
Novel Scaffolds for Modulation of NOD2 Identified by Pharmacophore-Based Virtual Screening




Abstract
:1. Introduction
2. Materials and Methods
2.1. Virtual Compound Library Preparation
2.2. Pharmacophore Modeling
2.3. Protein Preparation and Homology Modeling
2.4. Molecular Docking
2.5. Virtual Screening
2.6. Binding Free Energy Calculation
2.7. Analytical Procedures
2.8. Cell Culture
2.9. Cytotoxicity
2.10. NOD2-NF-κB Reporter Assay
2.11. Statistics
3. Results and Discussion
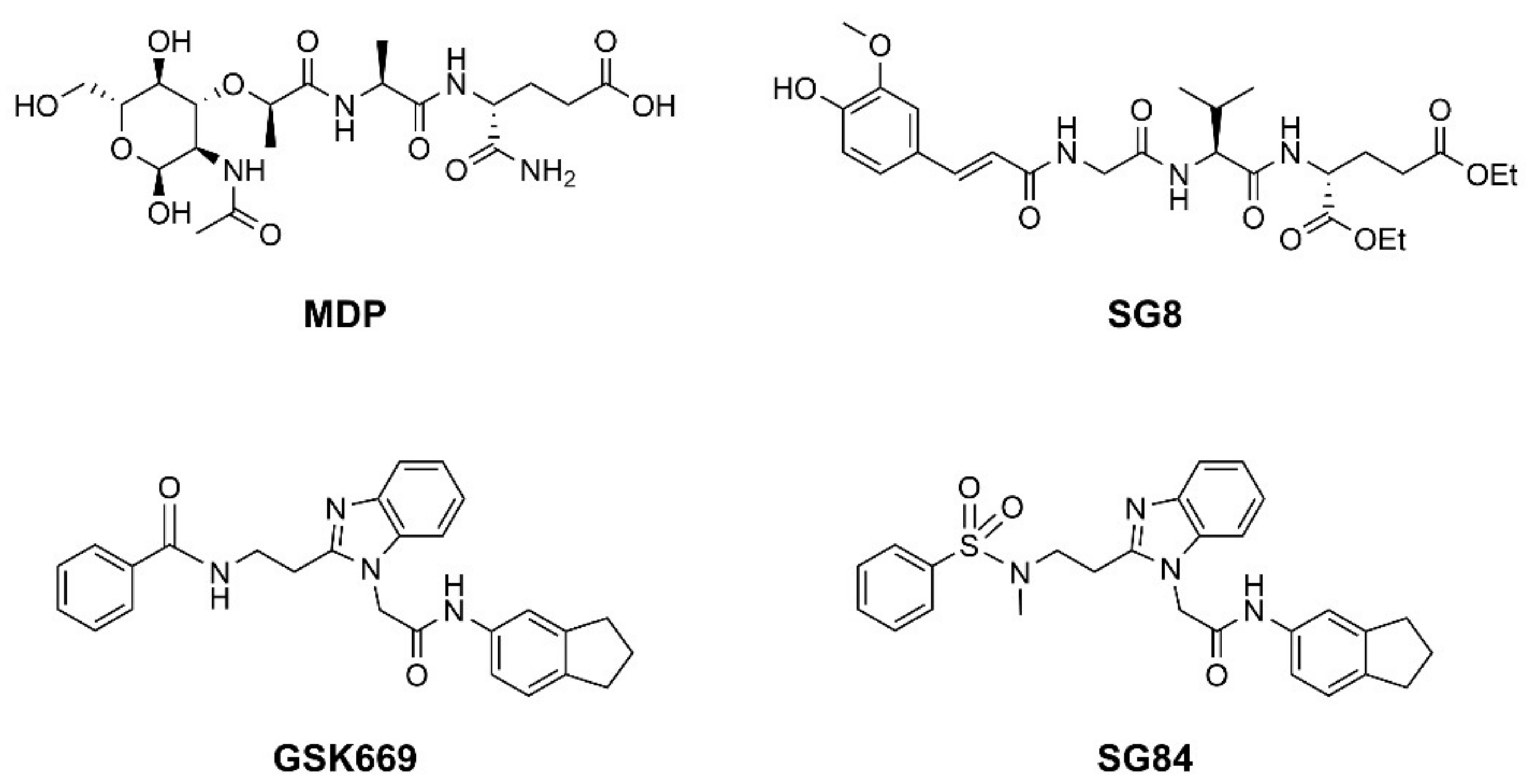
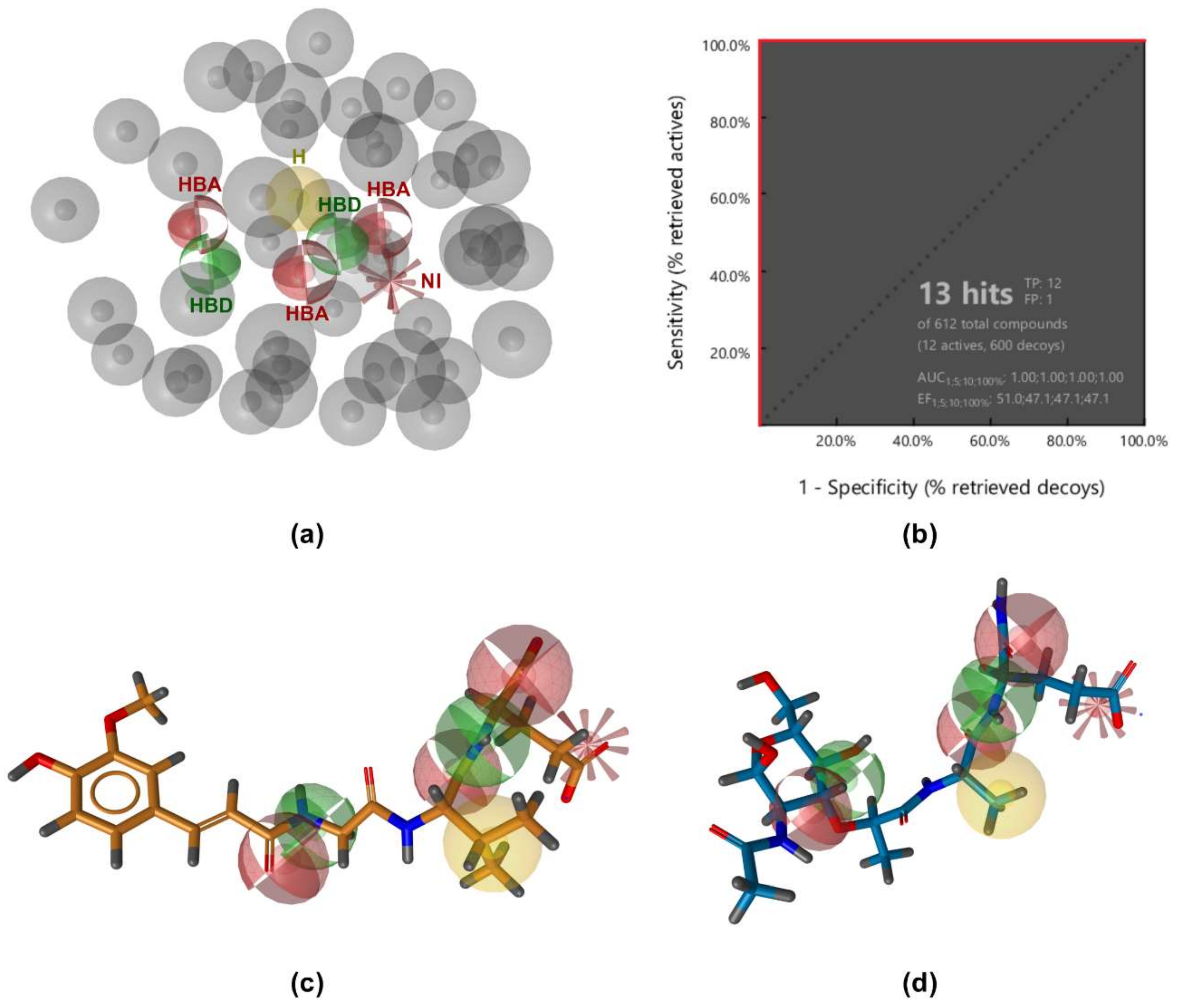
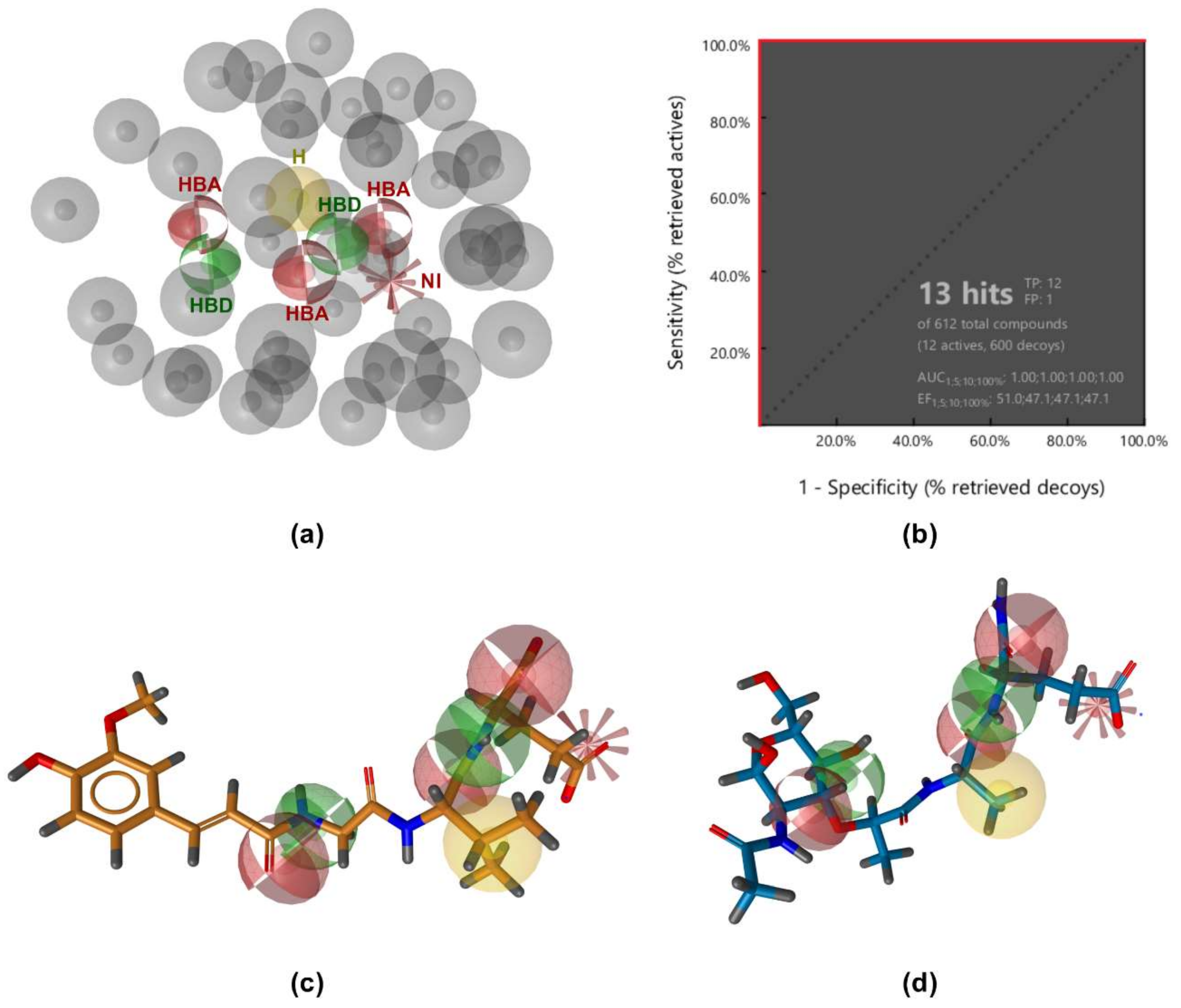
3.1. Ligand-Based Pharmacophore Modeling
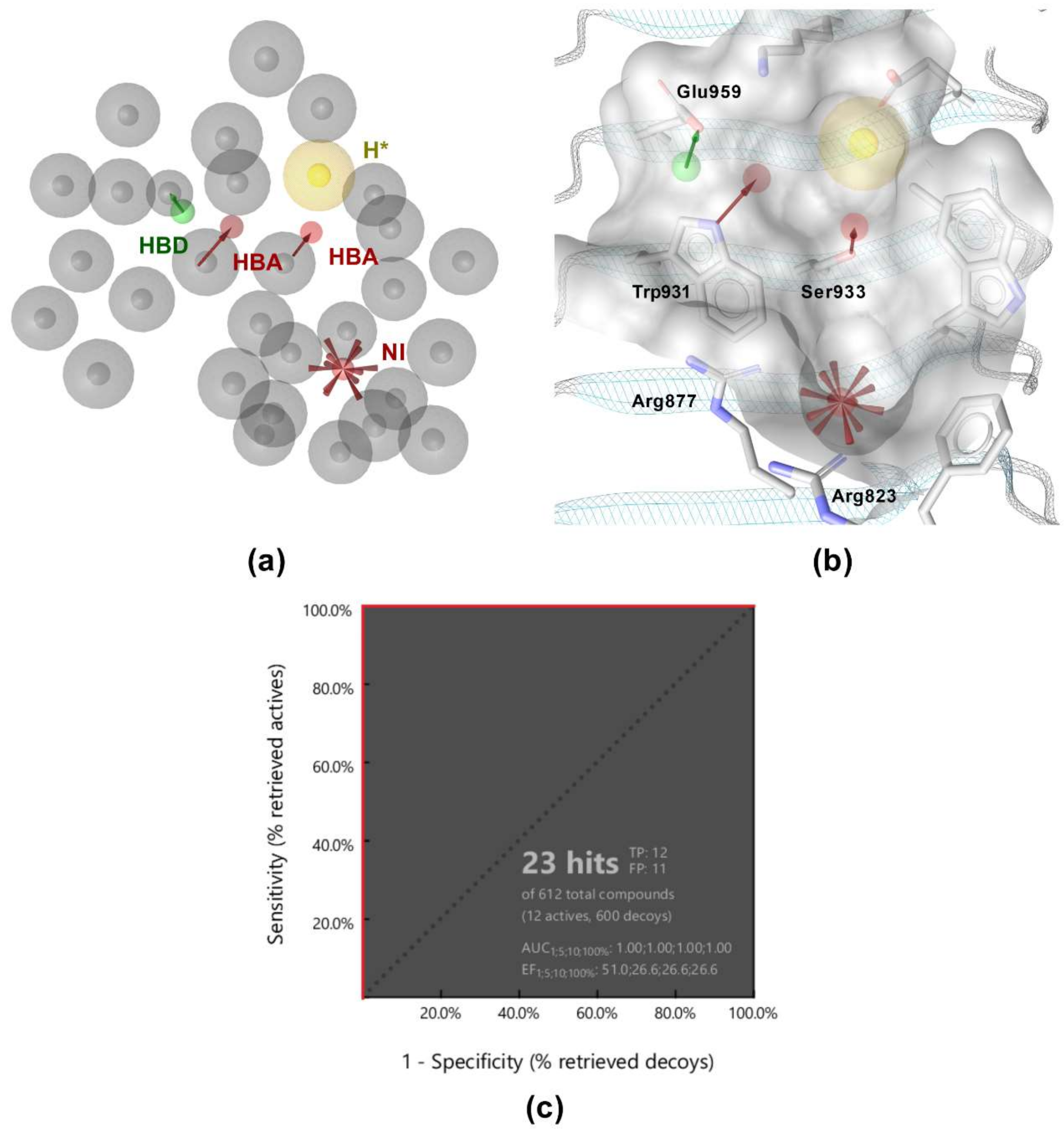
3.2. Structure-Based Pharmacophore Modeling
3.3. Virtual Screening
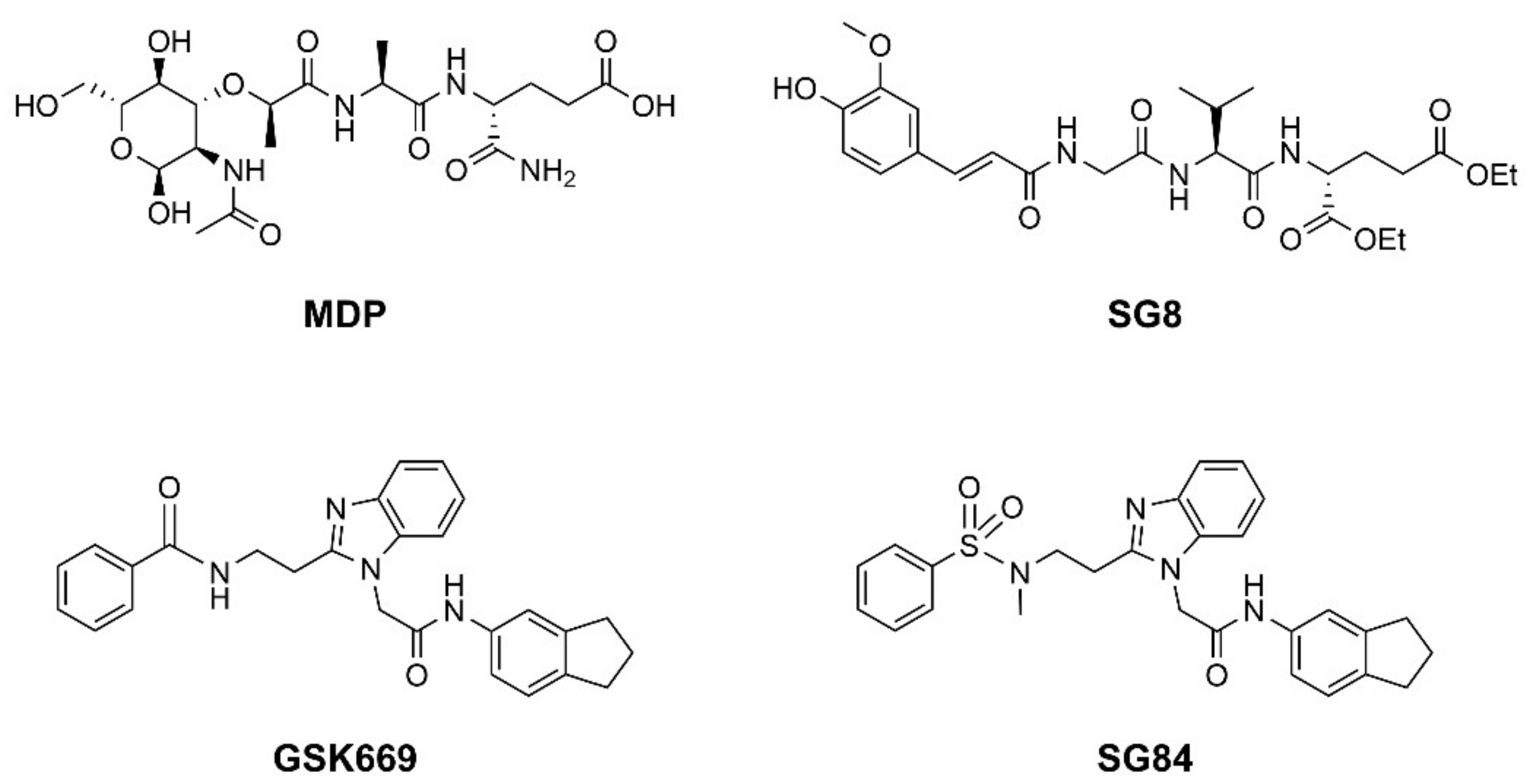
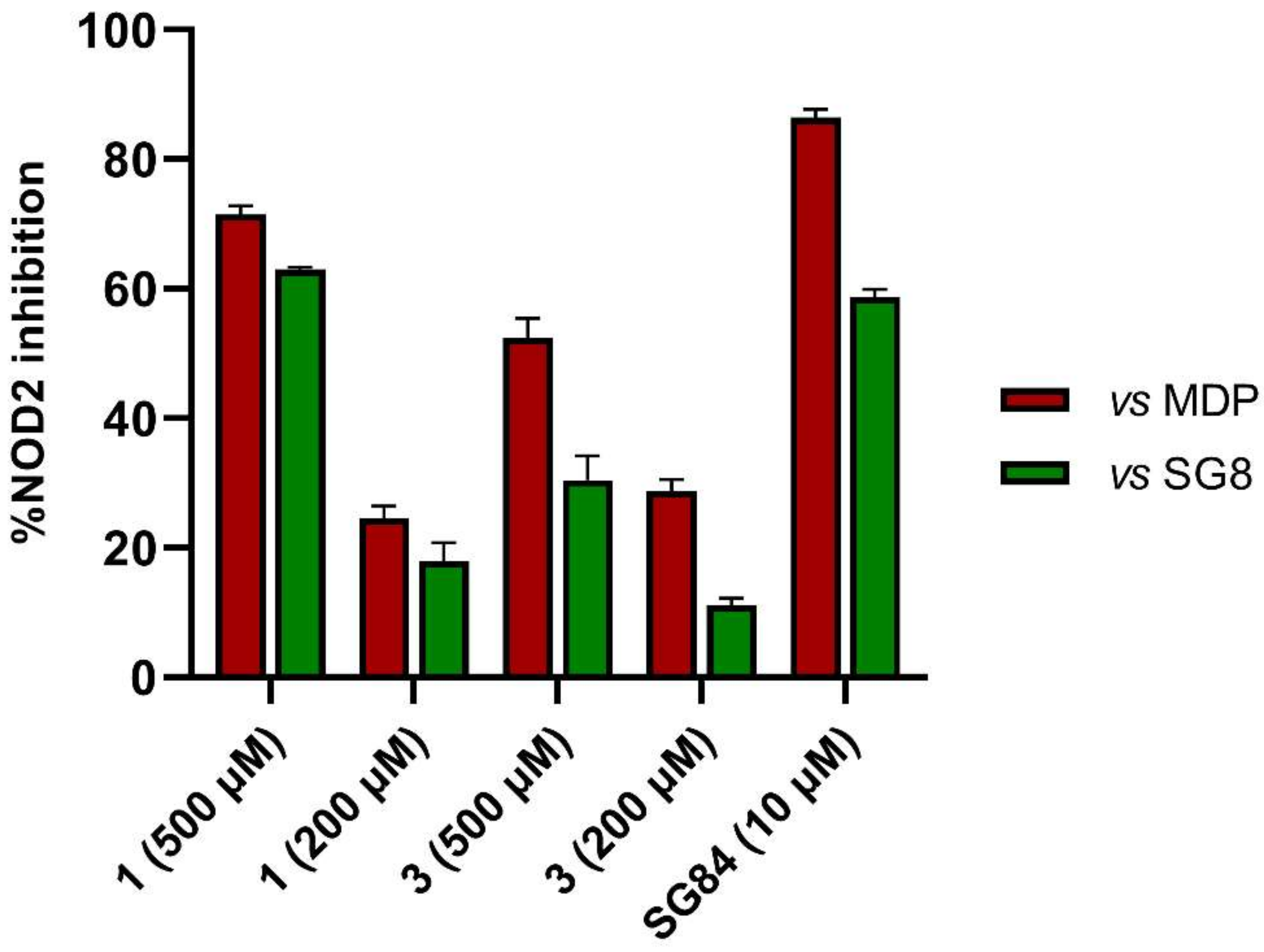
3.4. In Vitro Biological Evaluation of Virtual Screening Hits
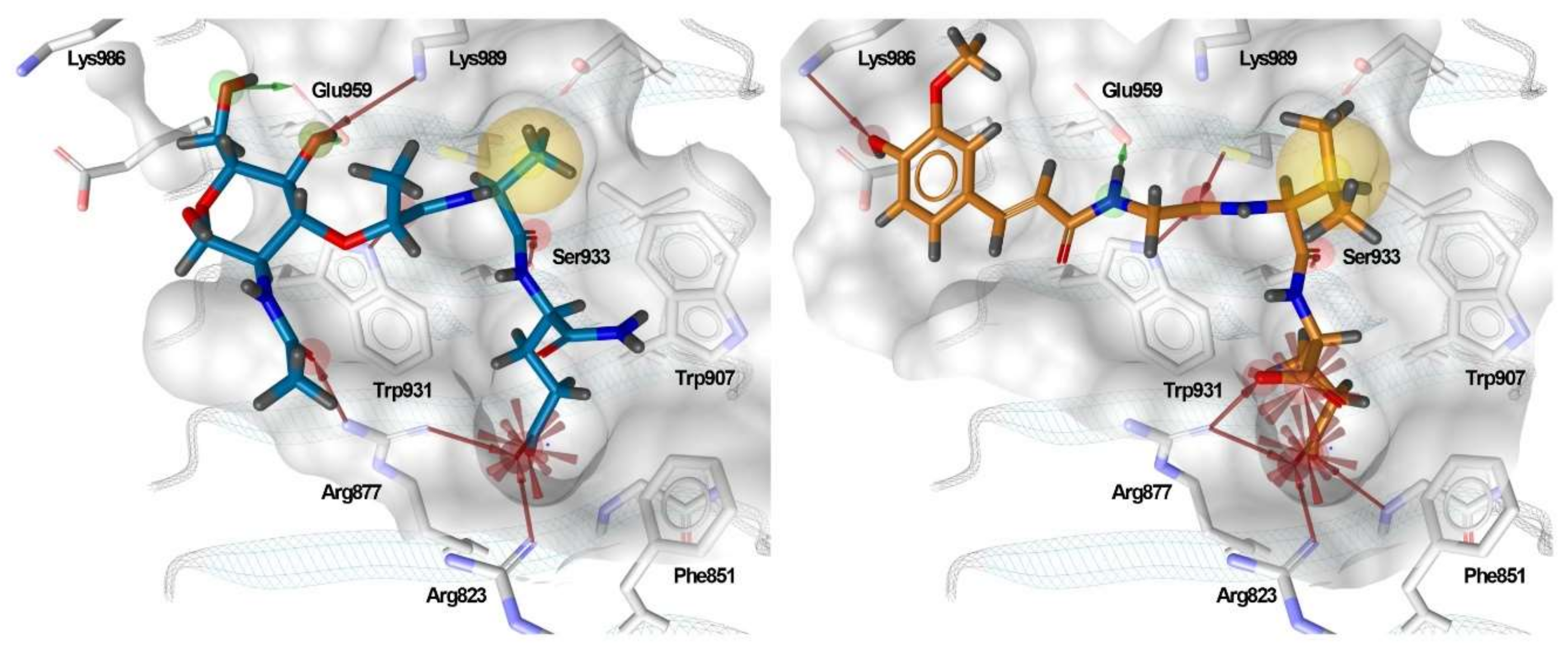
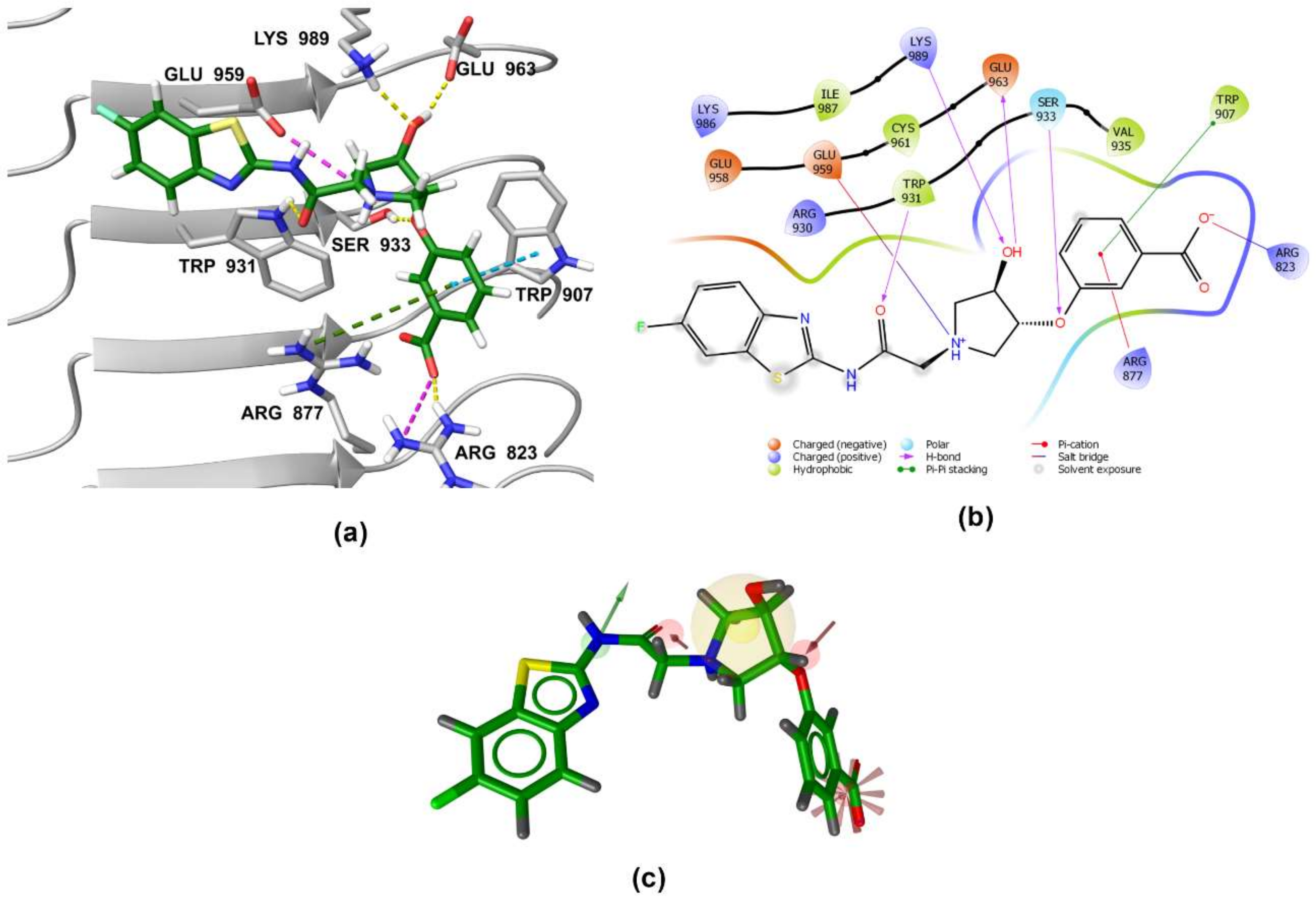
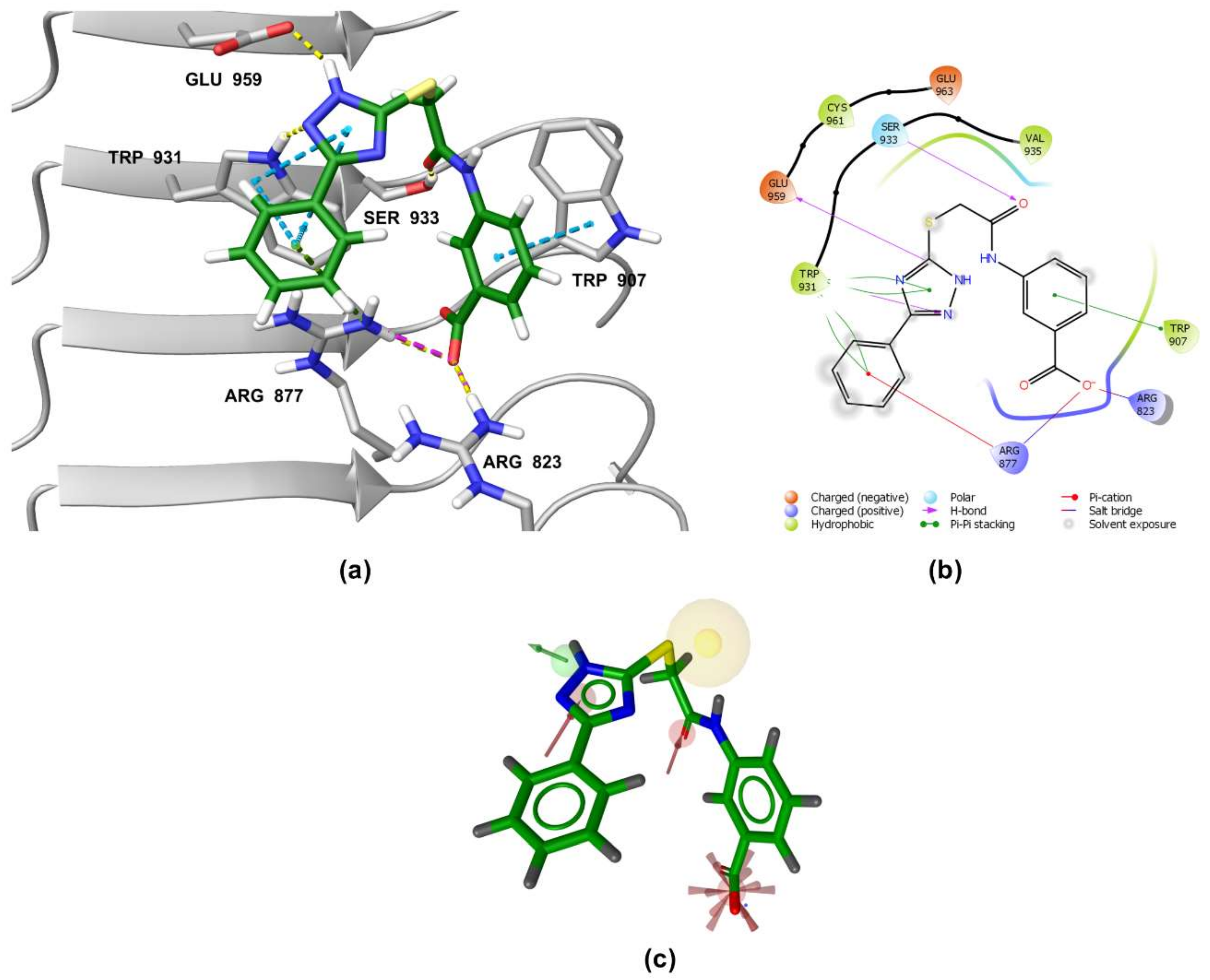
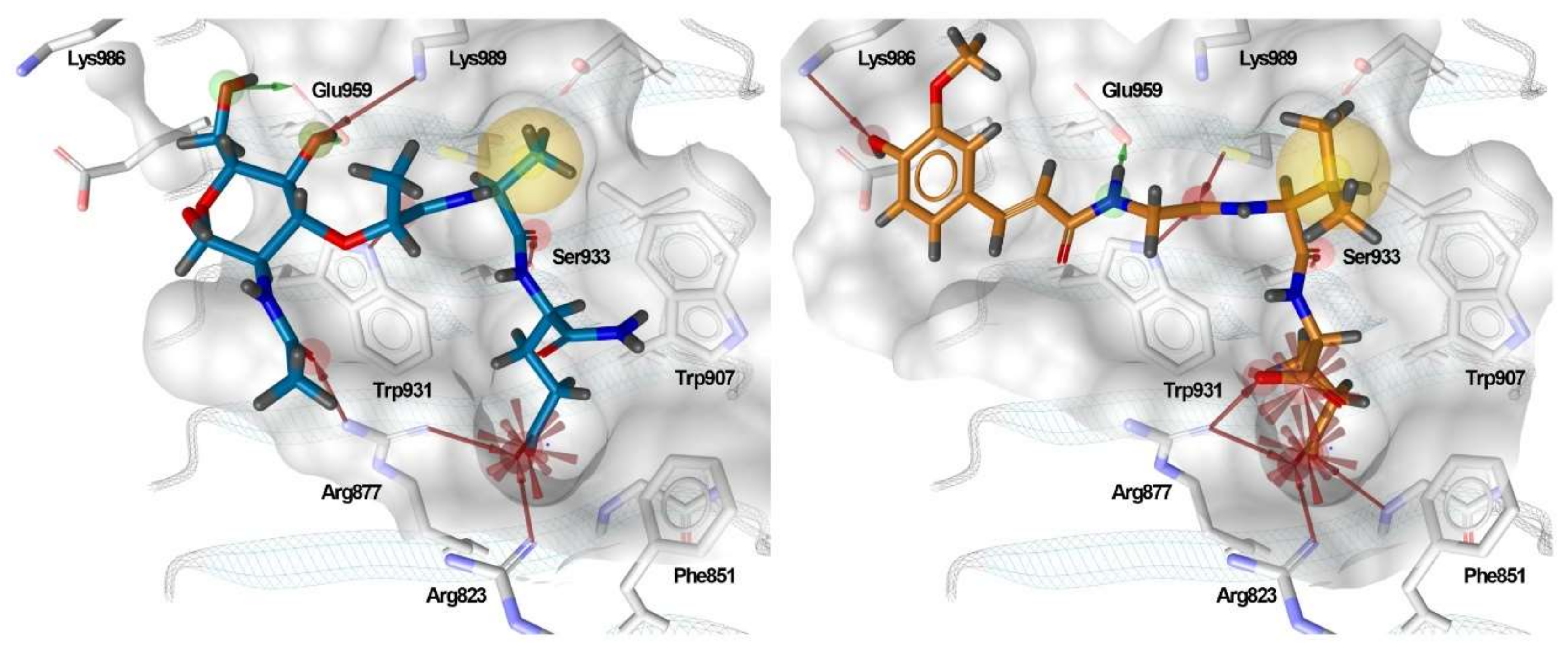
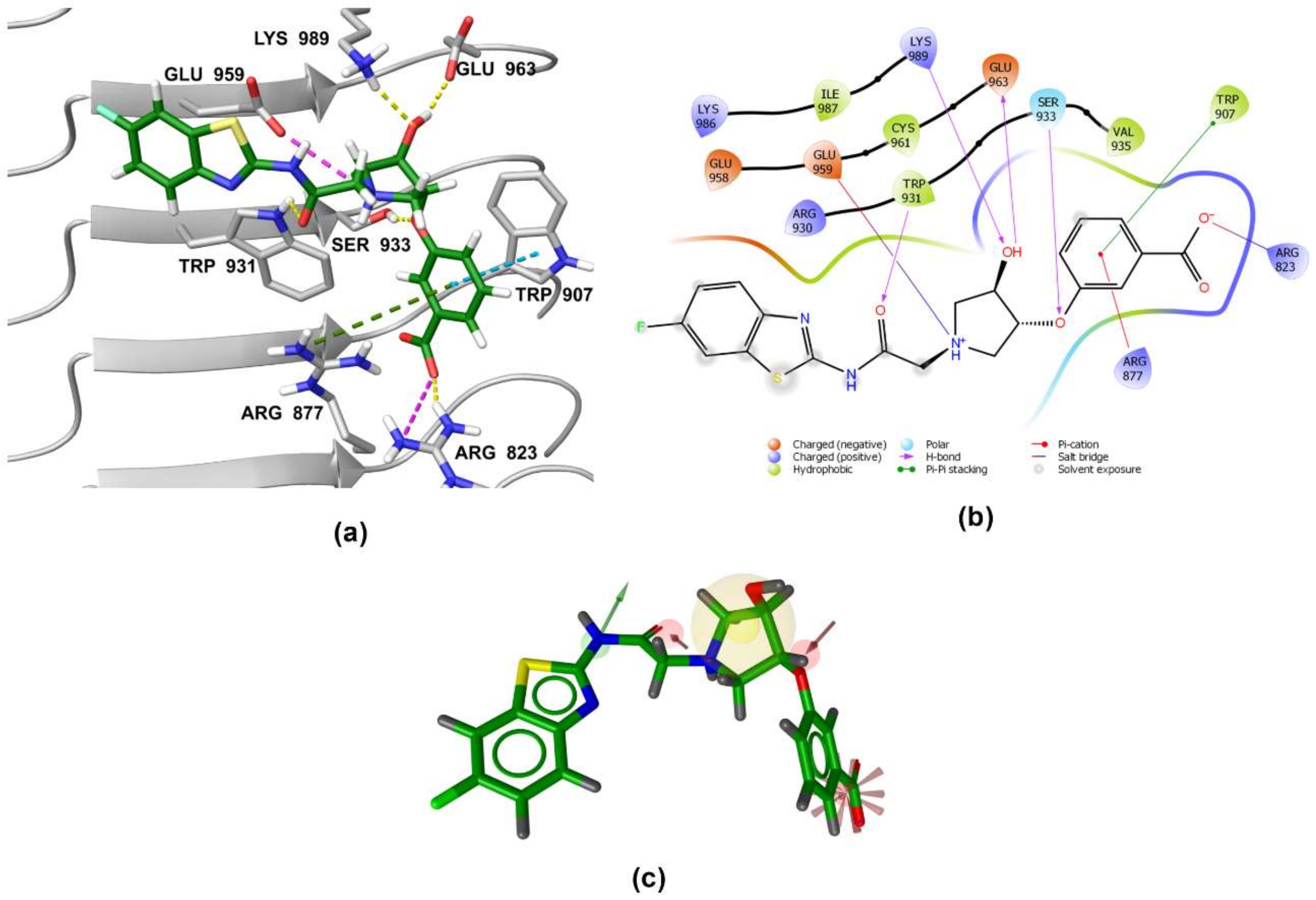
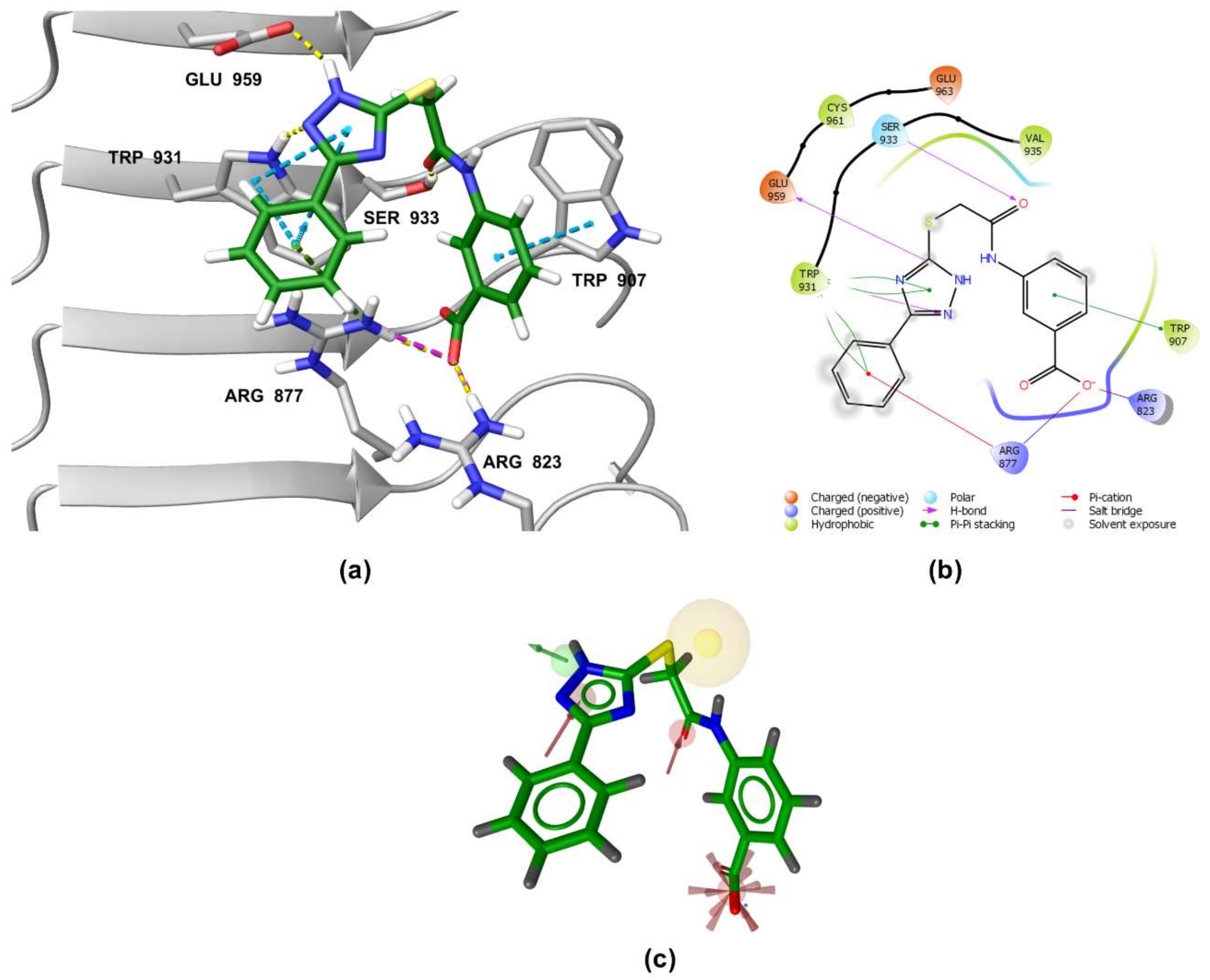
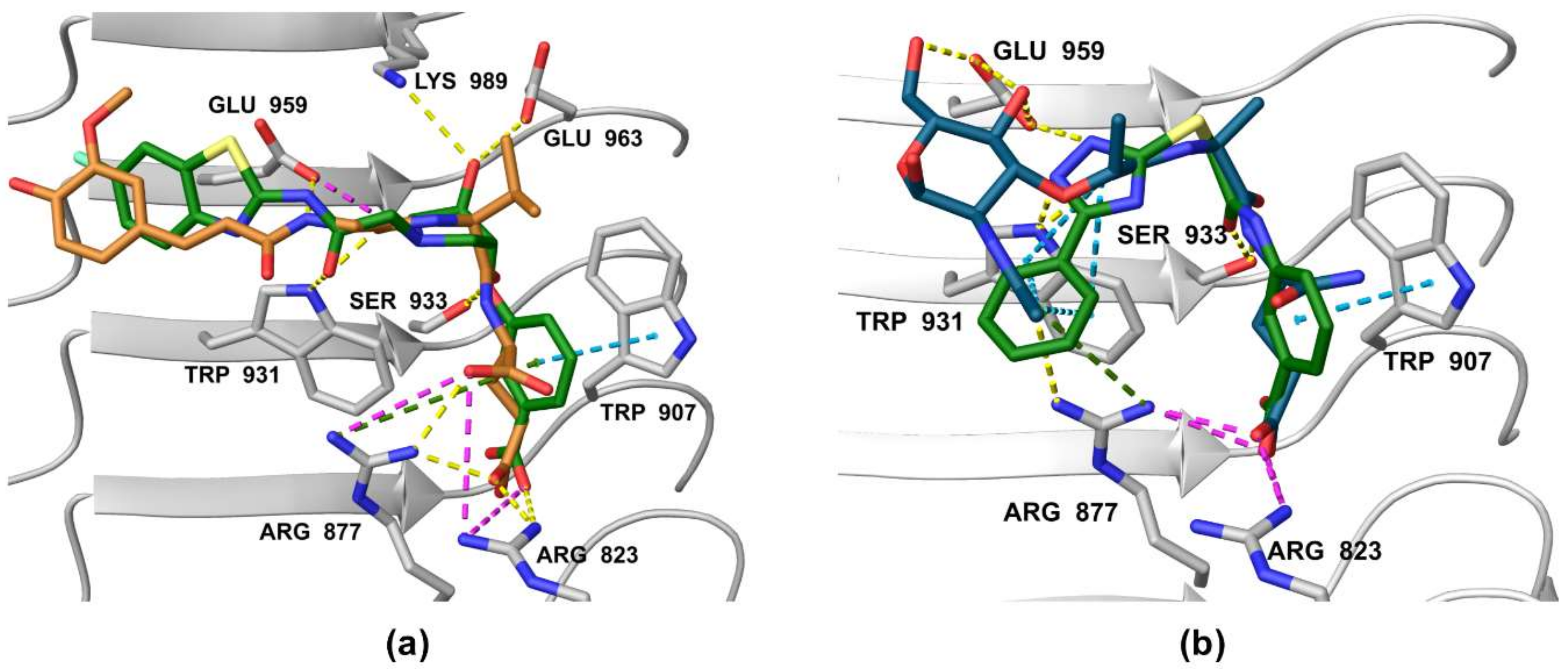
3.5. Binding Modes of Compounds 1 and 3 in the Putative NOD2 Ligand-Binding Pocket
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD Proteins: Regulators of Inflammation in Health and Disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate Immune Recognition. Annu Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 Is a General Sensor of Peptidoglycan through Muramyl Dipeptide (MDP) Detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host Recognition of Bacterial Muramyl Dipeptide Mediated through NOD2: Implications for Crohn’s Disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef] [Green Version]
- Al Nabhani, Z.; Dietrich, G.; Hugot, J.P.; Barreau, F. Nod2: The Intestinal Gate Keeper. PLoS Pathog. 2017, 13, e1006177. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.P.; Parkhouse, R.; Monie, T.P. Insights into the Molecular Basis of the Nod2 Signalling Pathway. Open Biol. 2014, 4, 140178. [Google Scholar] [CrossRef] [Green Version]
- Negroni, A.; Pierdomenico, M.; Cucchiara, S.; Stronati, L. NOD2 and Inflammation: Current Insights. J. Inflamm. Res. 2018, 11, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Jakopin, Ž. Nucleotide-Binding Oligomerization Domain (NOD) Inhibitors: A Rational Approach toward Inhibition of NOD Signaling Pathway. J. Med. Chem. 2014, 57, 6897–6918. [Google Scholar] [CrossRef]
- Geddes, K.; Magalhães, J.G.; Girardin, S.E. Unleashing the Therapeutic Potential of NOD-like Receptors. Nat. Rev. Drug Discov. 2009, 8, 465–479. [Google Scholar] [CrossRef]
- Ellouz, F.; Adam, A.; Ciorbaru, R.; Lederer, E. Minimal Structural Requirements for Adjuvant Activity of Bacterial Peptidoglycan Derivatives. Biochem. Biophys. Res. Commun. 1974, 59, 1317–1325. [Google Scholar] [CrossRef]
- Griffin, M.E.; Hespen, C.W.; Wang, Y.C.; Hang, H.C. Translation of Peptidoglycan Metabolites into Immunotherapeutics. Clin. Transl. Immunol. 2019, 8, e1095. [Google Scholar] [CrossRef]
- Gobec, M.; Mlinarič-Raščan, I.; Dolenc, M.S.; Jakopin, Ž. Structural Requirements of Acylated Gly- l -Ala- d -Glu Analogs for Activation of the Innate Immune Receptor NOD2. Eur. J. Med. Chem. 2016, 116, 1–12. [Google Scholar] [CrossRef]
- Gobec, M.; Tomašič, T.; Štimac, A.; Frkanec, R.; Trontelj, J.; Anderluh, M.; Mlinarič-Raščan, I.; Jakopin, Ž. Discovery of Nanomolar Desmuramylpeptide Agonists of the Innate Immune Receptor Nucleotide-Binding Oligomerization Domain-Containing Protein 2 (NOD2) Possessing Immunostimulatory Properties. J. Med. Chem. 2018, 61, 2707–2724. [Google Scholar] [CrossRef]
- Guzelj, S.; Nabergoj, S.; Gobec, M.; Pajk, S.; Klančič, V.; Slütter, B.; Frkanec, R.; Štimac, A.; Šket, P.; Plavec, J.; et al. Structural Fine-Tuning of Desmuramylpeptide NOD2 Agonists Defines Their In Vivo Adjuvant Activity. J. Med. Chem. 2021, 64, 7809–7838. [Google Scholar] [CrossRef]
- Liu, H.Q.; Zhang, X.Y.; Edfeldt, K.; Nijhuis, M.O.; Idborg, H.; Bäck, M.; Roy, J.; Hedin, U.; Jakobsson, P.J.; Laman, J.D.; et al. NOD2-Mediated Innate Immune Signaling Regulates the Eicosanoids in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2013, 33, 2193–2201. [Google Scholar] [CrossRef] [Green Version]
- Vlacil, A.K.; Schuett, J.; Ruppert, V.; Soufi, M.; Oberoi, R.; Shahin, K.; Wächter, C.; Tschernig, T.; Lei, Y.; Liu, F.; et al. Deficiency of Nucleotide-Binding Oligomerization Domain-Containing Proteins (NOD) 1 and 2 Reduces Atherosclerosis. Basic Res. Cardiol. 2020, 115, 47. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, L.; Tang, W.; Li, D.; Ma, L.; Liu, H.; Zhang, S.; Zhang, X.; Dong, L.; Shen, X.; et al. Hepatic NOD2 Promotes Hepatocarcinogenesis via a RIP2-Mediated Proinflammatory Response and a Novel Nuclear Autophagy-Mediated DNA Damage Mechanism. J. Hematol. Oncol. 2021, 14, 9. [Google Scholar] [CrossRef]
- Wang, S.; Yang, J.; Li, X.; Liu, Z.; Wu, Y.; Si, G.; Tao, Y.; Zhao, N.; Hu, X.; Ma, Y.; et al. Discovery of 1,4-Benzodiazepine-2,5-Dione (BZD) Derivatives as Dual Nucleotide Binding Oligomerization Domain Containing 1/2 (NOD1/NOD2) Antagonists Sensitizing Paclitaxel (PTX) to Suppress Lewis Lung Carcinoma (LLC) Growth in Vivo. J. Med. Chem. 2017, 60, 5162–5192. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, S.; Wang, C.; Li, Z.; Ma, Y.; Liu, G. Antagonizing NOD2 Signaling with Conjugates of Paclitaxel and Muramyl Dipeptide Derivatives Sensitizes Paclitaxel Therapy and Significantly Prevents Tumor Metastasis. J. Med. Chem. 2017, 60, 1219–1224. [Google Scholar] [CrossRef]
- Maekawa, S.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Crystal Structure of NOD2 and Its Implications in Human Disease. Nat. Commun. 2016, 7, 11813. [Google Scholar] [CrossRef] [Green Version]
- Parant, M.; Parant, F.; Chedid, L.; Yapo, A.; Petit, J.F.; Lederer, E. Fate of the Synthetic Immunoadjuvant, Muramyl Dipeptide (14C-Labelled) in the Mouse. Int. J. Immunopharmacol. 1979, 1, 35–41. [Google Scholar] [CrossRef]
- Harrison, J.; Fox, A. Degradation of Muramyl Dipeptide by Mammalian Serum. Infect. Immun. 1985, 50, 320–321. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Li, X.; Pei, Y.; Ye, J.; Wei, X.; Yang, J.; Si, G.; Tian, J.; Dong, Y.; Liu, G. Identification of Benzofused Five-Membered Sultams, Potent Dual NOD1/NOD2 Antagonists in Vitro and in Vivo. Eur. J. Med. Chem. 2020, 204, 112575. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, J.; Wei, X.; Pei, Y.; Ye, J.; Li, X.; Si, G.; Tian, J.; Dong, Y.; Liu, G. Nonpeptidic Quinazolinone Derivatives as Dual Nucleotide-Binding Oligomerization Domain-like Receptor 1/2 Antagonists for Adjuvant Cancer Chemotherapy. Eur. J. Med. Chem. 2020, 207, 112723. [Google Scholar] [CrossRef]
- Keček Plešec, K.; Urbančič, D.; Gobec, M.; Pekošak, A.; Tomašič, T.; Anderluh, M.; Mlinarič-Raščan, I.; Jakopin, Ž. Identification of Indole Scaffold-Based Dual Inhibitors of NOD1 and NOD2. Bioorganic Med. Chem. 2016, 24, 5221–5234. [Google Scholar] [CrossRef]
- Guzelj, S.; Gobec, M.; Urbančič, D.; Mlinarič-Raščan, I.; Corsini, E.; Jakopin, Ž. Structural Features and Functional Activities of Benzimidazoles as NOD2 Antagonists. Eur. J. Med. Chem. 2020, 190, 112089. [Google Scholar] [CrossRef]
- Rickard, D.J.; Sehon, C.A.; Kasparcova, V.; Kallal, L.A.; Zeng, X.; Montoute, M.N.; Chordia, T.; Poore, D.D.; Li, H.; Wu, Z.; et al. Identification of Benzimidazole Diamides as Selective Inhibitors of the Nucleotide-Binding Oligomerization Domain 2 (NOD2) Signaling Pathway. PLoS ONE 2013, 8, e69619. [Google Scholar] [CrossRef] [Green Version]
- Rubino, S.J.; Magalhaes, J.G.; Philpott, D.; Bahr, G.M.; Blanot, D.; Girardin, S.E. Identification of a Synthetic Muramyl Peptide Derivative with Enhanced Nod2 Stimulatory Capacity. Innate Immun. 2013, 19, 493–503. [Google Scholar] [CrossRef]
- Melnyk, J.E.; Mohanan, V.; Schaefer, A.K.; Hou, C.W.; Grimes, C.L. Peptidoglycan Modifications Tune the Stability and Function of the Innate Immune Receptor Nod2. J. Am. Chem. Soc. 2015, 137, 6987–6990. [Google Scholar] [CrossRef] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Studies in Classification, Data Analysis, and Knowledge Organization (GfKL 2007); Springer: Berlin, Germany, 2008; pp. 319–326. ISBN 978-3-540-78239-1. [Google Scholar]
- Poli, G.; Seidel, T.; Langer, T. Conformational Sampling of Small Molecules with ICon: Performance Assessment in Comparison With OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021–3; Prime, Schrödinger, LLC: New York, NY, USA, 2021.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2021–3; LigPrep, Schrödinger, LLC: New York, NY, USA, 2021.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PK a Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Lauro, M.L.; D’Ambrosio, E.A.; Bahnson, B.J.; Grimes, C.L. Molecular Recognition of Muramyl Dipeptide Occurs in the Leucine-Rich Repeat Domain of Nod2. ACS Infect. Dis. 2017, 3, 264–270. [Google Scholar] [CrossRef]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Schaefer, A.K.; Melnyk, J.E.; Baksh, M.M.; Lazor, K.M.; Finn, M.G.; Grimes, C.L. Membrane Association Dictates Ligand Specificity for the Innate Immune Receptor NOD2. ACS Chem. Biol. 2017, 12, 2216–2224. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Xu, S.; Wang, N.; Yang, H.; Yan, Z.; Cheng, G.; Liu, G. Chemical Conjugation of Muramyl Dipeptide and Paclitaxel to Explore the Combination of Immunotherapy and Chemotherapy for Cancer. Glycoconj. J. 2008, 25, 415–425. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Structure | Relative Pharmacophore Fit Score 1 | Docking Score (kcal/mol) | MM-GBSA Binding Energy (kcal/mol) |
|---|---|---|---|---|
| MDP | 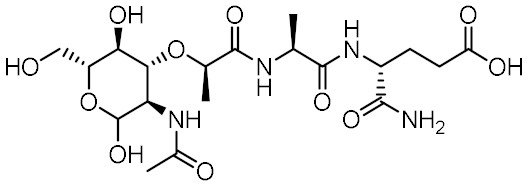 | 0.955 | −6.662 | −58.60 |
| SG8 |  | 0.976 | −6.677 | −53.68 |
| 1 | 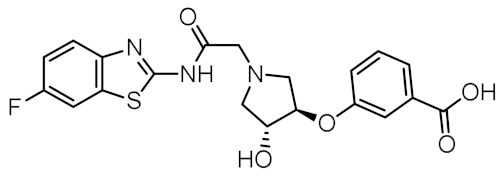 | 0.736 | −5.846 | −67.06 |
| 2 |  | 0.764 | −6.164 | −57.74 |
| 3 | 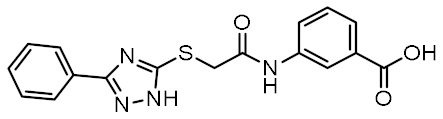 | 0.759 | −5.473 | −54.87 |
| 4 |  | 0.756 | −6.118 | −52.05 |
| 5 |  | 0.759 | −7.051 | −50.96 |
| 6 |  | 0.785 | −5.694 | −50.36 |
| 7 |  | 0.778 | −5.96 | −50.13 |
| 8 | 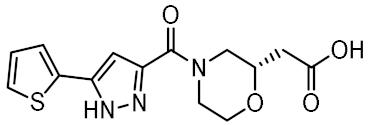 | 0.747 | −5.501 | −46.51 |
| 9 |  | 0.747 | −6.129 | −46.43 |
| 10 | 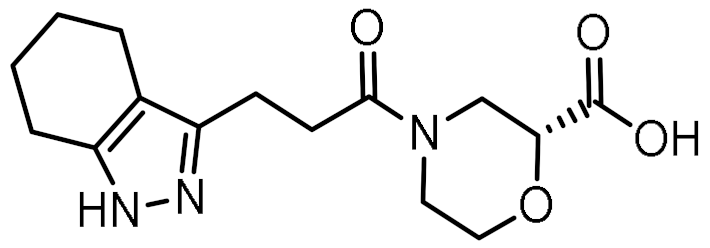 | 0.742 | −5.793 | −41.98 |
| 11 |  | 0.916 | −5.021 | −40.09 |
| 12 | 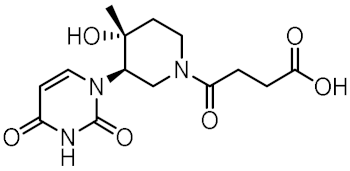 | 0.929 | −5.559 | −34.63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzelj, S.; Tomašič, T.; Jakopin, Ž. Novel Scaffolds for Modulation of NOD2 Identified by Pharmacophore-Based Virtual Screening. Biomolecules 2022, 12, 1054. https://doi.org/10.3390/biom12081054
Guzelj S, Tomašič T, Jakopin Ž. Novel Scaffolds for Modulation of NOD2 Identified by Pharmacophore-Based Virtual Screening. Biomolecules. 2022; 12(8):1054. https://doi.org/10.3390/biom12081054
Chicago/Turabian StyleGuzelj, Samo, Tihomir Tomašič, and Žiga Jakopin. 2022. "Novel Scaffolds for Modulation of NOD2 Identified by Pharmacophore-Based Virtual Screening" Biomolecules 12, no. 8: 1054. https://doi.org/10.3390/biom12081054
APA StyleGuzelj, S., Tomašič, T., & Jakopin, Ž. (2022). Novel Scaffolds for Modulation of NOD2 Identified by Pharmacophore-Based Virtual Screening. Biomolecules, 12(8), 1054. https://doi.org/10.3390/biom12081054