
Recognition of BRAF by CDC37 and Re-Evaluation of the Activation Mechanism for the Class 2 BRAF-L597R Mutant

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Protein Expression and Purification
2.2. Isothermal Titration Chromatography Kd Determinations
2.3. BRAF Kinase Assays
2.4. Thermal Shift Assay
2.5. Molecular Mass Determination
3. Results
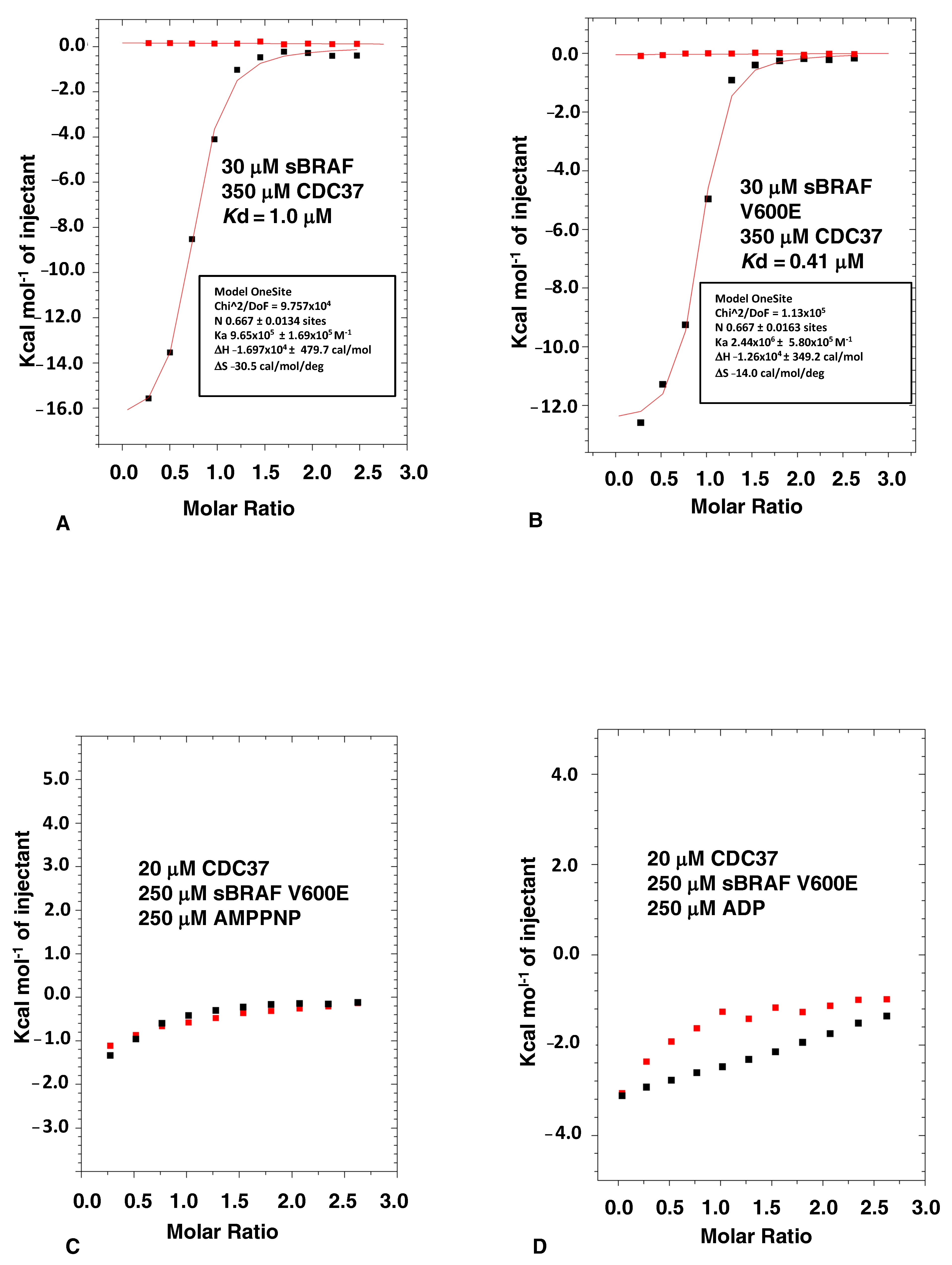
3.1. The Binding of CDC37 to sBRAF and sBRAF V600E Is Essentially Indistinguishable
3.2. Nucleotide Binding Prevents the CDC37-BRAF Interaction
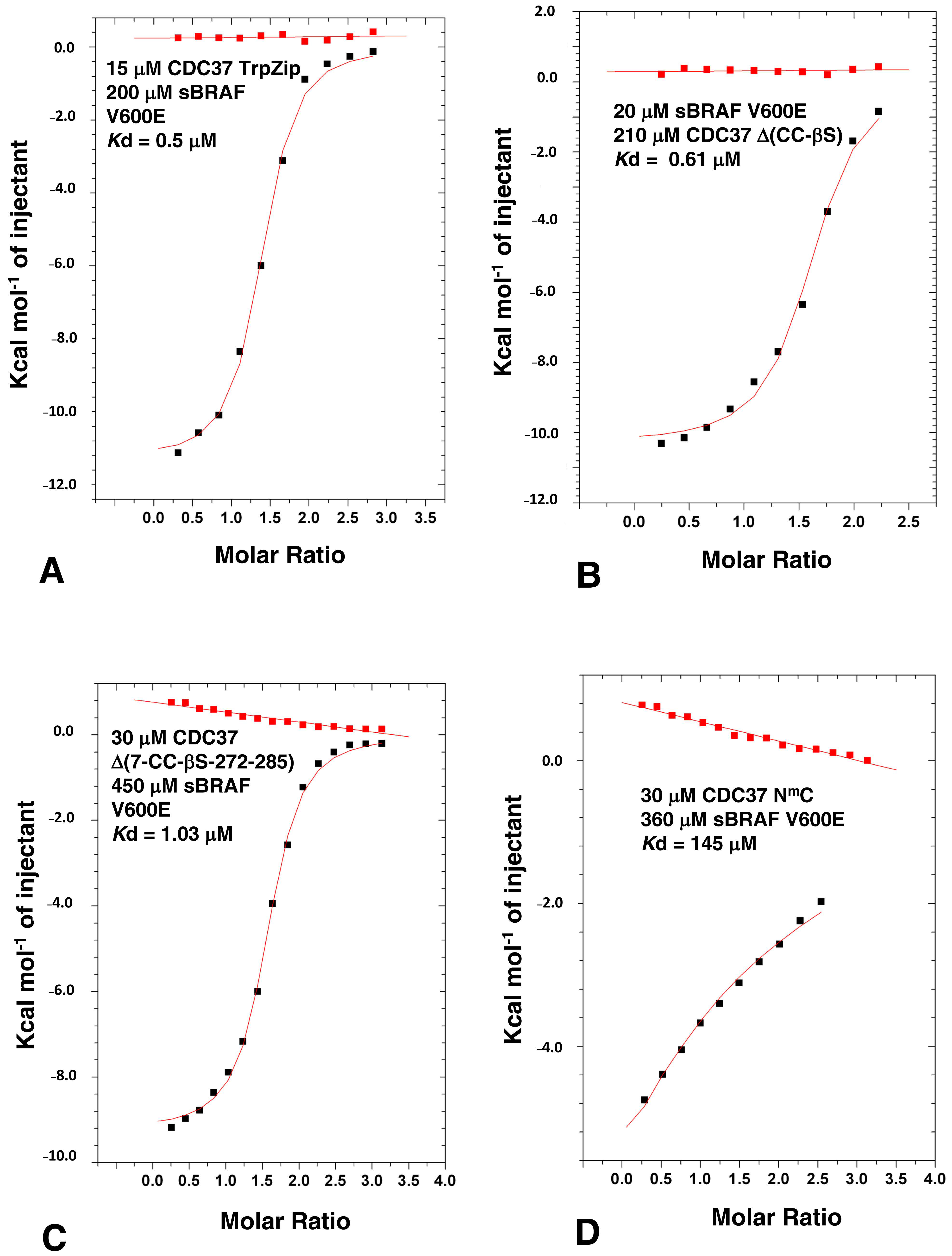
3.3. The n-terminal and c-terminal Domains of CDC37 Are Essential for Efficient Bipartite Binding to the Kinase Domain
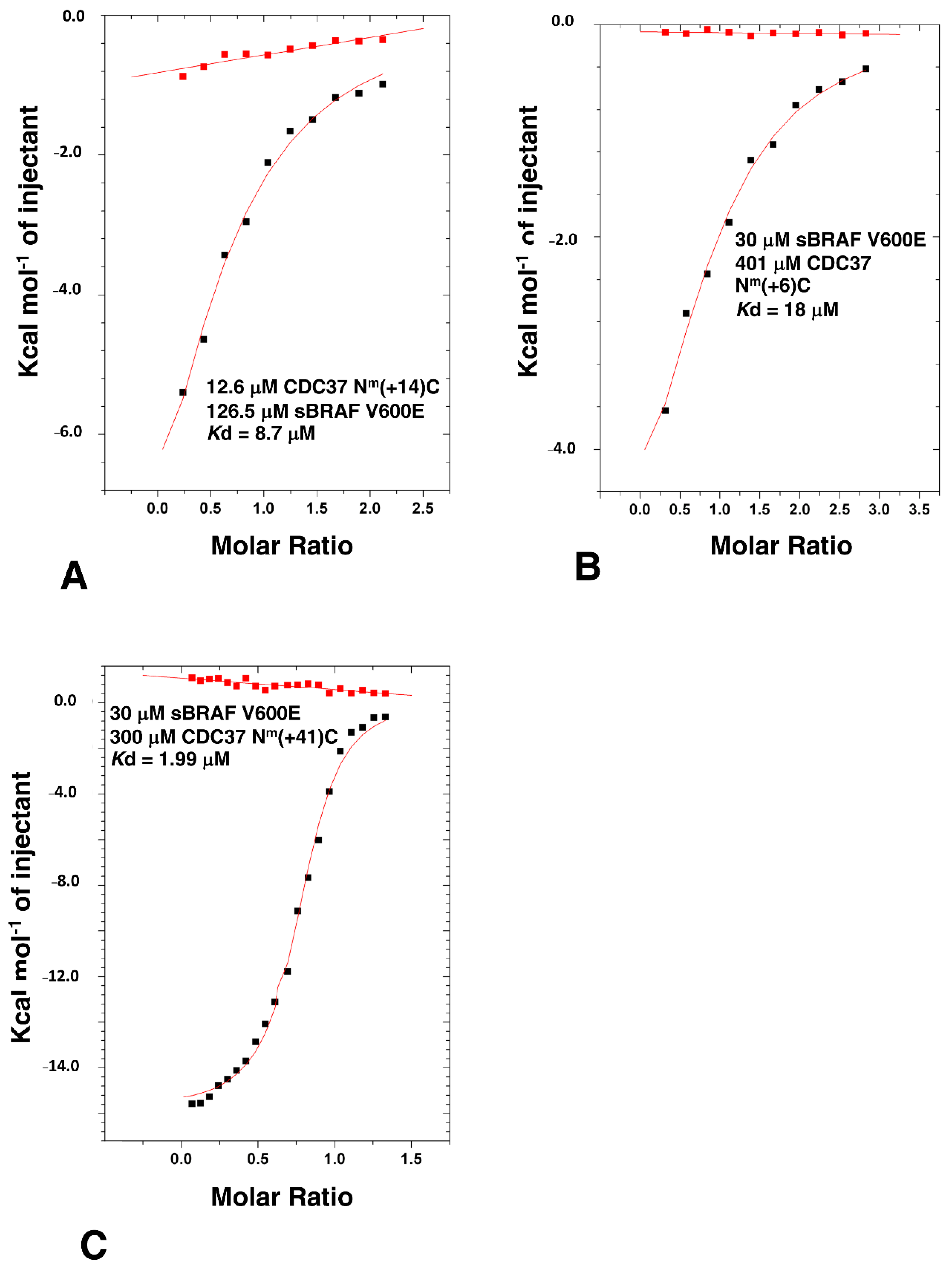
3.4. Determining the Minimal CDC37 Structure Required for High-Affinity Binding to sBRAF V600E
3.5. The CDC37 S13E-Phosphomimetic Mutation Does Not Affect Binding to BRAF
3.6. The CDC37 HPNI Amino Acid Motif Is Essential for High-Affinity Kinase Recognition
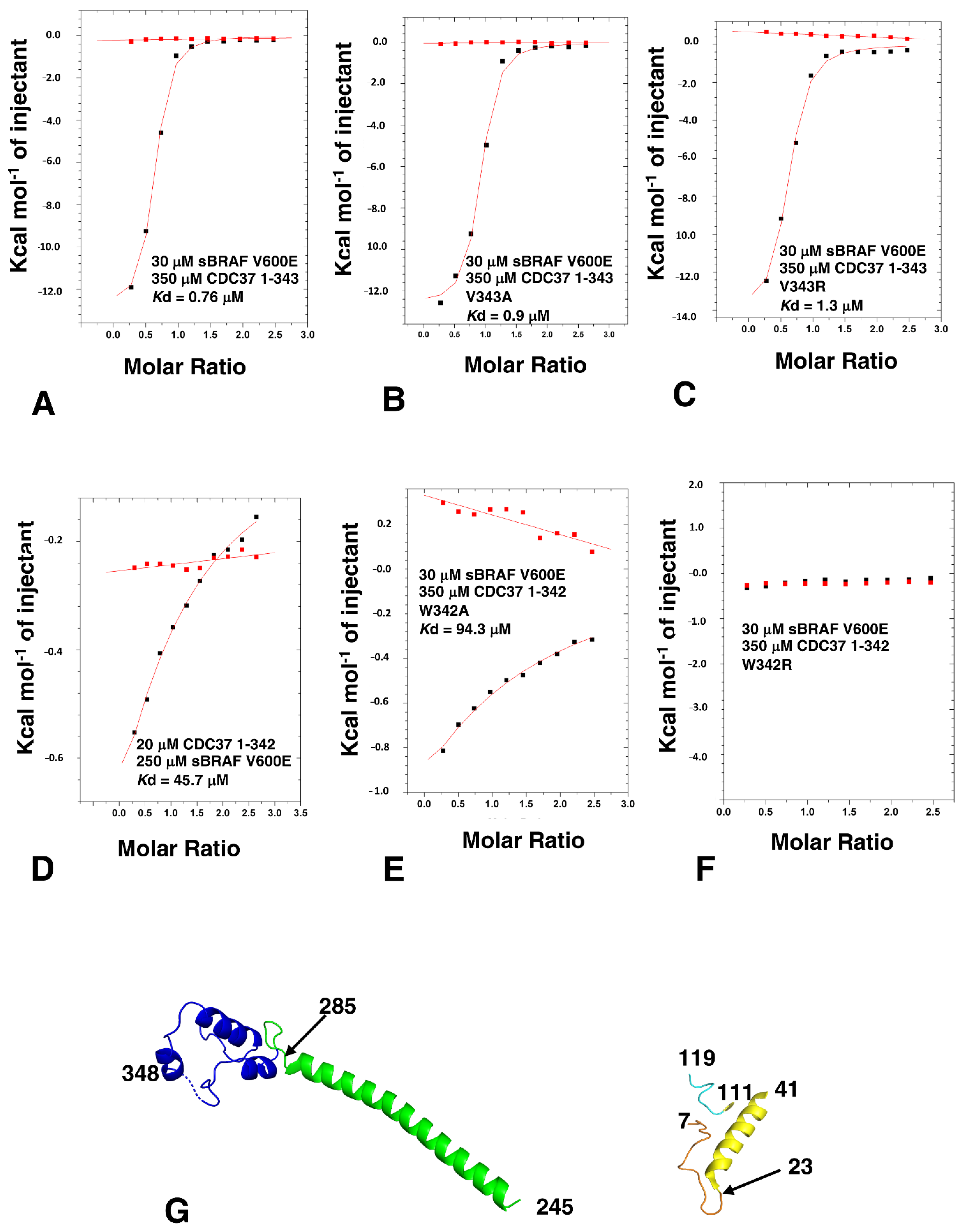
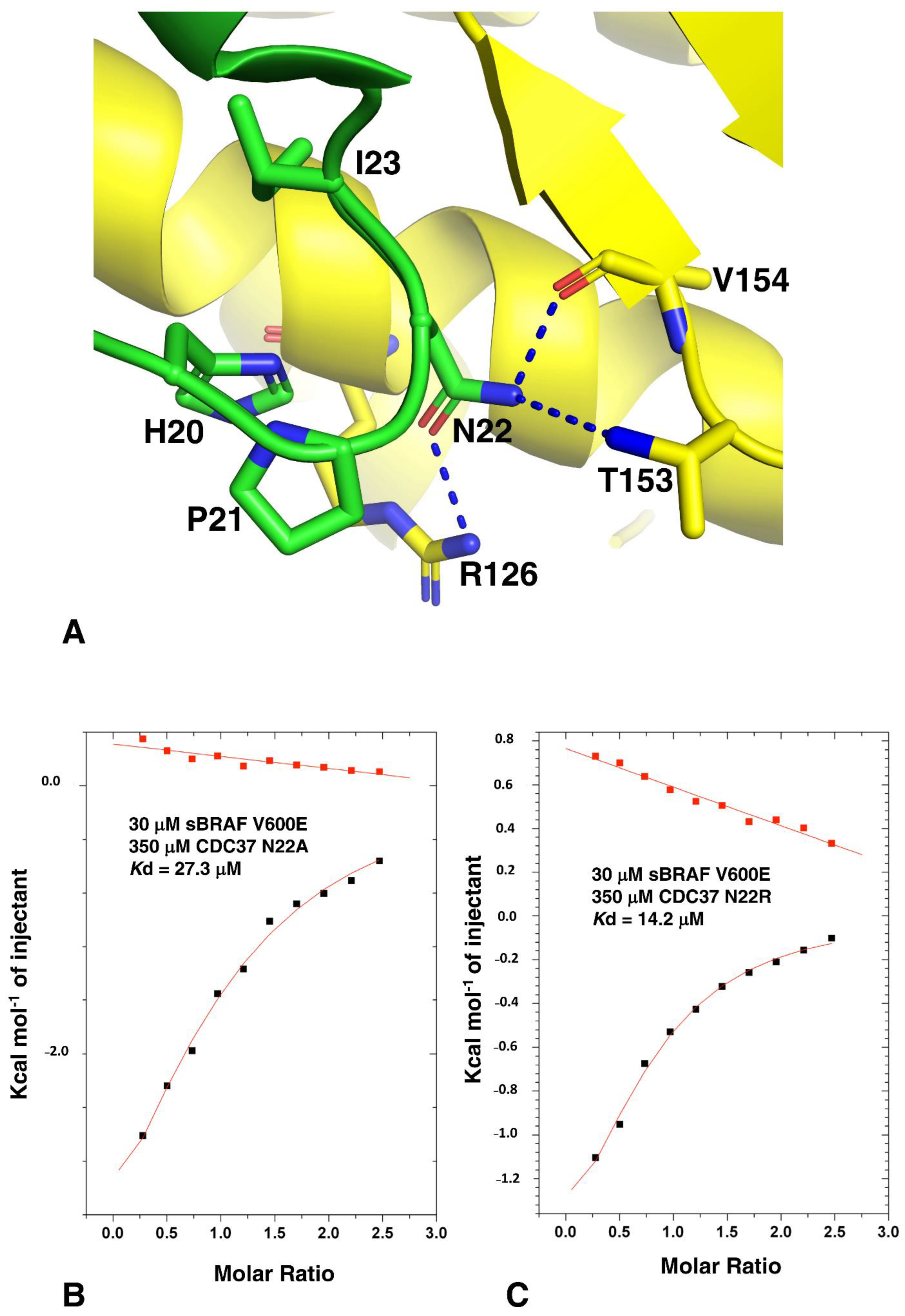
3.7. HPNI Is Part of a More Extensive CDC37 Binding Motif
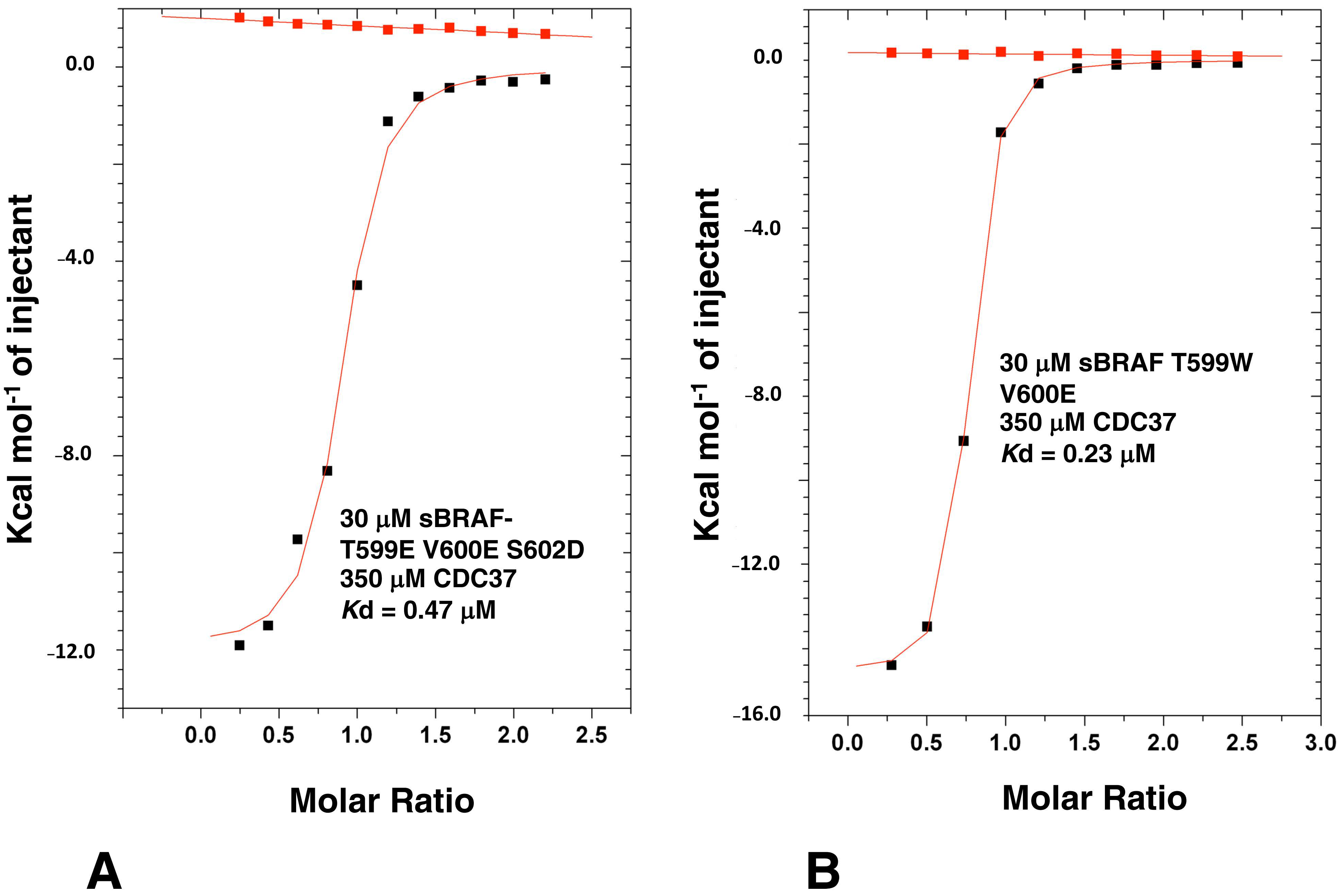
3.8. Activation Loop Mutants Do Not Influence BRAF Binding to CDC37
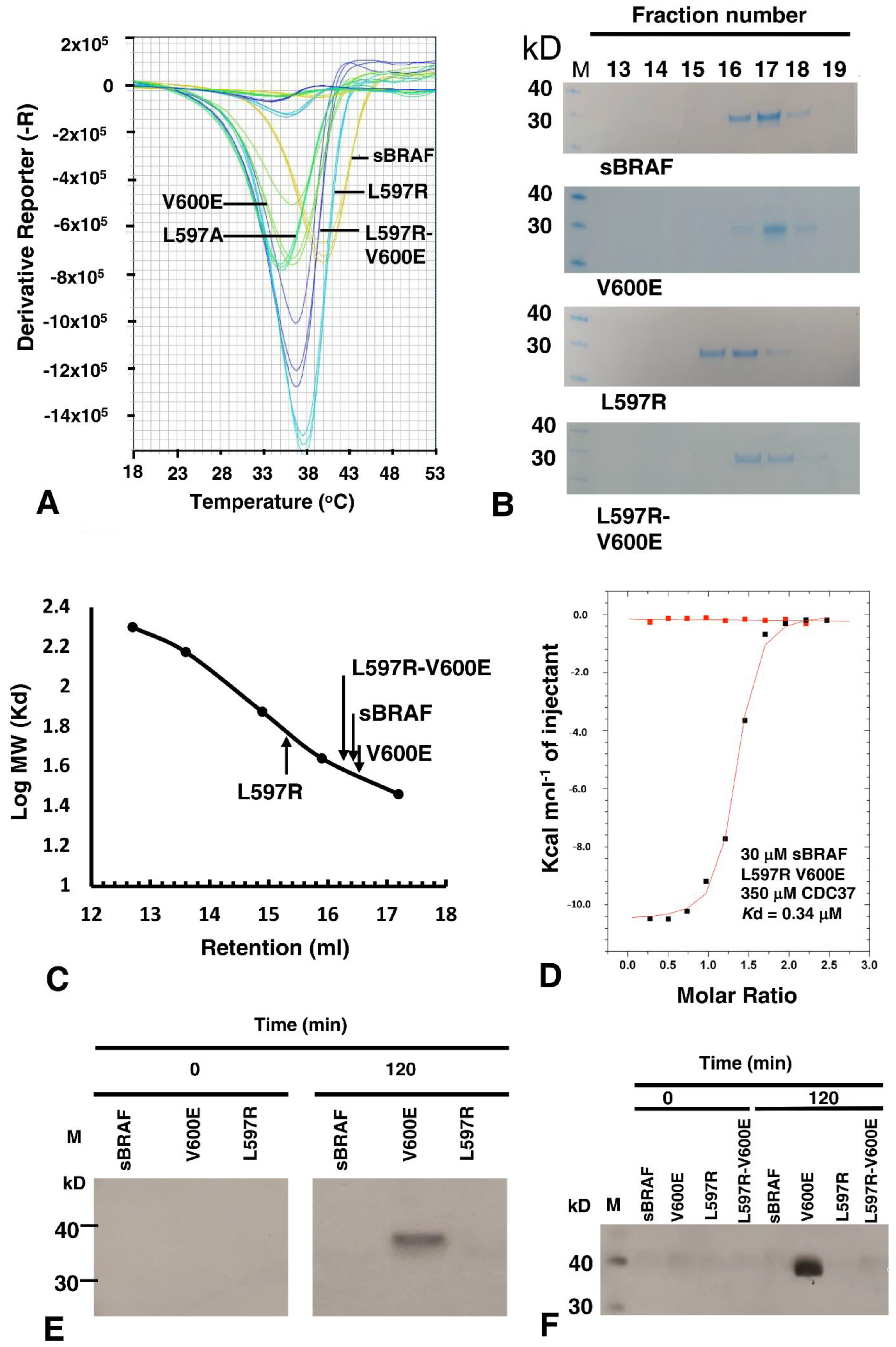
3.9. The BRAF Mutation L597R Compromises Binding to CDC37
3.10. sBRAF L597R and L597R-V600E Mutant Are Both Inactive
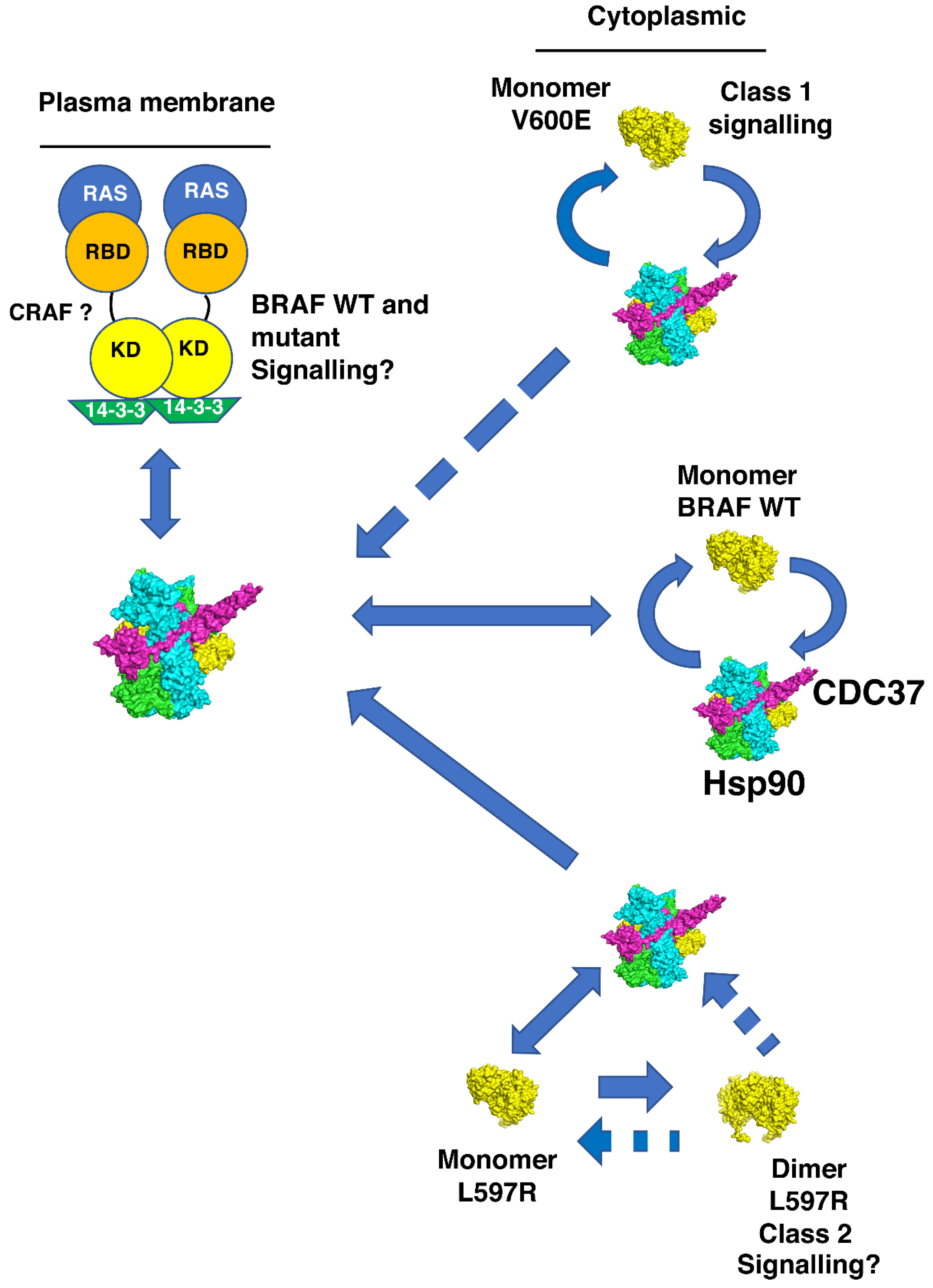
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keramisanou, D.; Aboalroub, A.; Zhang, Z.; Liu, W.; Marshall, D.; Diviney, A.; Larsen, R.W.; Landgraf, R.; Gelis, I. Molecular Mechanism of Protein Kinase Recognition and Sorting by the Hsp90 Kinome-Specific Cochaperone Cdc37. Mol. Cell 2016, 62, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H. ATP-Driven Nonequilibrium Activation of Kinase Clients by the Molecular Chaperone Hsp90. Biophys. J. 2020, 119, 1538–1549. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Poon, R.Y.C. Cdc37: A protein kinase chaperone? Trends Cell Biol. 1997, 7, 157–161. [Google Scholar] [CrossRef]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The Mechanism of Hsp90 Regulation by the Protein Kinase-Specific Cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Verba, K.A.; Wang, R.Y.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Keramisanou, D.; Dudhat, A.; Pare, M.; Gelis, I. The c-terminal domain of human Cdc37 studied by solution NMR. J. Biomol. NMR 2015, 63, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Sreeramulu, S.; Jonker, H.R.; Langer, T.; Richter, C.; Lancaster, C.R.; Schwalbe, H. The human Cdc37·Hsp90 complex studied by heteronuclear NMR spectroscopy. J. Biol. Chem. 2009, 284, 3885–3896. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Meharena, H.S.; Chang, P.; Keshwani, M.M.; Oruganty, K.; Nene, A.K.; Kannan, N.; Taylor, S.S.; Kornev, A.P. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013, 11, e1001680. [Google Scholar] [CrossRef] [Green Version]
- Kornev, A.P.; Taylor, S.S.; Ten Eyck, L.F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 14377–14382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Ahuja, L.G.; Meharena, H.S.; Kannan, N.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol. 2015, 35, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Ahuja, L.G.; Chao, F.A.; Xia, Y.; McClendon, C.L.; Kornev, A.P.; Taylor, S.S.; Veglia, G. A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv. 2017, 3, e1600663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, F.A.; Cook, S.J. Inhibition of RAF dimers: It takes two to tango. Biochem. Soc. Trans. 2021, 49, 237–251. [Google Scholar] [CrossRef]
- Perdew, G.H.; Wiegand, H.; VandenHeuvel, J.P.; Mitchell, C.; Singh, S.S. A 50 kilodalton protein associated with raf and pp(60v-src) protein kinases is a mammalian homolog of the cell cycle control protein cdc37. Biochemistry 1997, 36, 3600–3607. [Google Scholar] [CrossRef]
- Diedrich, B.; Rigbolt, K.T.; Roring, M.; Herr, R.; Kaeser-Pebernard, S.; Gretzmeier, C.; Murphy, R.F.; Brummer, T.; Dengjel, J. Discrete cytosolic macromolecular BRAF complexes exhibit distinct activities and composition. EMBO J. 2017, 36, 646–663. [Google Scholar] [CrossRef]
- Polier, S.; Samant, R.S.; Clarke, P.A.; Workman, P.; Prodromou, C.; Pearl, L.H. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat. Chem. Biol. 2013, 9, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Wartmann, M.; Davis, R.J. The native structure of the activated Raf protein kinase is a membrane-bound multi-subunit complex. J. Biol. Chem. 1994, 269, 6695–6701. [Google Scholar] [CrossRef]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Sharp, S.; Workman, P. Inhibitors of the HSP90 molecular chaperone: Current status. Adv. Cancer Res. 2006, 95, 323–348. [Google Scholar]
- Schulte, T.W.; Blagosklonny, M.V.; Ingui, C.; Neckers, L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995, 270, 24585–24588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.J.; Jang, H. Does Ras Activate Raf and PI3K Allosterically? Front. Oncol. 2019, 9, 1231. [Google Scholar] [CrossRef] [PubMed]
- Kohler, M.; Brummer, T. B-Raf activation loop phosphorylation revisited. Cell Cycle 2016, 15, 1171–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Guan, K.L. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000, 19, 5429–5439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Negrao, M.V.; Raymond, V.M.; Lanman, R.B.; Robichaux, J.P.; He, J.; Nilsson, M.B.; Ng, P.K.S.; Amador, B.E.; Roarty, E.B.; Nagy, R.J.; et al. Molecular Landscape of BRAF-Mutant NSCLC Reveals an Association Between Clonality and Driver Mutations and Identifies Targetable Non-V600 Driver Mutations. J. Thorac. Oncol. 2020, 15, 1611–1623. [Google Scholar] [CrossRef]
- Siligardi, G.; Panaretou, B.; Meyer, P.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Cochran, A.G.; Skelton, N.J.; Starovasnik, M.A. Tryptophan zippers: Stable, monomeric beta-hairpins. Proc. Natl. Acad. Sci. USA 2001, 98, 5578–5583. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Prince, T.; Hartson, S.D.; Matts, R.L. Phosphorylation of serine 13 is required for the proper function of the Hsp90 co-chaperone, Cdc37. J. Biol. Chem. 2003, 278, 38117–38120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, J.; Deepak, R.; Tian, Z.; Ng, W.H.; Goh, K.C.; Foo, A.; Tee, Z.H.; Mohanam, M.P.; Sim, Y.R.M.; Degirmenci, U.; et al. The stability of R-spine defines RAF inhibitor resistance: A comprehensive analysis of oncogenic BRAF mutants with in-frame insertion of alphaC-beta4 loop. Sci. Adv. 2021, 7, eabg0390. [Google Scholar] [CrossRef] [PubMed]
- Noeparast, A.; Teugels, E.; Giron, P.; Verschelden, G.; De Brakeleer, S.; Decoster, L.; De Greve, J. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 2017, 8, 60094–60108. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.K.; Li, J.; Jeong, K.J.; Shao, S.; Chen, H.; Tsang, Y.H.; Sengupta, S.; Wang, Z.; Bhavana, V.H.; Tran, R.; et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 2018, 33, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef] [Green Version]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Z.; Kannan, N. Altered conformational landscape and dimerization dependency underpins the activation of EGFR by alphaC-beta4 loop insertion mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E8162–E8171. [Google Scholar] [CrossRef] [Green Version]
- Zanetti-Domingues, L.C.; Korovesis, D.; Needham, S.R.; Tynan, C.J.; Sagawa, S.; Roberts, S.K.; Kuzmanic, A.; Ortiz-Zapater, E.; Jain, P.; Roovers, R.C.; et al. The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nat. Commun. 2018, 9, 4325. [Google Scholar] [CrossRef]
- Valley, C.C.; Arndt-Jovin, D.J.; Karedla, N.; Steinkamp, M.P.; Chizhik, A.I.; Hlavacek, W.S.; Wilson, B.S.; Lidke, K.A.; Lidke, D.S. Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol. Biol. Cell 2015, 26, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Taha, T.; Khoury, R.; Brenner, R.; Nasrallah, H.; Shofaniyeh, I.; Yousef, S.; Agbarya, A. Treatment of Rare Mutations in Patients with Lung Cancer. Biomedicines 2021, 9, 534. [Google Scholar] [CrossRef] [PubMed]
- Baik, C.S.; Myall, N.J.; Wakelee, H.A. Targeting BRAF-Mutant Non-Small Cell Lung Cancer: From Molecular Profiling to Rationally Designed Therapy. Oncologist 2017, 22, 786–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlman, K.B.; Xia, J.; Hutchinson, K.; Ng, C.; Hucks, D.; Jia, P.; Atefi, M.; Su, Z.; Branch, S.; Lyle, P.L.; et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012, 2, 791–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keramisanou, D.; Vasantha Kumar, M.V.; Boose, N.; Abzalimov, R.R.; Gelis, I. Assembly mechanism of early Hsp90-Cdc37-kinase complexes. Sci. Adv. 2022, 8, eabm9294. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Shigematsu, H.; Herz, J.; Minna, J.D. Mutations and addiction to EGFR: The Achilles ‘heal’ of lung cancers? Trends Mol. Med. 2004, 10, 481–486. [Google Scholar] [CrossRef]
- Johnson, B.E.; Janne, P.A. Epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Cancer Res. 2005, 65, 7525–7529. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef]
- Da Rocha Dias, S.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar] [PubMed]
- Zhao, Y.; Yu, H.; Ida, C.M.; Halling, K.C.; Kipp, B.R.; Geiersbach, K.; Rumilla, K.M.; Gupta, S.; Lin, M.T.; Zheng, G. Assessment of RAS Dependency for BRAF Alterations Using Cancer Genomic Databases. JAMA Netw. Open 2021, 4, e2035479. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Li, D.; Ji, H.; Haringsma, H.J.; Liniker, E.; Borgman, C.L.; Lowell, A.M.; Minami, Y.; McNamara, K.; Perera, S.A.; et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008, 68, 5827–5838. [Google Scholar] [CrossRef] [Green Version]
- Smalley, K.S.; Xiao, M.; Villanueva, J.; Nguyen, T.K.; Flaherty, K.T.; Letrero, R.; Van Belle, P.; Elder, D.E.; Wang, Y.; Nathanson, K.L.; et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene 2009, 28, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Montagut, C.; Sharma, S.V.; Shioda, T.; McDermott, U.; Ulman, M.; Ulkus, L.E.; Dias-Santagata, D.; Stubbs, H.; Lee, D.Y.; Singh, A.; et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008, 68, 4853–4861. [Google Scholar] [CrossRef] [Green Version]
- Jilaveanu, L.B.; Zito, C.R.; Aziz, S.A.; Conrad, P.J.; Schmitz, J.C.; Sznol, M.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. C-Raf is associated with disease progression and cell proliferation in a subset of melanomas. Clin. Cancer Res. 2009, 15, 5704–5713. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjorklund, D.M.; Morgan, R.M.L.; Oberoi, J.; Day, K.L.I.M.; Galliou, P.A.; Prodromou, C. Recognition of BRAF by CDC37 and Re-Evaluation of the Activation Mechanism for the Class 2 BRAF-L597R Mutant. Biomolecules 2022, 12, 905. https://doi.org/10.3390/biom12070905
Bjorklund DM, Morgan RML, Oberoi J, Day KLIM, Galliou PA, Prodromou C. Recognition of BRAF by CDC37 and Re-Evaluation of the Activation Mechanism for the Class 2 BRAF-L597R Mutant. Biomolecules. 2022; 12(7):905. https://doi.org/10.3390/biom12070905
Chicago/Turabian StyleBjorklund, Dennis M., R. Marc L. Morgan, Jasmeen Oberoi, Katie L. I. M. Day, Panagiota A. Galliou, and Chrisostomos Prodromou. 2022. "Recognition of BRAF by CDC37 and Re-Evaluation of the Activation Mechanism for the Class 2 BRAF-L597R Mutant" Biomolecules 12, no. 7: 905. https://doi.org/10.3390/biom12070905
APA StyleBjorklund, D. M., Morgan, R. M. L., Oberoi, J., Day, K. L. I. M., Galliou, P. A., & Prodromou, C. (2022). Recognition of BRAF by CDC37 and Re-Evaluation of the Activation Mechanism for the Class 2 BRAF-L597R Mutant. Biomolecules, 12(7), 905. https://doi.org/10.3390/biom12070905