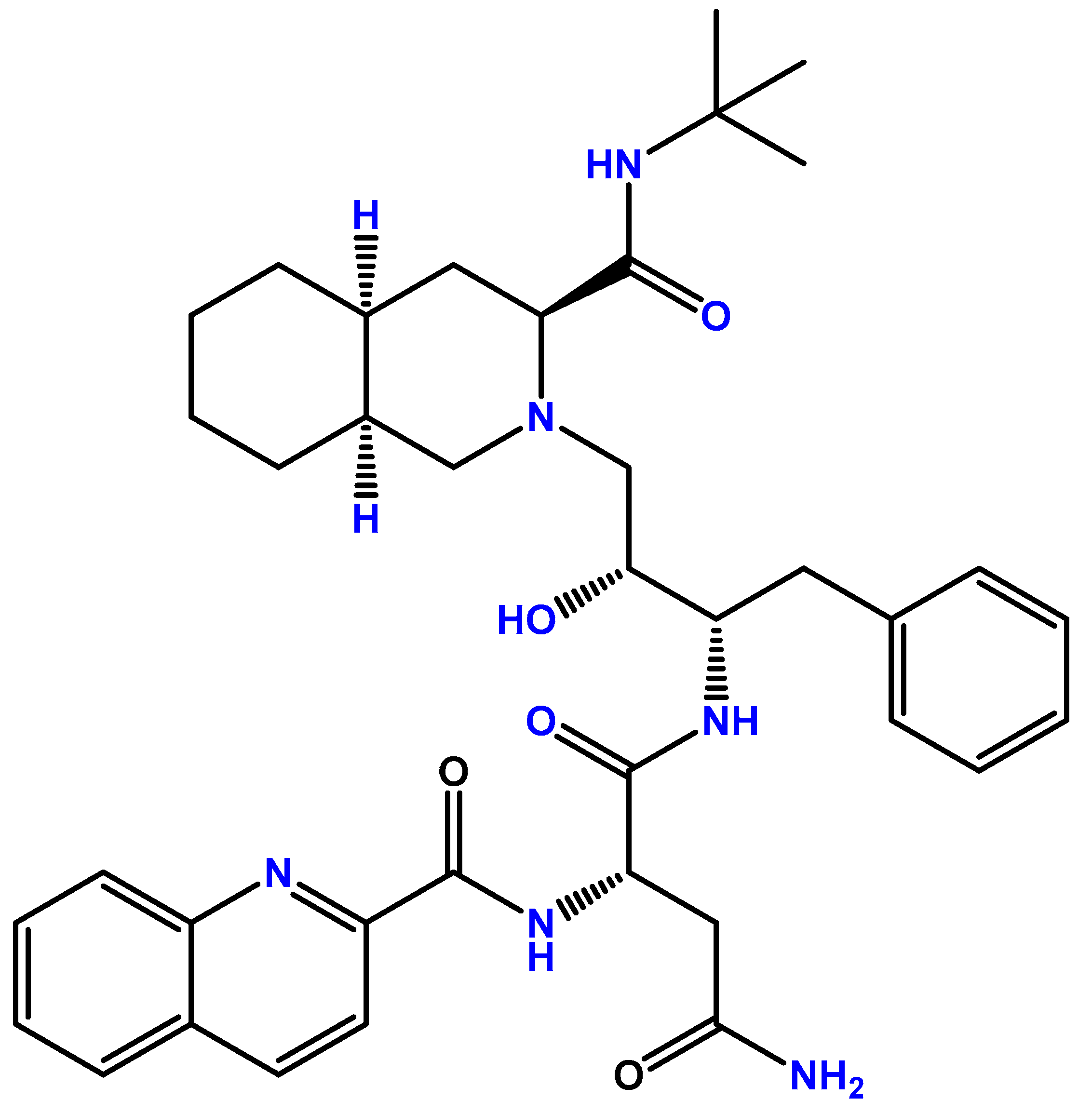
Saquinavir: From HIV to COVID-19 and Cancer Treatment



Abstract
:1. Introduction
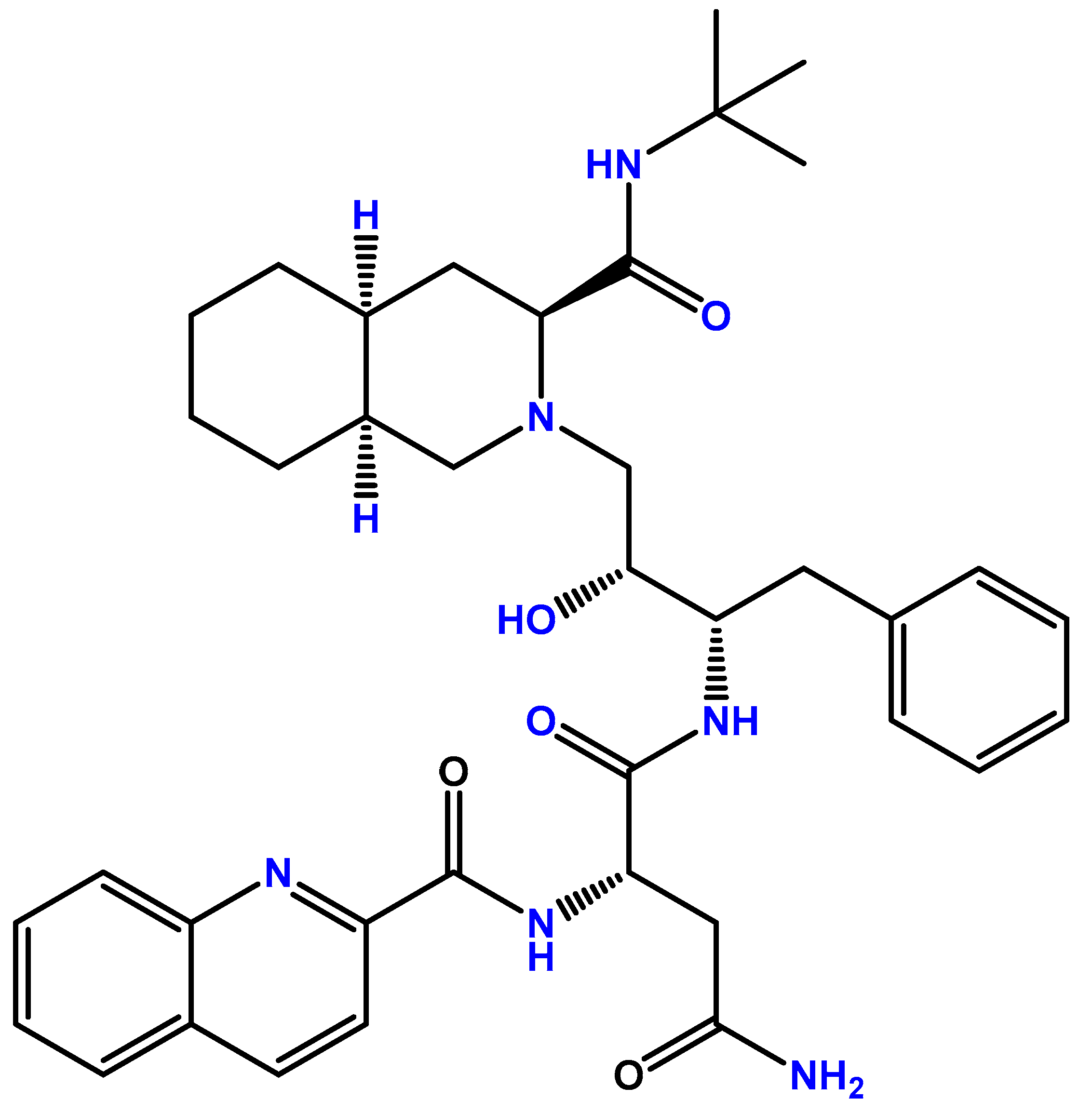
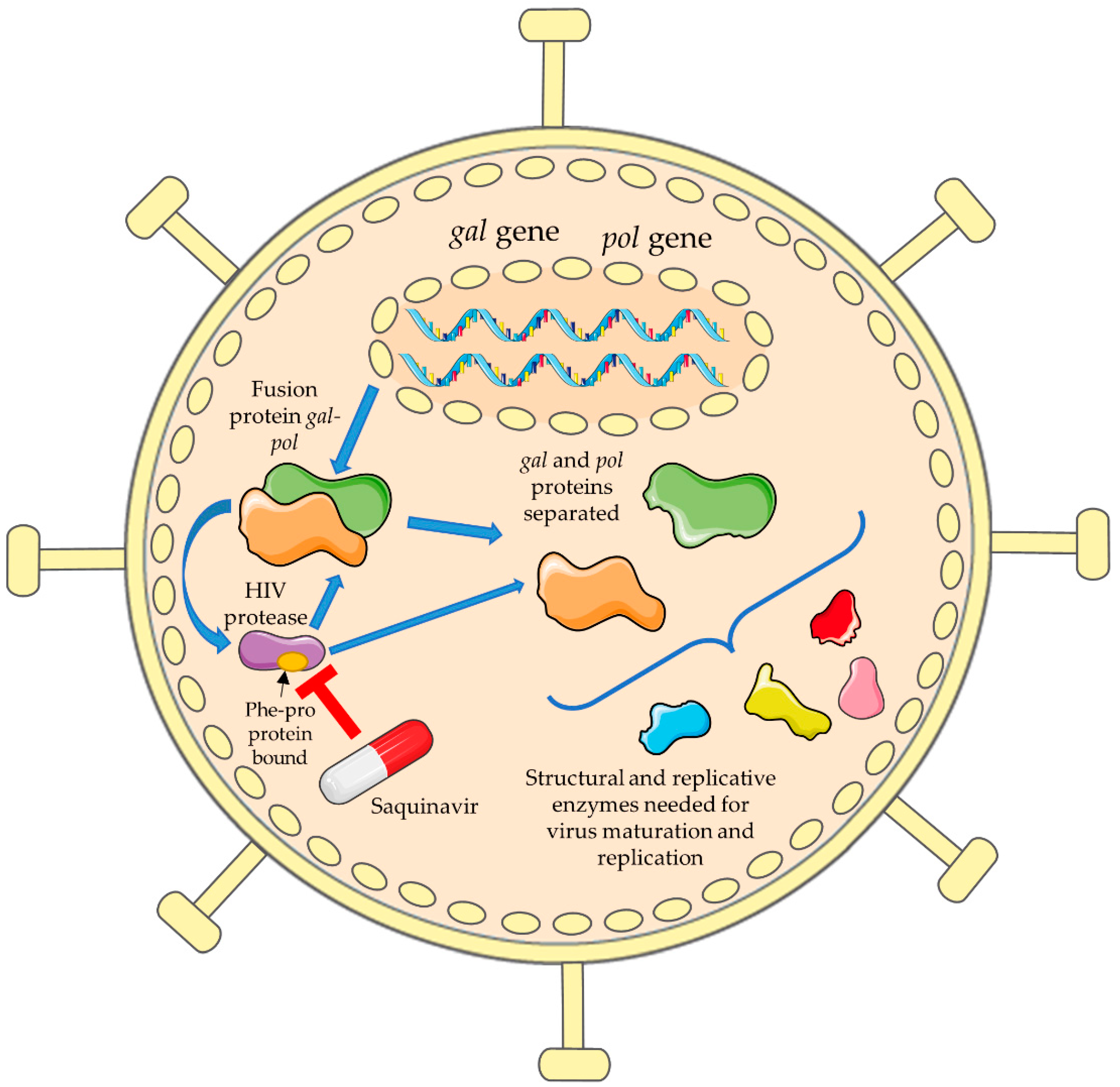
2. Saquinavir for the HIV
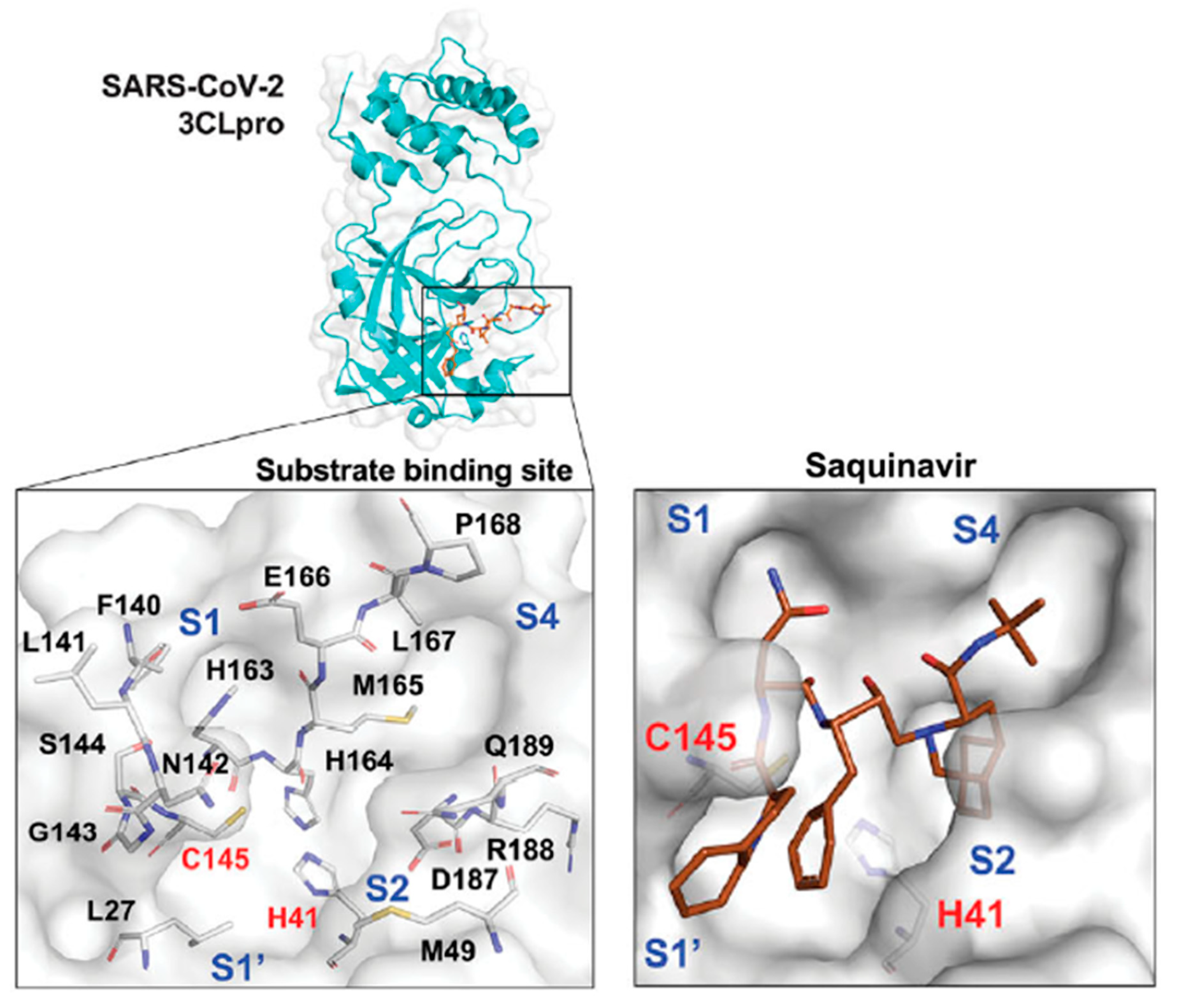
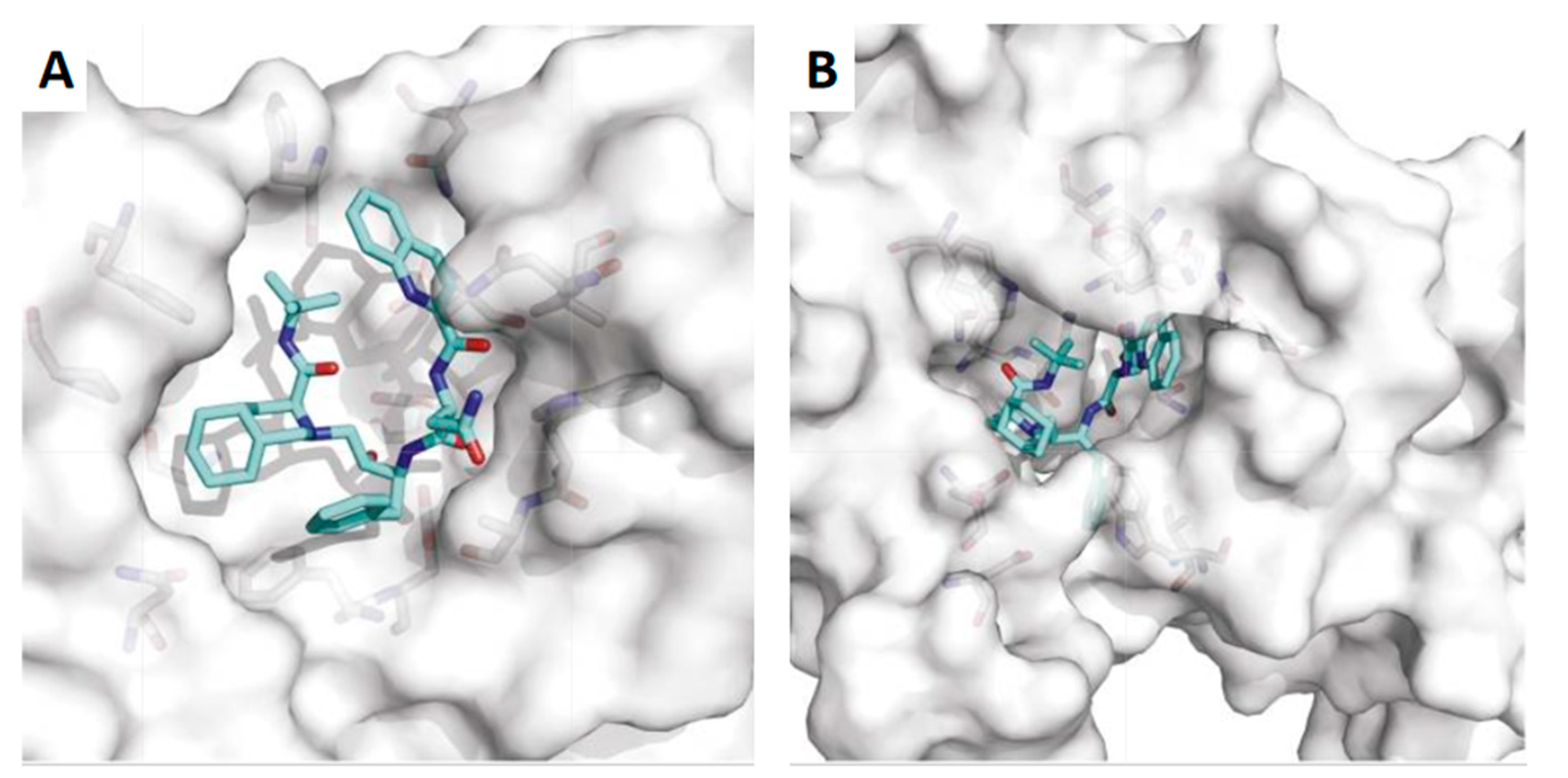
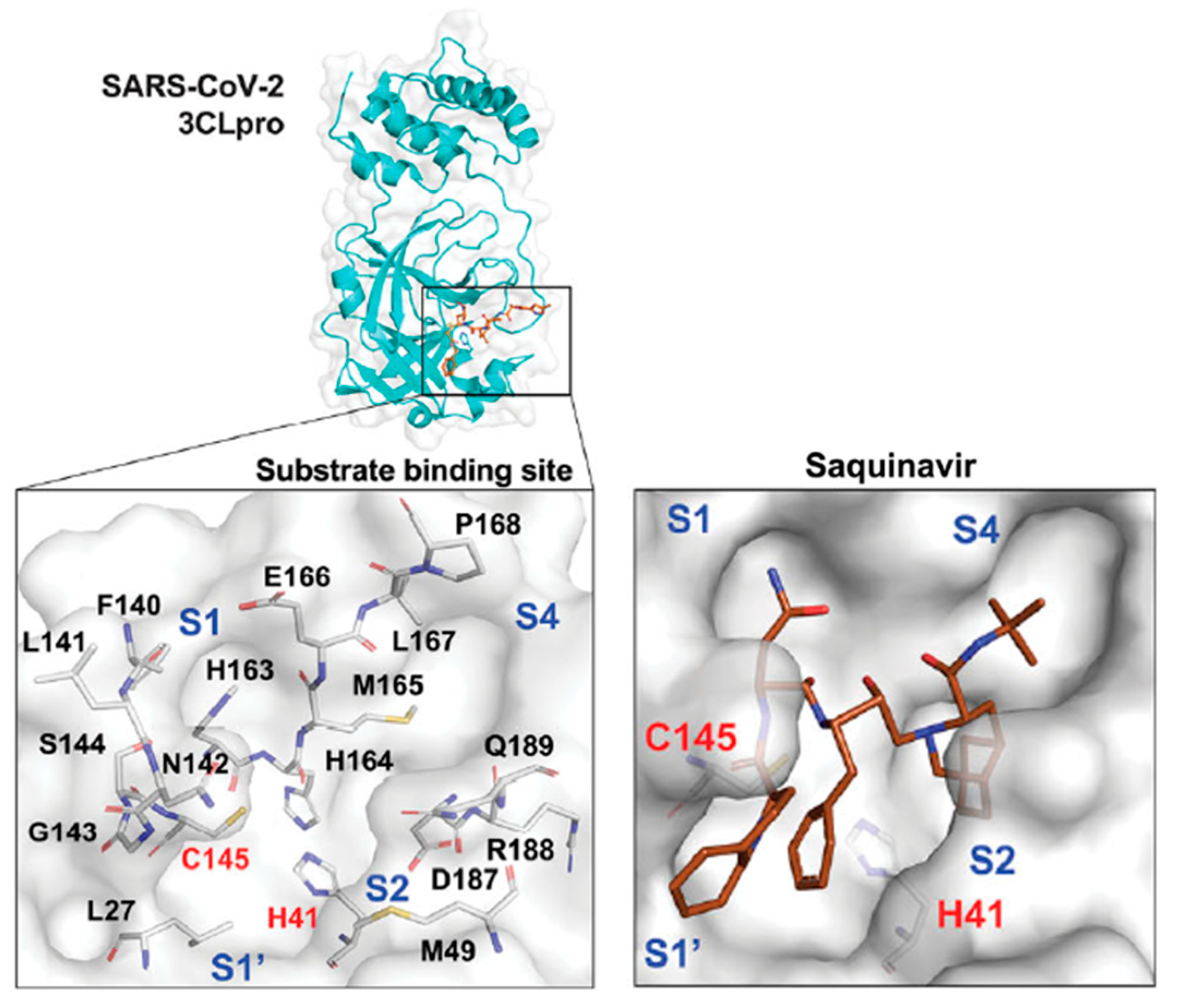
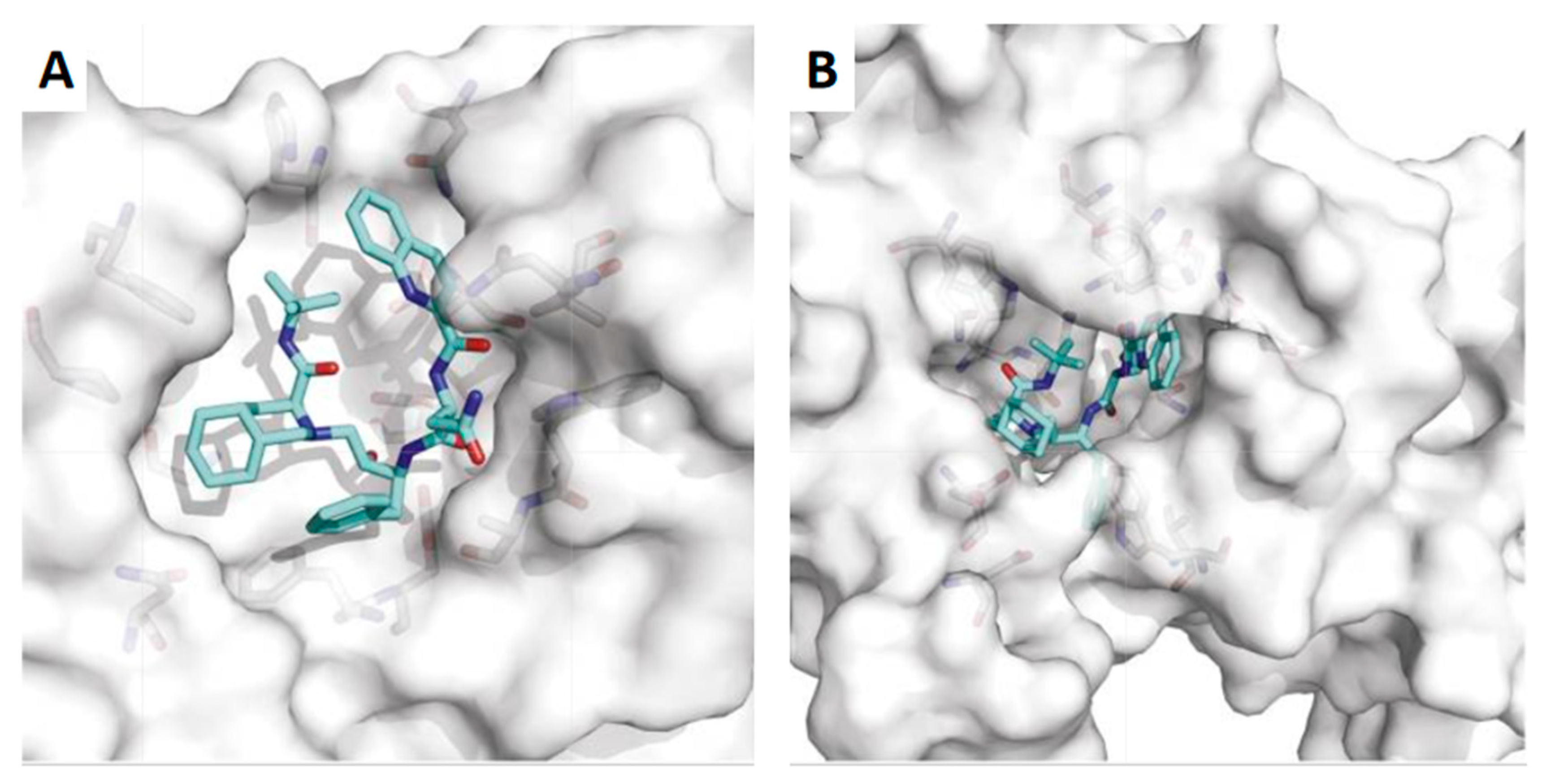
3. Saquinavir for COVID-19 Treatment
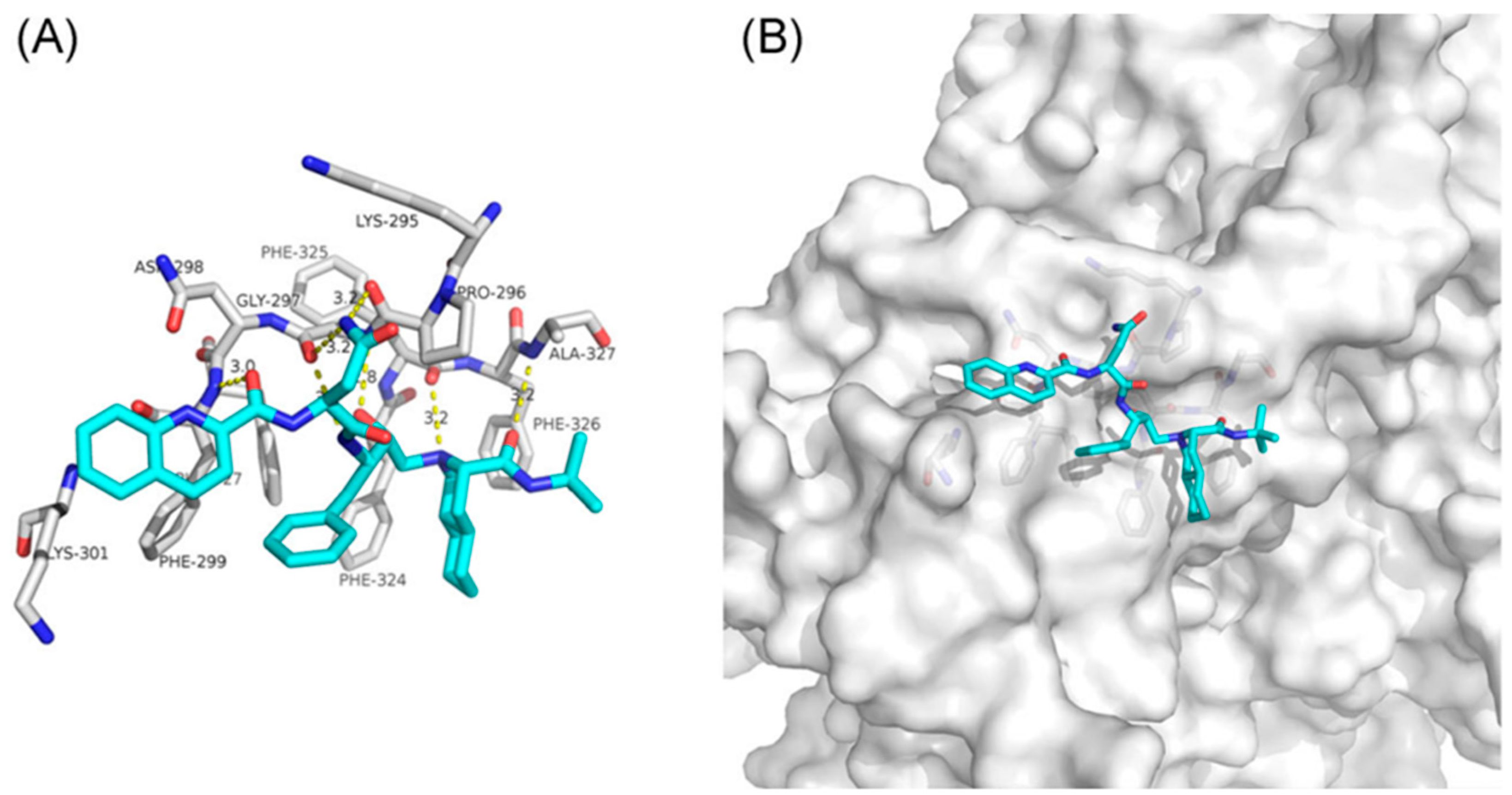
4. Repurposing of Saquinavir for Cancer Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Ist. Super. Sanita 2010, 46, 5–14. [Google Scholar]
- UNAIDS. Global HIV & AIDS Statistics—Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 10 May 2022).
- Fauci, A.S. The acquired immune deficiency syndrome. The ever-broadening clinical spectrum. JAMA 1983, 249, 2375–2376. [Google Scholar]
- De Cock, K.M.; Jaffe, H.W.; Curran, J.W. Reflections on 40 Years of AIDS. Emerg. Infect. Dis. 2021, 27, 1553–1560. [Google Scholar]
- Moir, S.; Chun, T.W.; Fauci, A.S. Pathogenic mechanisms of HIV disease. Annu. Rev. Pathol. 2011, 6, 223–248. [Google Scholar]
- Phanuphak, N.; Gulick, R.M. HIV treatment and prevention 2019: Current standards of care. Curr. Opin. HIV AIDS 2020, 15, 4–12. [Google Scholar]
- James, J.S. Saquinavir (Invirase): First protease inhibitor approved-reimbursement, information hotline numbers. AIDS Treat. News 1995, 237, 1–2. [Google Scholar]
- EMA. Invirase. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/invirase (accessed on 11 May 2022).
- U.S Food and Drug Admnistration (FDA). INVIRASE® (Saquinavir Mesylate) CAPSULES and TABLETS. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020628s032,021785s009lbl.pdf (accessed on 11 May 2022).
- EMA. Invirase: EPAR—Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/invirase-epar-product-information_en.pdf (accessed on 12 May 2022).
- Fitzsimmons, M.E.; Collins, J.M. Selective biotransformation of the human immunodeficiency virus protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4: Potential contribution to high first-pass metabolism. Drug Metab. Dispos. 1997, 25, 256–266. [Google Scholar]
- Washington, C.B.; Wiltshire, H.R.; Man, M.; Moy, T.; Harris, S.R.; Worth, E.; Weigl, P.; Liang, Z.; Hall, D.; Marriott, L.; et al. The disposition of saquinavir in normal and P-glycoprotein deficient mice, rats, and in cultured cells. Drug Metab. Dispos. 2000, 28, 1058–1062. [Google Scholar]
- Hsu, A.; Granneman, G.R.; Cao, G.; Carothers, L.; el-Shourbagy, T.; Baroldi, P.; Erdman, K.; Brown, F.; Sun, E.; Leonard, J.M. Pharmacokinetic interactions between two human immunodeficiency virus protease inhibitors, ritonavir and saquinavir. Clin. Pharmacol. Ther. 1998, 63, 453–464. [Google Scholar]
- Hoetelmans, R.M.; Meenhorst, P.L.; Mulder, J.W.; Burger, D.M.; Koks, C.H.; Beijnen, J.H. Clinical pharmacology of HIV protease inhibitors: Focus on saquinavir, indinavir, and ritonavir. Pharm. World Sci. 1997, 19, 159–175. [Google Scholar]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar]
- Noble, S.; Faulds, D. Saquinavir. A review of its pharmacology and clinical potential in the management of HIV infection. Drugs 1996, 52, 93–112. [Google Scholar]
- Ochani, R.; Asad, A.; Yasmin, F.; Shaikh, S.; Khalid, H.; Batra, S.; Sohail, M.R.; Mahmood, S.F.; Ochani, R.; Hussham Arshad, M.; et al. COVID-19 pandemic: From origins to outcomes. A comprehensive review of viral pathogenesis, clinical manifestations, diagnostic evaluation, and management. Infez. Med. 2021, 29, 20–36. [Google Scholar]
- Chen, B.; Tian, E.K.; He, B.; Tian, L.; Han, R.; Wang, S.; Xiang, Q.; Zhang, S.; El Arnaout, T.; Cheng, W. Overview of lethal human coronaviruses. Signal Transduct. Target. Ther. 2020, 5, 89. [Google Scholar]
- Singh, R.; Kang, A.; Luo, X.; Jeyanathan, M.; Gillgrass, A.; Afkhami, S.; Xing, Z. COVID-19: Current knowledge in clinical features, immunological responses, and vaccine development. FASEB J. 2021, 35, e21409. [Google Scholar]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar]
- Hall, D.C., Jr.; Ji, H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar]
- Adhikari, N.; Banerjee, S.; Baidya, S.K.; Ghosh, B.; Jha, T. Ligand-based quantitative structural assessments of SARS-CoV-2 3CL(pro) inhibitors: An analysis in light of structure-based multi-molecular modeling evidences. J. Mol. Struct. 2022, 1251, 132041. [Google Scholar]
- Raphael, V.P.; Shanmughan, S.K. Computational Evaluation of the Inhibition Efficacies of HIV Antivirals on SARS-CoV-2 (COVID-19) Protease and Identification of 3D Pharmacophore and Hit Compounds. Adv. Pharmacol. Pharm. Sci. 2020, 2020, 8818008. [Google Scholar]
- Talluri, S. Molecular Docking and Virtual Screening Based Prediction of Drugs for COVID-19. Comb. Chem. High Throughput Screen 2021, 24, 716–728. [Google Scholar]
- Wang, Q.; Zhao, Y.; Chen, X.; Hong, A. Virtual screening of approved clinic drugs with main protease (3CL(pro)) reveals potential inhibitory effects on SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 685–695. [Google Scholar]
- Chiou, W.C.; Hsu, M.S.; Chen, Y.T.; Yang, J.M.; Tsay, Y.G.; Huang, H.C.; Huang, C. Repurposing existing drugs: Identification of SARS-CoV-2 3C-like protease inhibitors. J. Enzyme Inhib. Med. Chem. 2021, 36, 147–153. [Google Scholar]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar]
- Liu, C.; Zhu, X.; Lu, Y.; Zhang, X.; Jia, X.; Yang, T. Potential treatment with Chinese and Western medicine targeting NSP14 of SARS-CoV-2. J. Pharm. Anal. 2021, 11, 272–277. [Google Scholar]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2021, 93, 389–400. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar]
- Radu, O.; Pantanowitz, L. Kaposi sarcoma. Arch. Pathol. Lab. Med. 2013, 137, 289–294. [Google Scholar]
- Stürzl, M.; Zietz, C.; Monini, P.; Ensoli, B. Human herpesvirus-8 and Kaposi’s sarcoma: Relationship with the multistep concept of tumorigenesis. Adv. Cancer Res. 2001, 81, 125–159. [Google Scholar]
- Sgadari, C.; Barillari, G.; Toschi, E.; Carlei, D.; Bacigalupo, I.; Baccarini, S.; Palladino, C.; Leone, P.; Bugarini, R.; Malavasi, L.; et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat. Med. 2002, 8, 225–232. [Google Scholar]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar]
- Bandiera, E.; Todeschini, P.; Romani, C.; Zanotti, L.; Erba, E.; Colmegna, B.; Bignotti, E.; Santin, A.D.; Sartori, E.; Odicino, F.E.; et al. The HIV-protease inhibitor saquinavir reduces proliferation, invasion and clonogenicity in cervical cancer cell lines. Oncol. Lett. 2016, 12, 2493–2500. [Google Scholar]
- McDade, T.P.; Perugini, R.A.; Vittimberga, F.J., Jr.; Callery, M.P. Ubiquitin-proteasome inhibition enhances apoptosis of human pancreatic cancer cells. Surgery 1999, 126, 371–377. [Google Scholar]
- Pajonk, F.; Pajonk, K.; McBride, W.H. Apoptosis and radiosensitization of hodgkin cells by proteasome inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2000, 47, 1025–1032. [Google Scholar]
- Pajonk, F.; Himmelsbach, J.; Riess, K.; Sommer, A.; McBride, W.H. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells. Cancer Res. 2002, 62, 5230–5235. [Google Scholar]
- Schmidtke, G.; Holzhütter, H.G.; Bogyo, M.; Kairies, N.; Groll, M.; de Giuli, R.; Emch, S.; Groettrup, M. How an inhibitor of the HIV-I protease modulates proteasome activity. J. Biol. Chem. 1999, 274, 35734–35740. [Google Scholar]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar]
- Osarogiagbon, U.R.; McGlave, P.B. Chronic myelogenous leukemia. Curr. Opin. Hematol. 1999, 6, 241–246. [Google Scholar]
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar]
- Reuther, J.Y.; Reuther, G.W.; Cortez, D.; Pendergast, A.M.; Baldwin, A.S., Jr. A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998, 12, 968–981. [Google Scholar]
- Timeus, F.; Crescenzio, N.; Ricotti, E.; Doria, A.; Bertin, D.; Saglio, G.; Tovo, P.A. The effects of saquinavir on imatinib-resistant chronic myelogenous leukemia cell lines. Haematologica 2006, 91, 711–712. [Google Scholar]
- Timeus, F.; Crescenzio, N.; Doria, A.; Foglia, L.; Pagliano, S.; Ricotti, E.; Fagioli, F.; Tovo, P.A.; Cordero di Montezemolo, L. In vitro anti-neuroblastoma activity of saquinavir and its association with imatinib. Oncol. Rep. 2012, 27, 734–740. [Google Scholar]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar]
- Beppu, K.; Jaboine, J.; Merchant, M.S.; Mackall, C.L.; Thiele, C.J. Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J. Natl. Cancer Inst. 2004, 96, 46–55. [Google Scholar]
- Bond, M.; Bernstein, M.L.; Pappo, A.; Schultz, K.R.; Krailo, M.; Blaney, S.M.; Adamson, P.C. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: A Children’s Oncology Group study. Pediatr. Blood Cancer 2008, 50, 254–258. [Google Scholar]
- Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, 2385. [Google Scholar]
- Grant, S. Enhancing proteotoxic stress as an anticancer strategy. Oncotarget 2011, 2, 284–286. [Google Scholar]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar]
- Workman, P.; Davies, F.E. A stressful life (or death): Combinatorial proteotoxic approaches to cancer-selective therapeutic vulnerability. Oncotarget 2011, 2, 277. [Google Scholar]
- Kraus, M.; Müller-Ide, H.; Rückrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar]
- Thompson, J.E.; Thompson, C.B. Putting the rap on Akt. J. Clin. Oncol. 2004, 22, 4217–4226. [Google Scholar]
- Xia, S.; Zhao, Y.; Yu, S.; Zhang, M. Activated PI3K/Akt/COX-2 pathway induces resistance to radiation in human cervical cancer HeLa cells. Cancer Biother. Radiopharm. 2010, 25, 317–323. [Google Scholar]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar]
- Plastaras, J.P.; Vapiwala, N.; Ahmed, M.S.; Gudonis, D.; Cerniglia, G.J.; Feldman, M.D.; Frank, I.; Gupta, A.K. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 2008, 7, 628–635. [Google Scholar]
- Imaizumi, K.; Miyoshi, K.; Katayama, T.; Yoneda, T.; Taniguchi, M.; Kudo, T.; Tohyama, M. The unfolded protein response and Alzheimer’s disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2001, 1536, 85–96. [Google Scholar]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar]
- Verbrugge, S.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther. 2015, 17, 17. [Google Scholar]
- Krishnan, K.M.; Williamson, K.C. The proteasome as a target to combat malaria: Hits and misses. Transl. Res. 2018, 198, 40–47. [Google Scholar]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium Tuberculosis Infection. Front. Immunol. 2021, 12, 647728. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer type | Model | Main results | Ref. |
|---|---|---|---|
| Kaposi sarcoma | In vitro models of KS tumor growth and lesion formation and nude mice | Inhibition of angiogenic lesions in vivo (reduction of angiogenesis and spindle cell growth), cell invasion, and antitumor growth effect | [33] |
| Cervical cancer | CC1, CC2, HeLa, CaSki, HT3, and C33a cell lines | High inhibitory effect in all cell lines (IC50 of 19 μM at 96 h in HeLa cells) independent of the proteasome. Interference with clonogenicity and cell invasion | [35] |
| Prostate cancer | PC-3, LnCap, and DU-145 prostate cancer cell lines | Blockage of NF-κB activation and stabilization of IκBα due to inhibition of the 20S and 26S proteasomes (at higher concentrations). Consequent induction of apoptosis (at the concentrations similar to the ones that cause proteasome inhibition). Radiosensitization to ionizing radiation | [38] |
| Bladder cancer | ECV 304 human bladder carcinoma cell line | Inhibition of the 20S and 26S proteasomes with a higher IC50 (50 μM) (since saquinavir is a substrate of P-gp) | |
| Leukemia | Jurkat leukemia cells | Induced apoptosis when using concentrations that inhibit the 20S and 26S proteasomes | |
| Erythroleukemia | K562 erythroleukemia cell line | ||
| Glioblastoma | U373 glioblastoma cell line | ||
| Chronic myelogenous leukemia | Sensitive and resistant CML cell lines | Time- and concentration-dependent inhibition of proliferation and induction of apoptosis (alone). Improvement of imatinib activity in all cell lines when in combination with saquinavir (decreasing its IC50) | [44] |
| Neuroblastoma | Neuroblastoma cell lines | Antiproliferative effect with low concentrations (alone) and enhancement of imatinib’s activity in combination (both antiproliferative and anti-invasive effect). Enhanced apoptotic effect of saquinavir when in combination. NF-κB and 26S proteasome inhibition | [45] |
| Acute myeloid leukemia | Acute myeloid leukemia cell lines | The cytotoxic effect at low concentrations and in all stages of differentiation. Synergic combination with bortezomib (PI), causing proteotoxic stress due to UPR activation | [53] |
| Head and neck cancer | SQ20B head and neck cell line | Decreased Akt phosphorylation in a time- and concentration-dependent way, with total loss at 20 min at lower concentrations at 24 h and with higher concentrations. Cell death effect | [57] |
| Bladder cancer cells | T24 bladder cancer cell line | Akt inhibition and consequent radiosensitization of cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, M.; Vale, N. Saquinavir: From HIV to COVID-19 and Cancer Treatment. Biomolecules 2022, 12, 944. https://doi.org/10.3390/biom12070944
Pereira M, Vale N. Saquinavir: From HIV to COVID-19 and Cancer Treatment. Biomolecules. 2022; 12(7):944. https://doi.org/10.3390/biom12070944
Chicago/Turabian StylePereira, Mariana, and Nuno Vale. 2022. "Saquinavir: From HIV to COVID-19 and Cancer Treatment" Biomolecules 12, no. 7: 944. https://doi.org/10.3390/biom12070944
APA StylePereira, M., & Vale, N. (2022). Saquinavir: From HIV to COVID-19 and Cancer Treatment. Biomolecules, 12(7), 944. https://doi.org/10.3390/biom12070944