
Zinc in Cognitive Impairment and Aging


Abstract
1. Introduction
2. Zinc and Neurobiology
2.1. Zinc and Neuronal Development
2.2. Zinc and Neurogenesis
3. Zinc and Aging
3.1. Zinc and Hypoxia
3.2. Zinc and Inflammation
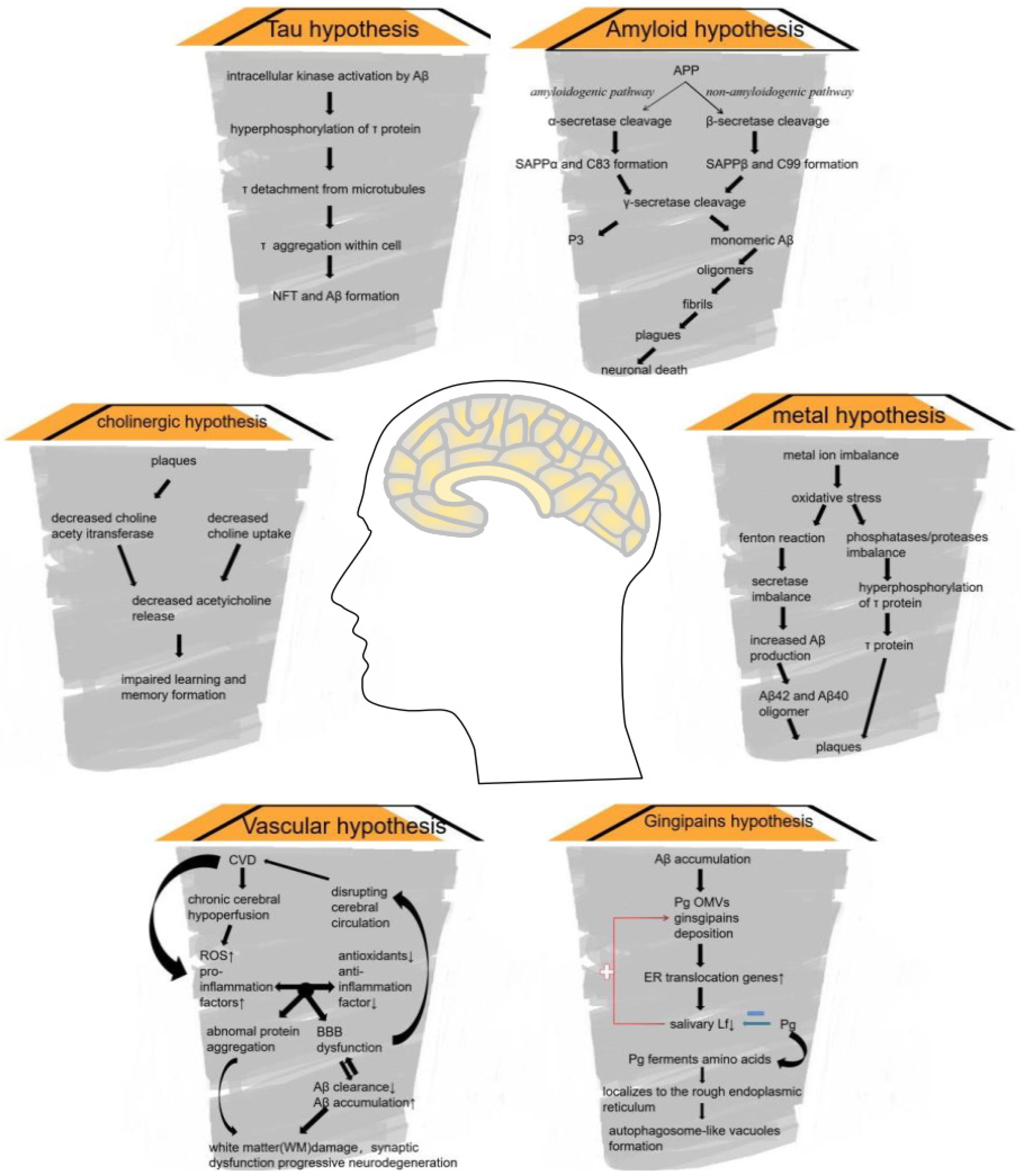
4. Zinc and AD
4.1. AD: Background
4.2. APP Processing
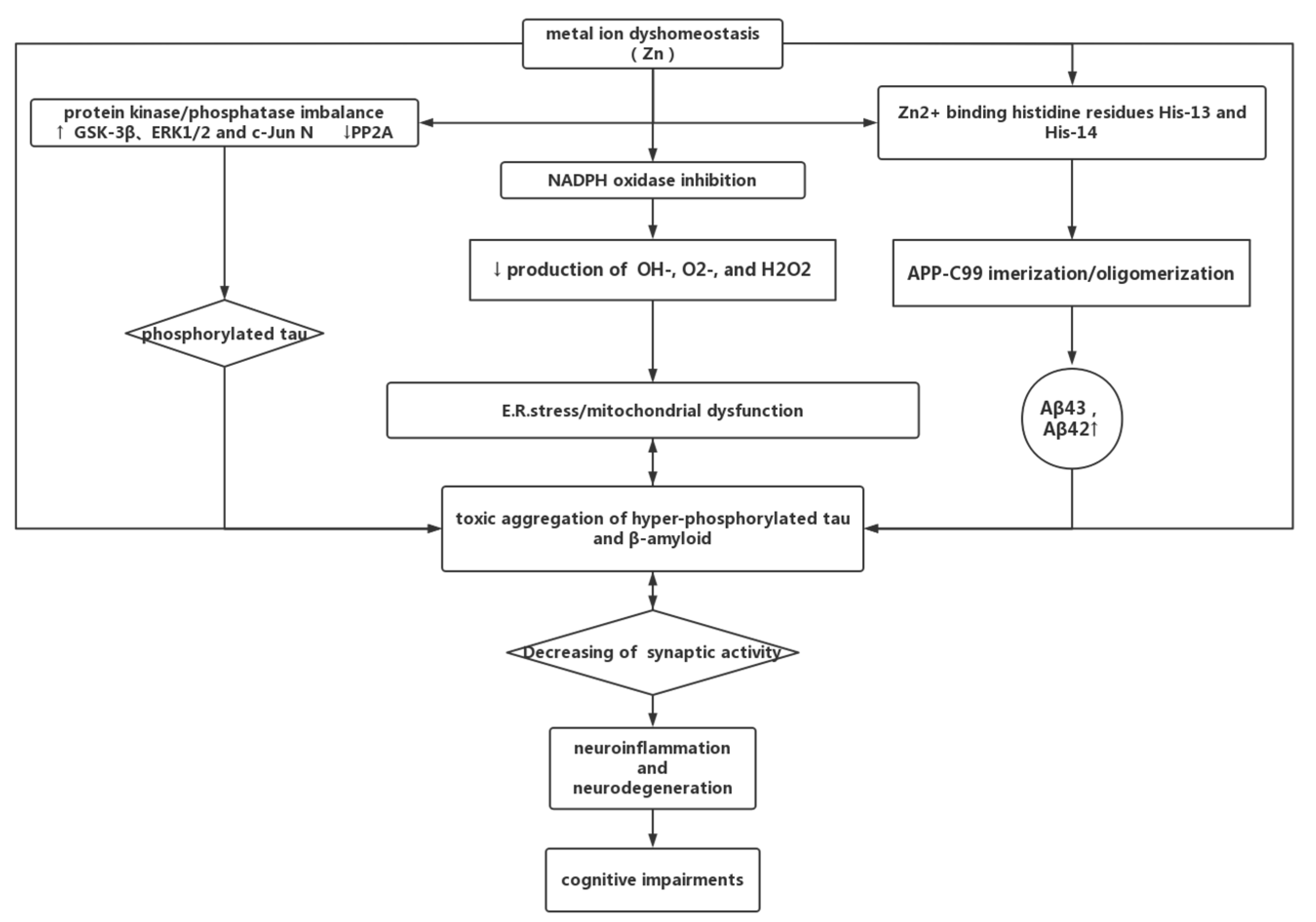
4.3. Zinc Dyshomeostasis and AD
4.3.1. Zinc in AD
4.3.2. Zinc Can Be a Double-Edged Sword in AD
5. Zinc and Vascular Dementia
5.1. Vascular Dementia
5.2. Risk Factors of VD
5.3. Metal Homeostasis and VD
6. Sleep Disorders
7. Molecular Mechanism of Zinc-Associated Aging and Cognitive Impairment
7.1. Zinc Transporters
7.1.1. Zinc Transporter-1
7.1.2. Zinc Transporter-3
7.1.3. Zinc Transporter-4
7.1.4. Zinc Transporter-6 (ZnT-6)
7.2. Metallothionein
7.2.1. Metallothionein-I/II
7.2.2. Metallothionein-III
8. ZFP and the Cellular Pathways
8.1. ZFP-1 (CIZ1)
8.2. Unkempt
8.3. Zinc Finger Protein 804A (ZFP804A)
9. Zinc and Autophagy
9.1. Extracellular Signal-Regulated Kinase 1/2 (ERK1/2)
9.2. MTs
9.3. Methylation
9.4. Zinc Homeostasis
10. Interventions for Zinc-Associated Aging and Cognitive Impairment
10.1. Clioquinol
10.2. Rapamycin
10.3. Zinc Supplements
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Min, C.J.; Young, K.J.; Ran, Y.H. Effects of oral zinc supplementation on zinc status and catch-up growth during the first 2 years of life in children with non-organic failure to thrive born preterm and at term. J. Pediatr. Neonatol. 2019, 60, 201–209. [Google Scholar]
- Akram Shahzad, K.; Carlsson-Skwirut, C.; Bhutta Zulfiqar, A.; Söder, O. Placental IGF-I, IGFBP-1, zinc, and iron, and maternal and infant anthropometry at birth. Acta Paediatr. 2011, 100, 1504–1509. [Google Scholar] [CrossRef]
- Merryana, A.; Bambang, W. The effect of adding zinc to vitamin A on IGF-1, bone age and linear growth in stunted children. J. Trace Elem. Med. Biol. 2014, 28, 431–435. [Google Scholar]
- Kanchan, B.; Abdul, M.A.B.; Atish, P. Possible role of metal ionophore against zinc induced cognitive dysfunction in D-galactose senescent mice. Biometals 2016, 29, 399–409. [Google Scholar]
- Seunghyuk, C.; Ki, H.D.; Young, C.B.; Won, S.S. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration, Role of zinc ion availability. Prog. Neurobiol. 2004, 75, 367–390. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Mizuno, D.; Kawahara, M. The molecular mechanisms of zinc neurotoxicity and the pathogenesis of vascular type senile dementia. Int. J. Mol. Sci. 2013, 14, 22067–22081. [Google Scholar] [CrossRef]
- Claudia, A.; Lucia, B.; Ivano, B.; Antonio, R. Counting the zinc-proteins encoded in the human genome. J. Proteome. Res. 2006, 5, 196–201. [Google Scholar]
- Rosanna, S.; Amit, P.; Mario, P.; Abofazl, A.; Mariacarla, V.; Mauro, C.R.; Tjaard, H. Zinc Therapy in Early Alzheimer’s Disease: Safety and Potential Therapeutic Efficacy. Biomolecules 2020, 10, 1164. [Google Scholar]
- McCarrey, A.C.; An, Y.; Kitner-Triolo, M.H.; Ferrucci, L.; Resnick, S.M. Sex differences in cognitive trajectories in clinically normal older adults. Psychol. Aging 2016, 31, 166–175. [Google Scholar] [CrossRef]
- Russell, J.K.; Jones, C.K.; Newhouse, P.A. The Role of Estrogen in Brain and Cognitive Aging. Neurotherapeutics 2019, 16, 649–665. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Banks, W.A.; Farr, S.A.; La Scola, M.E.; Morley, J.E. Intravenous human interleukin-1alpha impairs memory processing in mice: Dependence on blood-brain barrier transport into posterior division of the septum. J. Pharmacol. Exp. Ther. 2001, 299, 536–541. [Google Scholar]
- Hossain, Z.; Qasem, W.A.; Friel, J.K.; Omri, A. Effects of Total Enteral Nutrition on Early Growth, Immunity, and Neuronal Development of Preterm Infants. Nutrients 2021, 13, 2755. [Google Scholar] [CrossRef]
- Fenton, T.R.; Barbara, C.; Dena, G.; Roseann, N.; Belal, A.; Misha, E.; Hay, W.W.; Angela, H.; Diane, A.; Frank, B.; et al. “Extrauterine growth restriction” and “postnatal growth failure” are misnomers for preterm infants. J. Perinatol. 2020, 40, 704–714. [Google Scholar] [CrossRef]
- Halas, E.S.; Hunt, C.D.; Eberhardt, M.J. Learning and memory disabilities in young adult rats from mildly zinc deficient dams. Physiol. Behav. 1986, 37, 451–458. [Google Scholar] [CrossRef]
- Chua Joanne, S.C.; Cowley Carina, J.; Manavis, J.; Rofe Allan, M.; Coyle, P. Prenatal exposure to lipopolysaccharide results in neurodevelopmental damage that is ameliorated by zinc in mice. Brain Behav. Immun. 2012, 26, 326–336. [Google Scholar] [CrossRef]
- Alizadeh, F.; Davoodian, N.; Kazemi, H.; Ghasemi-Kasman, M.; Shaerzadeh, F. Prenatal zinc supplementation attenuates lipopolysaccharide-induced behavioral impairments in maternal immune activation model. Behav. Brain Res. 2020, 377, 112247. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; Chong, J.X.; Piton, A.; Popp, B.; Foss, K.; Guo, H.; Harripaul, R.; Xia, K.; Scheck, J.; Aldinger, K.A.; et al. De novo and inherited variants in ZNF292 underlie a neurodevelopmental disorder with features of autism spectrum disorder. Genet. Med. 2020, 22, 538–546. [Google Scholar] [CrossRef]
- Yukti, V.; Yewon, J.; Kevin, L.; Garner, C.C.; Montgomery, J.M. In vitro zinc supplementation alters synaptic deficits caused by autism spectrum disorder-associated Shank2 point mutations in hippocampal neurons. Mol. Brain 2021, 14, 95. [Google Scholar]
- Njoud, A.; Rafah, M.; Tomoshige, K. CH-Type Zinc Finger Proteins in Brain Development, Neurodevelopmental, and Other Neuropsychiatric Disorders: Systematic Literature-Based Analysis. Front. Neurol. 2020, 11, 32. [Google Scholar]
- Maruszak, A.; Pilarski, A.; Murphy, T.; Branch, N.; Thuret, S. Hippocampal neurogenesis in Alzheimer’s disease: Is there a role for dietary modulation? Alzheimers Dis. 2014, 38, 11–38. [Google Scholar] [CrossRef]
- Ping, H.; Matthias, S.; Rena, L.; Yong, S. Deficiency of patched 1-induced Gli1 signal transduction results in astrogenesis in Swedish mutated APP transgenic mice. Hum. Mol. Genet. 2014, 23, 6512–6527. [Google Scholar]
- Gamze, A.; Negin, R.; Pelin, Y.; Gizem, K.; Kahraman, D.C.; Saatci, Ö.; Şahin, Ö.; Çetin-Atalay, R.; Muyan, M. CXXC5 as an unmethylated CpG dinucleotide binding protein contributes to estrogen-mediated cellular proliferation. Sci. Rep. 2020, 10, 5971. [Google Scholar]
- Young, C.B.; Yeol, K.I.; Hee, K.J.; Eun, L.B.; Hee, L.S.; Ra, K.A.; Sohn, M.; Won, S.S. Zinc plus cyclo-(His-Pro) promotes hippocampal neurogenesis in rats. Neuroscience 2016, 339, 634–643. [Google Scholar]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Lee, B.E.; Lee, S.H.; Kho, A.R.; Sohn, M.; Suh, S.W. Administration of Zinc plus Cyclo-(His-Pro) Increases Hippocampal Neurogenesis in Rats during the Early Phase of Streptozotocin-Induced Diabetes. Int. J. Mol. Sci. 2017, 18, 73. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014, 28, 474–481. [Google Scholar] [CrossRef]
- Lee, B.E.; Choi, B.Y.; Hong, D.K.; Kim, J.H.; Lee, S.H.; Kho, A.R.; Kim, H.; Choi, H.C.; Suh, S.W. The cancer chemotherapeutic agent paclitaxel (Taxol) reduces hippocampal neurogenesis via down-regulation of vesicular zinc. Sci. Rep. 2017, 7, 11667. [Google Scholar] [CrossRef]
- Nam, S.M.; Kim, J.W.; Kwon, H.J.; Yoo, D.Y.; Jung, H.Y.; Kim, D.W.; Hwang, I.K.; Seong, J.K.; Yoon, Y.S. Differential Effects of Low- and High-dose Zinc Supplementation on Synaptic Plasticity and Neurogenesis in the Hippocampus of Control and High-fat Diet-fed Mice. Neurochem. Res. 2017, 42, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Hong, D.K.; Jeong, J.H.; Lee, B.E.; Koh, J.Y.; Suh, S.W. Zinc transporter 3 modulates cell proliferation and neuronal differentiation in the adult hippocampus. Stem Cells 2020, 38, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- George, J. Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. Biofactors 2012, 38, 107–113. [Google Scholar]
- Prasad Ananda, S. Zinc: An antioxidant and anti-inflammatory agent: Role of zinc in degenerative disorders of aging. J. Trace Elem. Med. Biol. 2014, 28, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fan, Z.; Gao, F.; Ou, L.; Li, M.; Zhou, X.; Luo, W.; Wei, P.; Miao, F. Emodin inhibits zinc-induced neurotoxicity in neuroblastoma SH-SY5Y cells. Biosci. Rep. 2019, 39, BSR20182378. [Google Scholar] [CrossRef]
- González-Mundo, I.; Pérez-Vielma, N.M.; Gómez-López, M.; Fleury, A.; Correa-Basurto, J.; Rosales-Hernández, M.C.; Sixto-López, Y.; Martínez-Godinez, M.L.Á.; Domínguez-López, A.; Miliar-García, A. DNA methylation of the RE-1 silencing transcription factor in peripheral blood mononuclear cells and gene expression of antioxidant enzyme in patients with late-onset Alzheimer disease. Exp. Gerontol. 2020, 136, 110951. [Google Scholar] [CrossRef]
- Datki, Z.; Galik-Olah, Z.; Janosi-Mozes, E.; Szegedi, V.; Kalman, J.; Hunya, Á.G.; Fulop, L.; Tamano, H.; Takeda, A.; Adlard, P.A.; et al. Alzheimer risk factors age and female sex induce cortical Aβ aggregation by raising extracellular zinc. Mol. Psychiatry 2020, 25, 2728–2741. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, Z.; Hu, C.; Zhang, J.; Sun, X.; Rong, C.; Jia, L. Antioxidant, antibacterial and anti-aging activities of intracellular zinc polysaccharides from Grifola frondosa SH-05. Int. J. Biol. Macromol. 2017, 95, 778–787. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Troche, C.; Kim, J.; Kim, M.H.; Teran, O.Y.; Leeuwenburgh, C.; Cousins, R.J. Aging amplifies multiple phenotypic defects in mice with zinc transporter Zip14 (Slc39a14) deletion. Exp. Gerontol. 2016, 85, 88–94. [Google Scholar] [CrossRef]
- Wong, C.P.; Magnusson, K.R.; Sharpton, T.J.; Ho, E. Effects of zinc status on age-related T cell dysfunction and chronic inflammation. Biometals 2021, 34, 291–301. [Google Scholar] [CrossRef]
- Hyun-Seo, P.; Heui, Y.M.; Jae-Young, K. Role of zinc dyshomeostasis in inflammasome formation in cultured cortical cells following lipopolysaccharide or oxygen-glucose deprivation/reperfusion exposure. Neurobiol. Dis. 2020, 137, 104771. [Google Scholar]
- Yin, X.; Zhang, Y.; Wen, Y.; Yang, Y.; Chen, H. Celecoxib alleviates zinc deficiency-promoted colon tumorigenesis through suppressing inflammation. Aging 2021, 13, 8320–8334. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wen, M.; Zhao, H.; Liu, G.; Chen, X.; Tian, G.; Cai, J.; Jia, G. Effect of zinc supplementation on growth performance, intestinal development, and intestinal barrier function in Pekin ducks with lipopolysaccharide challenge. Poult. Sci. 2021, 100, 101462. [Google Scholar] [CrossRef] [PubMed]
- Thonberg, H.; Chiang, H.H.; Lilius, L.; Forsell, C.; Lindström, A.K.; Johansson, C.; Björkström, J.; Thordardottir, S.; Sleegers, K.; Van Broeckhoven, C.; et al. Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol. Commun. 2017, 5, 43. [Google Scholar] [CrossRef]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef]
- Jung, S.J.; Jung, E.S.; Ha, K.C.; Baek, H.I.; Park, Y.K.; Han, S.K.; Chae, S.W.; Lee, S.O.; Chung, Y.C. Efficacy and Safety of Sesame Oil Cake Extract on Memory Function Improvement: A 12-Week, Randomized, Double-Blind, Placebo-Controlled Pilot Study. Nutrients 2021, 13, 2606. [Google Scholar] [CrossRef]
- Tofiq, A.; Zetterberg, H.; Blennow, K.; Basun, H.; Cederholm, T.; Eriksdotter, M.; Faxén-Irving, G.; Hjorth, E.; Jernerén, F.; Schultzberg, M.; et al. Effects of Peroral Omega-3 Fatty Acid Supplementation on Cerebrospinal Fluid Biomarkers in Patients with Alzheimer’s Disease: A Randomized Controlled Trial-The OmegAD Study. J. Alzheimers Dis. 2021, 83, 1291–1301. [Google Scholar] [CrossRef]
- Babić, L.M.; Jurasović, J.; Nikolac Perković, M.; Španić, E.; Sekovanić, A.; Orct, T.; Lukinović, Š.V.; Bačić, B.K.; Kiđemet-Piskač, S.; Vogrinc, Ž.; et al. The Association of Essential Metals with APOE Genotype in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 661–672. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Caragounis, A.; Du, T.; Laughton, K.M.; Volitakis, I.; Cherny, R.A.; Sharples, R.A.; Hill, A.F.; Li, Q.X.; Masters, C.L.; et al. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J. Biol. Chem. 2008, 283, 4568–4577. [Google Scholar] [CrossRef]
- Nara, P.L.; Sindelar, D.; Penn, M.S.; Potempa, J.; Griffin, W.S.T. Porphyromonas gingivalis Outer Membrane Vesicles as the Major Driver of and Explanation for Neuropathogenesis, the Cholinergic Hypothesis, Iron Dyshomeostasis, and Salivary Lactoferrin in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 1417–1450. [Google Scholar] [CrossRef]
- Scheffer, S.; Hermkens, D.A.M.; van der Weerd, L.; Vries, H.E.; Daemen, M.J.A.P. Vascular Hypothesis of Alzheimer Disease: Topical Review of Mouse Models. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef] [PubMed]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.K.; Kosman, D.J. Is brain iron trafficking part of the physiology of the amyloid precursor protein? J. Biol. Inorg. Chem. 2019, 24, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Liu, Y.; Yuan, Y.; Cui, W.; Zheng, F.; Ma, Y.; Piao, M. Isoflurane anesthesia exacerbates learning and memory impairment in zinc-deficient APP/PS1 transgenic mice. Neuropharmacology 2016, 111, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.; Wu, F.; Dimitrov, M.; Garcia, O.G.M.; Fraering, P.C. Zinc and Copper Differentially Modulate Amyloid Precursor Protein Processing by γ-Secretase and Amyloid-β Peptide Production. J. Biol. Chem. 2017, 292, 3751–3767. [Google Scholar] [CrossRef] [PubMed]
- August, A.; Schmidt, N.; Klingler, J.; Baumkotter, F.; Lechner, M.; Klement, J.; Eggert, S.; Vargas, C.; Wild, K.; Keller, S.; et al. Copper and zinc ions govern the transdirected dimerization of APP family members in multiple ways. J. Neurochem. 2019, 151, 1471–4159. [Google Scholar] [CrossRef]
- Liang, Y.; Raven, F.; Ward, J.F.; Zhen, S.; Zhang, S.; Sun, H.; Miller, S.J.; Choi, S.H.; Tanzi, R.E.; Zhang, C. Upregulation of Alzheimer’s Disease Amyloid-β Protein Precursor in Astrocytes Both in vitro and in vivo. J. Alzheimers Dis. 2020, 76, 1071–1082. [Google Scholar] [CrossRef]
- Gotoh, N.; Saito, Y.; Hata, S.; Saito, H.; Ojima, D.; Murayama, C.; Shigeta, M.; Abe, T.; Konno, D.; Matsuzaki, F.; et al. Amyloidogenic processing of amyloid β protein precursor (APP) is enhanced in the brains of alcadein α-deficient mice. J. Biol. Chem. 2020, 295, 9650–9662. [Google Scholar] [CrossRef]
- Fu, C.X.; Dai, L.; Yuan, X.Y.; Xu, Y.J. Effects of Fish Oil Combined with Selenium and Zinc on Learning and Memory Impairment in Aging Mice and Amyloid Precursor Protein Processing. Biol. Trace Elem. Res. 2021, 199, 1855–1863. [Google Scholar] [CrossRef]
- Yoshida, K.; Gi, M.; Fujioka, M.; Teramoto, I.; Wanibuchi, H. Long-term administration of excess zinc impairs learning and memory in aged mice. J. Toxicol. Sci. 2019, 44, 681–691. [Google Scholar] [CrossRef]
- Boström, F.; Hansson, O.O.; Gerhardsson, L.; Lundh, T.; Minthon, L.; Stomrud, E.; Zetterberg, H.; Londos, E. CSF Mg and Ca as diagnostic markers for dementia with Lewy bodies. Neurobiol. Aging 2009, 30, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wu, H.; Zhao, J. Multifunctional roles of zinc in Alzheimer’s disease. Neurotoxicology 2020, 80, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Religa, D.; Strozyk, D.; Cherny, R.A.; Volitakis, I.; Haroutunian, V.; Winblad, B.; Naslund, J.; Bush, A.I. Elevated cortical zinc in Alzheimer disease. Neurology 2006, 67, 69–75. [Google Scholar] [CrossRef]
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollut. Res. Int. 2020, 27, 43542–43552. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Agrón, E.; Launer Lenore, J.; Grodstein, F.; Bernstein, P.S.; Age-Related Eye Disease Study 2 (AREDS2) Research Group. Effect of Omega-3 Fatty Acids, Lutein/Zeaxanthin, or Other Nutrient Supplementation on Cognitive Function: The AREDS2 Randomized Clinical Trial. JAMA 2015, 314, 791–801. [Google Scholar] [CrossRef]
- Seo, B.R.; Lee, S.J.; Cho, K.S.; Yoon, Y.H.; Koh, J.Y. The zinc ionophore clioquinol reverses autophagy arrest in chloroquine-treated ARPE-19 cells and in APP/mutant presenilin-1-transfected Chinese hamster ovary cells. Neurobiol. Aging 2015, 36, 3228–3238. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Figueira, A.J.; Carapeto, A.P.; Rodrigues, M.S.; Cardoso, I.; Gomes, C.M. The S100B Alarmin Is a Dual-Function Chaperone Suppressing Amyloid-β Oligomerization through Combined Zinc Chelation and Inhibition of Protein Aggregation. ACS Chem. Neurosci. 2020, 11, 2753–2760. [Google Scholar] [CrossRef]
- Hu, J.Y.; Zhang, D.L.; Liu, X.L.; Li, X.S.; Cheng, X.Q.; Chen, J.; Du, H.N.; Liang, Y. Pathological concentration of zinc dramatically accelerates abnormal aggregation of full-length human Tau and thereby significantly increases Tau toxicity in neuronal cells. Biochim. Biophys Acta Mol. Basis Dis. 2017, 1863, 414–427. [Google Scholar] [CrossRef]
- Yue, C.; Shan, Z.; Tan, Y.; Yao, C.; Liu, Y.; Liu, Q. His-Rich Domain of Selenoprotein P Ameliorates Neuropathology and Cognitive Deficits by Regulating TrkB Pathway and Zinc Homeostasis in an Alzheimer Model of Mice. ACS Chem. Neurosci. 2020, 11, 4098–4110. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cho, E.; Seo, J.W.; Hwang, J.J.; Koh, J.Y. Alteration of the cerebral zinc pool in a mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Yoshigai, E.; Ohashi, T.; Fukada, T. Zinc transporters as potential therapeutic targets: An updated review. J Pharmacol. Sci. 2022, 148, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Vilella, A.; Belletti, D.; Sauer, A.K.; Hagmeyer, S.; Sarowar, T.; Masoni, M.; Stasiak, N.; Mulvihill, J.J.E.; Ruozi, B.; Forni, F.; et al. Reduced plaque size and inflammation in the APP23 mouse model for Alzheimer’s disease after chronic application of polymeric nanoparticles for CNS targeted zinc delivery. J. Trace Elem. Med. Biol. 2018, 49, 210–221. [Google Scholar] [CrossRef]
- Blanco-Alvarez, V.M.; Soto-Rodriguez, G.; Gonzalez-Barrios, J.A.; Martinez-Fong, D.; Brambila, E.; Torres-Soto, M.; Aguilar-Peralta, A.K.; Gonzalez-Vazquez, A.; Tomás-Sanchez, C.; Limón, I.D.; et al. Prophylactic Subacute Administration of Zinc Increases CCL2, CCR2, FGF2, and IGF-1 Expression and Prevents the Long-Term Memory Loss in a Rat Model of Cerebral Hypoxia-Ischemia. Neural. Plast. 2015, 2015, 375391. [Google Scholar] [CrossRef]
- Giacconi, R.; Costarelli, L.; Malavolta, M.; Cardelli, M.; Galeazzi, R.; Piacenza, F.; Gasparini, N.; Basso, A.; Mariani, E.; Fulop, T.; et al. Effect of ZIP2 Gln/Arg/Leu (rs2234632) polymorphism on zinc homeostasis and inflammatory response following zinc supplementation. Biofactors 2015, 41, 414–423. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H. Cognitive decline due to excess synaptic Zn(2+) signaling in the hippocampus. Front. Aging Neurosci. 2014, 6, 26. [Google Scholar] [CrossRef]
- Cao, Q.; Tan, C.C.; Xu, W.; Hu, H.; Cao, X.P.; Dong, Q.; Tan, L.; Yu, J.T. The Prevalence of Dementia: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2020, 73, 1157–1166. [Google Scholar] [CrossRef]
- Babusikova, E.; Dobrota, D.; Turner, A.J.; Hu, H.; Cao, X.P.; Dong, Q.; Tan, L.; Yu, J.T. Effect of Global Brain Ischemia on Amyloid Precursor Protein Metabolism and Expression of Amyloid-Degrading Enzymes in Rat Cortex: Role in Pathogenesis of Alzheimer’s Disease. Biochemistry 2021, 86, 680–692. [Google Scholar] [CrossRef]
- Shetty, M.S.; Sharma, M.; Sajikumar, S. Chelation of hippocampal zinc enhances long-term potentiation and synaptic tagging/capture in CA1 pyramidal neurons of aged rats: Implications to aging and memory. Aging Cell 2017, 16, 136–148. [Google Scholar] [CrossRef]
- Higashi, Y.; Aratake, T.; Shimizu, T.; Shimizu, S.; Saito, M. Protective Role of Glutathione in the Hippocampus after Brain Ischemia. Int. J. Mol. Sci. 2021, 22, 7765. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, Y.-J. Effect of Obesity on Cognitive Impairment in Vascular Dementia Rat Model via BDNF-ERK-CREB Pathway. Biol. Res. Nurs. 2021, 23, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Mur, J.; McCartney, D.L.; Chasman, D.I.; Visscher, P.M.; Muniz-Terrera, G.; Cox, S.R.; Russ, T.C.; Marioni, R.E. Variation in VKORC1 Is Associated with Vascular Dementia. J. Alzheimer’s Dis. 2021, 80, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Papanastasiou, C.A.; Theochari, C.A.; Zareifopoulos, N.; Arfaras-Melainis, A.; Giannakoulas, G.; Karamitsos, T.D.; Palaiodimos, L.; Ntaios, G.; Avgerinos, K.I.; Kapogiannis, D.; et al. Atrial Fibrillation Is Associated with Cognitive Impairment, All-Cause Dementia, Vascular Dementia, and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Gen. Intern. Med. 2021, 36, 3122–3135. [Google Scholar] [CrossRef]
- Shams, M.; Martola, J.; Charidimou, A.; Granberg, T.; Ferreira, D.; Westman, E.; Wintermark, M.; Iv, M.; Larvie, M.; Kristoffersen, W.M.; et al. Cerebrospinal Fluid Metals and the Association with Cerebral Small Vessel Disease. J. Alzheimers Dis. 2020, 78, 1229–1236. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.-I.; Kato-Negishi, M. Copper as a Collaborative Partner of Zinc-Induced Neurotoxicity in the Pathogenesis of Vascular Dementia. Int. J. Mol. Sci. 2021, 22, 7242. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Kawahara, M. Copper Enhances Zinc-Induced Neurotoxicity and the Endoplasmic Reticulum Stress Response in a Neuronal Model of Vascular Dementia. Front. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef]
- Hu, Z.; Hu, K.; Wang, R.; Gu, Y.; Ouyang, W.; Zhou, J.; Wen, Y. Differentially expressed genes accompanying neurobehavioral deficits in a modified rat model of vascular dementia. Neurosci. Lett. 2021, 750, 135774. [Google Scholar] [CrossRef]
- Choi, H.; Choi, N.Y.; Park, H.H.; Lee, K.Y.; Lee, Y.J.; Koh, S.H. Sublethal Doses of Zinc Protect Rat Neural Stem Cells Against Hypoxia Through Activation of the PI3K Pathway. Stem. Cells Dev. 2019, 28, 769–780. [Google Scholar] [CrossRef]
- Kawahara, M.; Sadakane, Y.; Mizuno, K.; Kato-Negishi, M.; Tanaka, K.I. Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia. Int. J. Mol. Sci. 2020, 21, 2570. [Google Scholar] [CrossRef]
- Cui, S.Y.; Song, J.Z.; Cui, X.Y.; Hu, X.; Ma, Y.N.; Shi, Y.T.; Luo, Y.; Ge, Y.R.; Ding, H.; Ye, H.; et al. Intracerebroventricular streptozotocin-induced Alzheimer’s disease-like sleep disorders in rats: Role of the GABAergic system in the parabrachial complex. CNS Neurosci. Ther. 2018, 24, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wei, S.; Gao, F.; Gao, L.; Dang, L.; Shang, S.; Chen, C.; Huo, K.; Wang, J.; Wang, J.; et al. Sleep Disturbance is Associated with Higher Plasma Aβ Levels in Cognitively Normal Adults-A Population-Based Cross-Sectional Study. Front Aging Neurosci. 2020, 12, 615838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Liu, P.; Wei, M.; Li, Y.; Liu, J.; Ma, L.; Shang, S.; Jiang, Y.; Huo, K.; Wang, J.; et al. Chronic Sleep Restriction Induces Aβ Accumulation by Disrupting the Balance of Aβ Production and Clearance in Rats. Neurochem. Res. 2019, 44, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Rondanelli, M.; Opizzi, A.; Monteferrario, F.; Antoniello, N.; Manni, R.; Klersy, C. The effect of melatonin, magnesium, and zinc on primary insomnia in long-term care facility residents in Italy: A double-blind, placebo-controlled clinical trial. J. Am. Geriatr. Soc. 2011, 59, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Samad, N.; Yasmin, F.; Naheed, S.; Bari, A.Z.; Ayaz, M.M.; Zaman, A.; Akbar, A.; Saeed, S.; Ullah, N. Serum levels of leptin, zinc and tryptophan in obese subjects with sleep deficits. Pak. J. Pharm. Sci. 2017, 30, 1431–1438. [Google Scholar]
- Villa, C.; Ferini-Strambi, L.; Combi, R. The Synergistic Relationship between Alzheimer’s Disease and Sleep Disorders: An Update. J. Alzheimers Dis. 2015, 46, 571–580. [Google Scholar] [CrossRef]
- Leng, Y.; Blackwell, T.; Stone, K.L.; Hoang, T.D.; Redline, S.; Yaffe, K. Periodic Limb Movements in Sleep are Associated with Greater Cognitive Decline in Older Men without Dementia. Sleep 2016, 39, 1807–1810. [Google Scholar] [CrossRef]
- Yaffe, K.; Laffan, A.M.; Harrison, S.L.; Redline, S.; Spira, A.P.; Ensrud, K.E.; Ancoli-Israel, S.; Stone, K.L. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 2011, 306, 613–619. [Google Scholar] [CrossRef]
- Chen, T.-I.; Chen, M.; Yu, C. Zinc Is Indispensable in Exercise-Induced Cardioprotection against Intermittent Hypoxia-Induced Left Ventricular Function Impairment in Rats. PLoS ONE 2016, 11, e0168600. [Google Scholar] [CrossRef]
- Levenson, C.W.; Tassabehji, N.M. Role and Regulation of Copper and Zinc Transport Proteins in the Central Nervous System. In Handbook of Neurochemistry and Molecular Neurobiology; Springer: New York, NY, USA, 2007; pp. 257–284. [Google Scholar]
- Assaf, S.Y.; Shin, H.C. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984, 308, 734–736. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.-Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2014, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Smith, J.L.; Xiong, S.; Markesbery, W.R. Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage alzheimer’s disease. Neurotox. Res. 2004, 7, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Cotrim, C.A.; Russell, J.; Jarrott, J.; Martin, L.; Drew, D. A structural overview of the zinc transporters in the cation diffusion facilitator family. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 357–367. [Google Scholar] [CrossRef]
- Andrews, G.K.; Wang, H.; Dey, S.K.; Palmiter, R.D. Mousezinc transporter 1 gene provides an essential function during early embryonic development. Genesis 2003, 40, 74–81. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Findley, S.D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995, 14, 639–649. [Google Scholar] [CrossRef]
- Ohana, E.; Sekler, I.; Kaisman, T.; Kahn, N.; Cove, J.; Silverman, W.F.; Amsterdam, A.; Hershfinkel, M. Silencing of ZnT-1 expression enhances heavy metal influx and toxicity. J. Mol. Med. 2006, 84, 753–763. [Google Scholar] [CrossRef]
- Nitzan, Y.B.; Sekler, I.; Hershfinkel, M.; Moran, A.; Silverman, W.F. Postnatal regulation of ZnT-1 expression in the mouse brain. Dev. Brain Res. 2002, 137, 149–157. [Google Scholar] [CrossRef]
- Sekler, I.; Moran, A.; Hershfinkel, M.; Dori, A.; Margulis, A.; Birenzweig, N.; Nitzan, Y.; Silverman, W.F. Distribution of the zinc transporter ZnT-1 in comparison with chelatable zinc in the mouse brain. J. Comp. Neurol. 2002, 447, 201–209. [Google Scholar] [CrossRef]
- Sindreu, C.; Bayés, À.; Altafaj, X.; Pérez-Clausell, J. Zinc transporter-1 concentrates at the postsynaptic density of hippocampal synapses. Mol. Brain 2013, 7, 16. [Google Scholar] [CrossRef]
- Medvedeva, Y.V.; Lin, B.; Shuttleworth, C.W.; Weiss, J.H. Intracellular Zn2+ Accumulation Contributes to Synaptic Failure, Mitochondrial Depolarization, and Cell Death in an Acute Slice Oxygen-Glucose Deprivation Model of Ischemia. J. Neurosci. 2008, 29, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Gore, A.; Sekler, I.; Kresse, W.; Hershfinkel, M.; Hoffmann, A.; Kettenmann, H.; Moran, A. ZnT-1 expression in astroglial cells protects against zinc toxicity and slows the accumulation of intracellular zinc. Glia 2003, 48, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Smith, J.L.; Markesbery, W.R. Elevated Zinc Transporter-6 in Mild Cognitive Impairment, Alzheimer Disease, and Pick Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 489–498. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baulac, S.; LaVoie, M.J.; Kimberly, W.T.; Strahle, J.; Wolfe, M.S.; Selkoe, D.J.; Xia, W. Functional γ-secretase complex assembly in Golgi/trans-Golgi network: Interactions among presenilin, nicastrin, Aph1, Pen-2, and γ-secretase substrates. Neurobiol. Dis. 2002, 14, 194–204. [Google Scholar] [CrossRef]
- Siman, R.; Velji, J. Localization of presenilin-nicastrin complexes and γ-secretase activity to the trans-Golgi network. J. Neurochem. 2002, 84, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.; Robert, D.M.; Kari, M.K.; Rudolph, E.T. Response: Zinc and Alzheimer’s Disease. Science 1995, 268, 1921–1923. [Google Scholar] [CrossRef]
- Upmanyu, N.; Jin, J.; von der Emde, H.; Ganzella, M.; Bösche, L.; Malviya, V.N.; Zhuleku, E.; Politi, A.Z.; Ninov, M.; Silbern, I.; et al. Colocalization of different neurotransmitter transporters on synaptic vesicles is sparse except for VGLUT1 and ZnT3. Neuron 2021, 110, 1483–1497. [Google Scholar] [CrossRef]
- Minckley, T.F.; Zhang, C.; Fudge, D.H.; Dischler, A.M.; LeJeune, K.D.; Xu, H.; Qin, Y. Sub-nanomolar sensitive GZnP3 reveals TRPML1-mediated neuronal Zn2+ signals. Nat. Commun. 2019, 10, 4806. [Google Scholar] [CrossRef]
- Sanford, L.; Carpenter, M.C.; Palmer, A.E. Intracellular Zn2+ transients modulate global gene expression in dissociated rat hippocampal neurons. Sci. Rep. 2019, 9, 9411. [Google Scholar] [CrossRef]
- Kawahara, M.; Midori, K.-N.; Tanaka, K. Amyloids: Regulators of Metal Homeostasis in the Synapse. Molecules 2019, 25, 1441. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. A Proposed Mechanism for Neurodegeneration in Movement Disorders Characterized by Metal Dyshomeostasis and Oxidative Stress. Cell Chem. Biol. 2018, 25, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Sindreu, C.; Palmiter, R.D.; Storm, D.R. Zinc transporter ZnT-3 regulates pre-synaptic Erk1/2 signaling and hippocampus-dependent memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3366–3370. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.L.; Bonaventura, J.; Keighron, J.; Wright, K.M.; Marable, D.L.; Rodriguez, L.A.; Lam, S.; Carlton, M.L.; Ellis, R.J.; Jordan, C.J.; et al. Synaptic Zn2+ potentiates the effects of cocaine on striatal dopamine neurotransmission and behavior. Transl. Psychiatry 2021, 11, 570. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinctransporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Olesen, R.H.; Hyde, T.M.; Kleinman, J.E.; Smidt, K.; Rungby, J.; Larsen, A. Obesity and age-related alterations in the gene expression of zinc-transporter proteins in the human brain. Transl. Psychiatry 2016, 6, e838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Wang, X.; Zheng, Z.H.; Ren, H.; Stoltenberg, M.; Danscher, G.; Huang, L.; Rong, M.; Wang, Z.Y. Altered expression and distribution of zinc transporters in APP/PS1 transgenic mouse brain. Neurobiol. Aging 2010, 31, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Colvin, R.A.; Fontaine, C.P.; Laskowski, M.; Thomas, D. Zn2+ transporters and Zn2+ homeostasis in neurons. Eur. J. Pharmacol. 2003, 479, 171–185. [Google Scholar] [CrossRef]
- Hong, N.; Li, C.; Feng, X.; Cen, J.N. Effects of forced running exercise on cognitive function and its relation to zinc homeostasis-related gene expression in rat hippocampus. Biol. Trace Elem. Res. 2011, 142, 704–712. [Google Scholar]
- Beyer, N.; Coulson, D.T.; Heggarty, S.; Ravid, R.; Hellemans, J.; Irvine, G.B.; Johnston, J.A. Zinc transporter mRNA levels in Alzheimer’s disease postmortem brain. J. Alzheimer’s Dis. JAD 2012, 29, 863–873. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef]
- Shie, F.-S.; Leboeuf, R.C.; Jin, L.-W.; LeBoeur, R.C. Early intraneuronal Abeta deposition in the hippocampus of APP transgenic mice. Neuroreport 2003, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Xiong, S.; Markesbery, W.R.; Lovell, M.A. Altered expression of zinc transporters-4 and -6 in mild cognitive impairment, early and late Alzheimer’s disease brain. Neuroscience 2006, 140, 879–888. [Google Scholar] [CrossRef]
- Wang, P.; Pan, R.; Weaver, J.; Jia, M.; Yang, X.; Yang, T.; Liang, J.; Liu, K.J. MicroRNA-30a regulates acute cerebral ischemia-induced blood-brain barrier damage through ZnT4/zinc pathway. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2021, 41, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Manabe, Y.; Kubo, N.; Morino, N.; Yuasa, H.; Shiotsu, M.; Tsuji, T.; Sugawara, T.; Kambe, T. Early secretory pathway-resident Zn transporter proteins contribute to cellular sphingolipid metabolism through activation of sphingomyelin phosphodiesterase 1. Am. J. Physiol. Cell Physiol. 2022, 322, C948–C959. [Google Scholar] [CrossRef]
- Huang, L.; Kirschke, C.P.; Gitschier, J. Functional characterization of a novel mammalian zinc transporter, ZnT6. J. Biol. Chem. 2002, 277, 26389–26395. [Google Scholar] [CrossRef]
- Maret, W. Molecular aspects of zinc signals. In Zinc Signals in Cellular Functions and Disorders; Fukuda, T., Kambe, T., Eds.; Springer: Tokyo, Japan, 2014; pp. 7–26. [Google Scholar]
- Tamano, H.; Koike, Y.; Nakada, H.; Shakushi, Y.; Takeda, A. Significance of synaptic Zn2+ signaling in zincergic and non-zincergic synapses in the hippocampus in cognition. J. Trace. Elem. Med. Biol. 2016, 38, 93–98. [Google Scholar] [CrossRef]
- Vasak, M.; Meloni, G. Chemistry and biology of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1067–1078. [Google Scholar] [CrossRef]
- Hardyman, J.E.; Tyson, J.; Jackson, K.A.; Aldridge, C.; Cockell, S.J.; Wakeling, L.A.; Valentine, R.A.; Ford, D. Zinc sensing by metal-responsive transcription factor 1 (MTF1) controls metallothionein and ZnT1 expression to buffer the sensitivity of the transcriptome response to zinc. Metallomics 2016, 8, 337–343. [Google Scholar] [CrossRef]
- Artells, E.; Palacios, O.; Capdevila, M.; Atrian, S. Mammalian MT1 and MT2 metallothioneins differ in their metal binding abilities. Metallomics 2013, 5, 1397–1410. [Google Scholar] [CrossRef]
- Juárez-Rebollar, D.; Rios, C.; Nava-Ruíz, C.; Méndez-Armenta, M. Metallothionein in Brain Disorders. Oxidative Med. Cell. Longev. 2017, 2017, 5828056. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Murphy, M.; Garwood, C.J.; Jennings, L.; Heath, P.R.; Chambers, A.; Matthews, F.E.; Brayne, C.; Ince, P.G.; Wharton, S.B.; et al. Cognitive Function and Ageing Neuropathology Study Group. Metallothionein-I/II expression associates with the astrocyte DNA damage response and not Alzheimer-type pathology in the aging brain. Glia 2018, 66, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.R.; Schwarz, M.; Woo, E.S.; Yee, E.; Wasserloos, K.; Tran, S.; Lazo, J.S. Overexpression of metallothionein decreases sensitivity of pulmonary endothelial cells to oxidant injury. Am. J. Physiol. 1997, 273, L856. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Pi, J.; Waalkes, M.P. Metallothionein blocks oxidative DNA damage in vitro. Arch. Toxicol. 2013, 87, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Jong-Heon, K.; Nam, Y.P.; Jeon, S.M.; Han, H.S.; Suk, K. Amyloid neurotoxicity is attenuated by metallothionein: Dual mechanisms at work. J. Neurochem. 2012, 121, 751–762. [Google Scholar]
- Hidalgo, J.; Penkowa, M.; Espejo, C.; Martínez-Cáceres, E.M.; Carrasco, J.; Quintana, A.; Molinero, A.; Florit, S.; Giralt, M.; Ortega-Aznar, A. Expression of metallothionein-I, -II, and -III in Alzheimer disease and animal models of neuroinflammation. Exp. Biol. Med. 2006, 231, 1450–1458. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- McAuliffe, J.J.; Joseph, B.; Hughes, E.; Miles, L.; Vorhees, C.V. Metallothionein I,II deficient mice do not exhibit significantly worse long-term behavioral outcomes following neonatal hypoxia-ischemia: MT-I,II deficient mice have inherent behavioral impairments. Brain Res. 2008, 1190, 175–185. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Hozumi, H.; Miyoshi, K.; Sogawa, N. Protective effects of metallothionein against dopamine quinone-induced dopaminergic neurotoxicity. FEBS Lett. 2007, 581, 5003–5008. [Google Scholar] [CrossRef]
- Penkowa, M. Metallothioneins are multipurpose neuroprotectants during brain pathology. FEBS J. 2006, 273, 1857–1870. [Google Scholar] [CrossRef]
- Uchida, Y.; Takio, K.; Titani, K.; Ihara, Y.; Tomonaga, M. The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron 1991, 7, 337–347. [Google Scholar] [CrossRef]
- Tsuji, S.; Kobayashi, H.; Uchida, Y.; Ihara, Y.; Miyatake, T. Molecular cloning of human growth inhibitory factor cDNA and its down-regulation in Alzheimer’s disease. EMBO J. 1992, 11, 4843–4850. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Lukiw, W.J.; Bergeron, C.; Niznik, H.B.; Fraser, P.E. Metallothionein III is reduced in Alzheimer’s disease. Brain Res. 2001, 894, 37–45. [Google Scholar] [CrossRef]
- Hidalgo, J. Metallothioneins and brain injury: What transgenic mice tell us. Environ. Health Prev. Med. 2004, 9, 87–94. [Google Scholar] [CrossRef]
- Yoshifumi, I.; Keung, W.M. Anti-amyloid beta activity of metallothionein-III is different from its neuronal growth inhibitory activity: Structure-activity studies. Brain Res. 2003, 960, 228–234. [Google Scholar]
- Miyazaki, I.; Sogawa, C.A.; Asanuma, M.; Higashi, Y.; Tanaka, K.I.; Nakanishi, T.; Ogawa, N. Expression of metallothionein-III mRNA and its regulation by levodopa in the basal ganglia of hemi-parkinsonian rats. Neurosci. Lett. 2000, 293, 65–68. [Google Scholar] [CrossRef]
- Uchida, Y.; Gomi, F.; Masumizu, T.; Miura, Y. Growth inhibitory factor prevents neurite extension and the death of cortical neurons caused by high oxygen exposure through hydroxyl radical scavenging. J. Biol. Chem. 2002, 277, 32353–32359. [Google Scholar] [CrossRef]
- Jen, J.; Wang, Y.C. Zinc finger proteins in cancer progression. J. Biomed. Sci. 2016, 23, 53. [Google Scholar] [CrossRef]
- Wolfe, S.A.; Nekludova, L.; Pabo, C.O. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 183–212. [Google Scholar] [CrossRef]
- Lee, S.J.; Michel, S.L.J. Structural metal sites in nonclassical zinc finger proteins involved in transcriptional and translational regulation. Acc. Chem. Res. 2014, 47, 2643–2650. [Google Scholar] [CrossRef]
- Michalek, J.L.; Besold, A.N.; Michel, S.L. Cysteine and histidine shuffling: Mixing and matching cysteine and histidine residues in zinc finger proteins to afford different folds and function. Dalton Trans. 2011, 40, 12619–12632. [Google Scholar] [CrossRef] [PubMed]
- Blackshear, P.J. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 2002, 30 Pt 6, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Cavallaro, G. Minimal functional sites allow a classification of zinc sites in proteins. PLoS ONE 2011, 6, e26325. [Google Scholar] [CrossRef]
- Decaria, L.; Bertini, I.; Williams, R.J. Zinc proteomes, phylogenetics and evolution. Met. Integr. Biomet. Sci. 2010, 2, 706–709. [Google Scholar] [CrossRef]
- Brandis, J.E.P.; Zalesak, S.M.; Kane, M.A.; Michel, S. Cadmium Exchange with Zinc in the Non-Classical Zinc Finger Protein Tristetraprolin. Inorg. Chem. 2021, 60, 7697–7707. [Google Scholar] [CrossRef] [PubMed]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Farmiloe, G.; Lodewijk, G.A.; Robben, S.F.; van Bree, E.J.; Jacobs, F.M.J. Widespread correlation of KRAB zinc finger protein binding with brain-developmental gene expression patterns. Philos. Trans. R. Soc. Lond. B Biol. 2020, 375, 20190333. [Google Scholar] [CrossRef] [PubMed]
- Bu, S.; Lv, Y.; Liu, Y.; Qiao, S.; Wang, H. Zinc Finger Proteins in Neuro-Related Diseases Progression. Front. Neurosci. 2021, 15, 760567. [Google Scholar] [CrossRef]
- Bosch, D.G.; Boonstra, F.N.; Gonzaga-Jauregui, C.; Xu, M.; de Ligt, J.; Jhangiani, S.; Wiszniewski, W.; Muzny, D.M.; Yntema, H.G.; Pfundt, R.; et al. NR2F1 mutations cause optic atrophy with intellectual disability. Am. J. Hum. Genet. 2014, 94, 303–309. [Google Scholar] [CrossRef]
- Mitsui, K.; Matsumoto, A.; Ohtsuka, S.; Ohtsubo, M.; Yoshimura, A. Cloning and characterization of a novel p21(Cip1/Waf1)-interacting zinc finger protein, ciz1. Biochem. Biophys. Res. Commun. 1999, 264, 457–464. [Google Scholar] [CrossRef]
- Coverley, D.; Marr, J.; Ainscough, J. Ciz1 promotes mammalian DNA replication. J. Cell Sci. 2005, 118, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Xiao, J.; Patel, D.; LeDoux, M.S. DNA damage and neurodegenerative phenotypes in aged Ciz1 null mice. Neurobiol. Aging. 2018, 62, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cohen, M.L.; Lerner, A.J.; Yang, Y.; Herrup, K. DNA damage and cell cycle events implicate cerebellar dentate nucleus neurons as targets of Alzheimer’s disease. Mol. Neurodegener. 2010, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.R.; Santos, A.C.; Farfel, J.M.; Grinberg, L.T.; Ferretti, R.E.; Campos, A.H.; Cunha, I.W.; Begnami, M.D.; Rocha, R.M.; Carraro, D.M.; et al. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS ONE 2014, 9, e99897. [Google Scholar] [CrossRef]
- Dahmcke, C.M.; Büchmann-Møller, S.; Jensen, N.A.; Mitchelmore, C. Altered splicing in exon 8 of the DNA replication factor CIZ1 affects subnuclear distribution and is associated with Alzheimer’s disease. Mol. Cell. Neurosci. 2008, 38, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Avet-Rochex, A.; Carvajal, N.; Christoforou, C.P.; Yeung, K.; Maierbrugger, K.T.; Hobbs, C.; Lalli, G.; Cagin, U.; Plachot, C.; McNeill, H.; et al. Unkempt is negatively regulated by mTOR and uncouples neuronal differentiation from growth control. PLoS Genet. 2014, 10, e1004624. [Google Scholar] [CrossRef]
- Bateman, J.M. Mechanistic insights into the role of mTOR signaling in neuronal differentiation. Neurogenesis 2015, 2, e1058684. [Google Scholar] [CrossRef][Green Version]
- Li, N.; Liu, Q.; Xiong, Y.; Yu, J. Headcase and Unkempt Regulate Tissue Growth and Cell Cycle Progression in Response to Nutrient Restriction. Cell Rep. 2019, 26, 733–747.e3. [Google Scholar] [CrossRef]
- Maierbrugger, K.T.; Sousa-Nunes, R.; Bateman, J.M. The mTOR pathway component Unkempt regulates neural stem cell and neural progenitor cell cycle in the Drosophila central nervous system. Dev. Biol. 2020, 461, 55–65. [Google Scholar] [CrossRef]
- Vinsland, E.; Baskaran, P.; Mihaylov, S.R.; Baskaran, P.; Mihaylov, S.R.; Hobbs, C.; Wood, H.; Bouybayoune, I.; Shah, K.; Houart, C.; et al. The zinc finger/RING domain protein Unkempt regulates cognitive flexibility. Sci. Rep. 2021, 11, 16299. [Google Scholar] [CrossRef]
- Huber, K.M.; Klann, E.; Costa-Mattioli, M.; Zukin, R.S. Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J. Neurosci. 2015, 35, 13836–13842. [Google Scholar] [CrossRef] [PubMed]
- Murn, J.; Teplova, M.; Zarnack, K.; Shi, Y.; Patel, D.J. Recognition of distinct RNA motifs by the clustered CCCH zinc fingers of neuronal protein Unkempt. Nat. Struct. Mol. Biol. 2016, 23, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Halloran, J.; Hussong, S.A.; Burbank, R.; Podlutskaya, N.; Fischer, K.E.; Sloane, L.B.; Austad, S.N.; Strong, R.; Richardson, A.; Hart, M.J.; et al. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience 2012, 223, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.L.; Lu, G.L.; Chiou, L.C.; Lin, W.S.; Cheng, Y.Y.; Hsueh, T.E.; Huang, Y.C.; Hwang, N.H.; Yeh, J.W.; Liao, R.M.; et al. Serotonin receptor HTR6-mediated mTORC1 signaling regulates dietary restriction-induced memory enhancement. PLoS Biol. 2019, 17, e2007097. [Google Scholar] [CrossRef] [PubMed]
- Glatter, T.; Schittenhelm, R.B.; Rinner, O.; Roguska, K.; Wepf, A.; Jünger, M.A.; Köhler, K.; Jevtov, I.; Choi, H.; Schmidt, A.; et al. Modularity and hormone sensitivity of the Drosophila melanogaster insulin receptor/target of rapamycin interaction proteome. Mol. Syst. Biol. 2011, 7, 547. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E.; Srinivasan, R.; Khankhanian, P.; Okuda, D.T.; Nelson, S.J.; Matthews, P.M.; Hauser, S.L.; Oksenberg, J.R.; Pelletier, D. Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 2010, 133, 2603–2611. [Google Scholar] [CrossRef]
- Deans, P.J.M.; Raval, P.; Sellers, K.J.; Gatford, N.; Halai, S.; Duarte, R.; Shum, C.; Warre-Cornish, K.; Kaplun, V.E.; Cocks, G.; et al. Psychosis Risk Candidate ZNF804A Localizes to Synapses and Regulates Neurite Formation and Dendritic Spine Structure. Biol. Psychiatry 2017, 82, 49–61. [Google Scholar] [CrossRef]
- Hess, J.L.; Quinn, T.P.; Akbarian, S.; Glatt, S.J. Bioinformatic analyses and conceptual synthesis of evidence linking ZNF804A to risk for schizophrenia and bipolar disorder. Am. J. Med. Genetics. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2015, 168B, 14–35. [Google Scholar] [CrossRef]
- Tao, R.; Cousijn, H.; Jaffe, A.E.; Burnet, P.W.; Edwards, F.; Eastwood, S.L.; Shin, J.H.; Lane, T.A.; Walker, M.A.; Maher, B.J.; et al. Expression of ZNF804A in human brain and alterations in schizophrenia, bipolar disorder, and major depressive disorder: A novel transcript fetally regulated by the psychosis risk variant rs1344706. JAMA Psychiatry 2014, 71, 1112–1120. [Google Scholar] [CrossRef]
- Chang, E.H.; Kirtley, A.; Chandon, T.S.; Borger, P.; Husain-Krautter, S.; Vingtdeux, V.; Malhotra, A.K. Postnatal neurodevelopmental expression and glutamate-dependent regulation of the ZNF804A rodent homologue. Schizophr. Res. 2015, 168, 402–410. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, F.; Lanz, T.A.; Reinhart, V.; Li, M.; Liu, L.; Zou, J.; Xi, H.S.; Mao, Y. Interactome analysis reveals ZNF804A, a schizophrenia risk gene, as a novel component of protein translational machinery critical for embryonic neurodevelopment. Mol. Psychiatry 2018, 23, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.J.; Jeffries, A.R.; Dobson, R.J.; Price, J.; Bray, N.J. Knockdown of the psychosis susceptibility gene ZNF804A alters expression of genes involved in cell adhesion. Hum. Mol. Genet. 2012, 21, 1018–1024. [Google Scholar] [CrossRef]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 2014, 71, 1323–1331. [Google Scholar] [CrossRef]
- Paternoster, V.; vanborg, M.; Edhager, A.V.; Rajkumar, A.P.; Eickhardt, E.A.; Pallesen, J.; Grove, J.; Qvist, P.; Fryland, T.; Wegener, G.; et al. Brain proteome changes in female Brd1+/- mice unmask dendritic spine pathology and show enrichment for schizophrenia risk. Neurobiol. Dis. 2019, 124, 479–488. [Google Scholar] [CrossRef]
- van Spronsen, M.; Hoogenraad, C.C. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar] [CrossRef]
- Dong, F.; Mao, J.; Chen, M.; Yoon, J.; Mao, Y. Schizophrenia risk ZNF804A interacts with its associated proteins to modulate dendritic morphology and synaptic development. Mol. Brain 2021, 14, 12. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Zhou, Q.X.; Yang, C.X.; Yang, C.P.; Mei, W.Y.; Zhang, L.; Zhang, Q.; Hu, L.; Hu, Y.Q.; et al. ZFP804A mutant mice display sex-dependent schizophrenia-like behaviors. Mol. Psychiatry 2021, 26, 2514–2532. [Google Scholar] [CrossRef]
- Becker, J.; Czamara, D.; Hoffmann, P.; Landerl, K.; Blomert, L.; Brandeis, D.; Vaessen, A.; Maurer, U.; Moll, K.; Ludwig, K.U.; et al. Evidence for the involvement of ZNF804A in cognitive processes of relevance to reading and spelling. Transl. Psychiatry 2012, 2, e136. [Google Scholar] [CrossRef]
- Cocchella, A.; Malacarne, M.; Forzano, F.; Marciano, C.; Pierluigi, M.; Perroni, L.; Faravelli, F.; Di Maria, E. The refinement of the critical region for the 2q31.2q32.3 deletion syndrome indicates candidate genes for mental retardation and speech impairment. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2010, 153B, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Vellutino, F.R.; Fletcher, J.M.; Snowling, M.J.; Scanlon, D.M. Specific reading disability (dyslexia): What have we learned in the past four decades? J. Child Psychol. Psychiatry Allied Discip. 2004, 45, 2–40. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Pazos, R. Interplay Between Autophagy and Zinc. J. Trace Elem. Med. Biol. 2020, 62, 126636. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Rubinsztein Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Truong-Tran, A.Q.; Carter, J.; Ruffin, R.E.; Zalewski, P.D. The role of zinc in caspase activation and apoptotic cell death. Biometals 2001, 14, 315–330. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, Y.; Schoene, N.W.; Han, C.T.; Shih, R.S.; Lei, K.Y. Zinc deficiency depresses p21 gene expression: Inhibition of cell cycle progression is independent of the decrease in p21 protein level in HepG2 cells. Am. J. Physiol. Cell Physiol. 2007, 292, C2175–C2184. [Google Scholar] [CrossRef]
- Lee, S.J.; Koh, J.Y. Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain 2010, 3, 30. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Yoo, C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol. Trace Elem. Res. 2013, 156, 350–356. [Google Scholar] [CrossRef]
- Hwang, J.J.; Kim, H.N.; Kim, J.; Cho, D.H.; Kim, M.J.; Kim, Y.S.; Kim, Y.; Park, S.J.; Koh, J.Y. Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals 2010, 23, 997–1013. [Google Scholar] [CrossRef]
- Hung, H.H.; Huang, W.P.; Pan, C.Y. Dopamine- and zinc-induced autophagosome formation facilitates PC12 cell survival. Cell Biol. Toxicol. 2013, 29, 415–429. [Google Scholar] [CrossRef]
- Sánchez, C.; Sánchez, M.; López-Jurado, E.; Planells, J.; Llopis, P. Aranda Assessment of iron and Zn intake and related biochemical parameters in an adult Mediterranean population from southern Spain: Influence of lifestyle factors. J. Nutr. Biochem. 2009, 20, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. Regulation of the aging process by autophagy. Trends Mol. Med. 2009, 15, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Moloudizargari, M.; Asghari, M.H.; Ghobadi, E.; Fallah, M.; Rasouli, S.; Abdollahi, M. Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res. Rev. 2017, 40, 64–74. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Karabiyik, C.; Licitra, F.; et al. Rubinsztein Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Schafe, G.E.; Atkins, C.M.; Swank, M.W.; Bauer, E.P.; Sweatt, J.D.; LeDoux, J.E. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 8177–8187. [Google Scholar] [CrossRef]
- Perry, G.; Roder, H.; Nunomura, A.; Takeda, A.; Friedlich, A.L.; Zhu, X.; Raina, A.K.; Holbrook, N.; Siedlak, S.L.; Harris, P.L.; et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10, 2411–2415. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, H.; An, W.L.; Winblad, B.; Cowburn, R.F.; Iqbal, K.; Grundke-Iqbal, I. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain research. Mol. Brain Res. 2002, 109, 45–55. [Google Scholar] [CrossRef]
- Pei, J.J.; Gong, C.X.; An, W.L.; Winblad, B.; Cowburn, R.F.; Grundke-Iqbal, I.; Iqbal, K. Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 845–858. [Google Scholar] [CrossRef]
- Bian, X.; Teng, T.; Zhao, H.; Qin, J.; Qiao, Z.; Sun, Y.; Liun, Z.; Xu, Z. Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells. Free Radic. Res. 2018, 52, 80–91. [Google Scholar] [CrossRef]
- Zhu, B.; Zhou, Y.; Xu, F.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 induces autophagy via the AMPK/ERK/TSC2/mTOR signaling pathway in PK-15 cells. J. Virol. 2012, 86, 12003–12012. [Google Scholar] [CrossRef]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, J.R.; Oteiza, P.I. Zinc and the ERK kinases in the developing brain. Neurotox. Res. 2012, 21, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Botti, J.; Djavaheri-Mergny, M.; Pilatte, Y.; Codogno, P. Autophagy signaling and the cogwheels of cancer. Autophagy 2006, 2, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, R.; Guttieri, L.; Gilardini Montani, M.S.; Santarelli, R.; Bassetti, E.; D’Orazi, G.; Cirone, M. Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation. Biology 2022, 11, 132. [Google Scholar] [CrossRef]
- Lichtlen, P.; Schaffner, W. The metal transcription factor MTF-1: Biological facts and medical implications. Swiss Med. Wkly. 2001, 131, 647–652. [Google Scholar]
- Xiao, W.; Wang, J.; Wang, X.; Cai, S.; Guo, Y.; Ye, L.; Li, D.; Hu, A.; Jin, S.; Yuan, B.; et al. Therapeutic targeting of the USP2-E2F4 axis inhibits autophagic machinery essential for zinc homeostasis in cancer progression. Autophagy 2022, 1–21. [Google Scholar] [CrossRef]
- Ramnarine, T.J.S.; Grath, S.; Parsch, J. Natural variation in the transcriptional response of Drosophila melanogaster to oxidative stress. G3 2022, 12, jkab366. [Google Scholar] [CrossRef]
- Actis, C.; Muzio, G.; Autelli, R. Autophagy Triggers Tamoxifen Resistance in Human Breast Cancer Cells by Preventing Drug-Induced Lysosomal Damage. Cancers 2021, 13, 1252. [Google Scholar] [CrossRef]
- Cho, Y.H.; Lee, S.H.; Lee, S.J.; Kim, H.N.; Koh, J.Y. A role of metallothionein-3 in radiation-induced autophagy in glioma cells. Sci. Rep. 2020, 10, 2015. [Google Scholar] [CrossRef]
- Sharif, R.; Thomas, P.; Zalewski, P.; Fenech, M. The role of zinc in genomic stability. Mutat. Res. 2012, 733, 111–112. [Google Scholar] [CrossRef]
- Meiser, N.; Mench, N.; Hengesbach, M. RNA secondary structure dependence in METTL3-METTL14 mRNA methylation is modulated by the N-terminal domain of METTL3. Biol. Chem. 2020, 402, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Chen, Y.S.; Ping, X.L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jin, X.; Nie, Q.; Chen, M.; Guo, W.; Chen, L.; Li, Y.; Chen, X.; Zhang, W.; Chen, H.; et al. YTHDF3 facilitates triple-negative breast cancer progression and metastasis by stabilizing ZEB1 mRNA in an m6A-dependent manner. Ann. Transl. Med. 2022, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Bodi, Z.; Mackinnon, K.; Zhong, S.; Archer, N.; Mongan, N.P.; Simpson, G.G.; Fray, R.G. Two zinc finger proteins with functions in m6A writing interact with HAKAI. Nat. Commun. 2022, 13, 1127. [Google Scholar] [CrossRef]
- Bodi, Z.; Zhong, S.; Mehra, S.; Song, J.; Graham, N.; Li, H.; May, S.; Fray, R.G. Adenosine Methylation in Arabidopsis mRNA is Associated with the 3’ End and Reduced Levels Cause Developmental Defects. Front. Plant Sci. 2012, 3, 48. [Google Scholar] [CrossRef]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef]
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. TIG 2008, 24, 604–612. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Li, C. Autophagy plays a positive role in zinc-induced apoptosis in intestinal porcine epithelial cells. Toxicol. Vitro 2017, 44, 392–402. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, S.J.; Byun, H.R.; Kim, Y.; Oh, Y.J.; Koh, J.J. Hwang Clioquinol induces autophagy in cultured astrocytes and neurons by acting as a zinc ionophore. Neurobiol. Dis. 2011, 42, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Speirs, C.K.; Jung, D.K.; Lu, B. The zinc ionophore PCI-5002 radiosensitizes non-small cell lung cancer cells by enhancing autophagic cell death. J. Thorac. Oncol. 2011, 6, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Yoon, Y.H.; Choi, J.A.; Lee, S.J.; Koh, J.Y. Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5344–5353. [Google Scholar] [CrossRef]
- Lee, S.J.; Cho, K.S.; Koh, J.Y. Oxidative injury triggers autophagy in astrocytes: The role of endogenous zinc. Glia 2009, 57, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Li, Y.; Shen, X.; Wang, Z.; Zhang, K.; Zhang, J.; Mei, X. Protective Effects of Zinc on Spinal Cord Injury. J. Mol. Neurosci. MN 2021, 71, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Juan, S.M.A.; Adlard, P.A. Ageing and Cognition. Subcell. Biochem. 2019, 91, 107–122. [Google Scholar]
- Paoletti, P.; Vergnano, A.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef]
- Lee, J.Y.; Friedman, J.E.; Angel, I.; Kozak, A.; Koh, J.Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol. Aging 2004, 25, 1315–1321. [Google Scholar] [CrossRef]
- Ding, W.Q.; Yu, H.J.; Lind, S.E. Zinc-binding compounds induce cancer cell death via distinct modes of action. Cancer Lett. 2008, 271, 251–259. [Google Scholar] [CrossRef]
- Colvin, R.A.; Bush, A.I.; Volitakis, I.; Fontaine, C.P.; Thomas, D.; Kikuchi, K.; Holmes, W.R. Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Cell Physiol. 2008, 294, C726–C742. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef]
- Linkous, D.H.; Flinn, J.M.; Koh, J.Y.; Lanzirotti, A.; Bertsch, P.M.; Jones, B.F.; Giblin, L.J.; Frederickson, C.J. Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J. Histochem. Cytochem. 2008, 56, 3–6. [Google Scholar] [CrossRef]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Sedjahtera, A.; Gunawan, L.; Bray, L.; Hare, D.; Lear, J.; Doble, P.; Bush, A.I.; Finkelstein, D.I.; Cherny, R.A. A novel approach to rapidly prevent age-related cognitive decline. Aging Cell 2014, 13, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.; Lal, V.; James, S.; Hare, D.; Doble, P.; Finkelstein, D.I.; Bush, A.I. Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol. Dis. 2015, 81, 196–202. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.A. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di, D.F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef]
- Oddo, S. The role of mTOR signaling in Alzheimer disease. Front. Biosci. 2012, 4, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.-J.; Jacques, H. mTOR-dependent signalling in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Barone, E.; Butterfield, D.A. mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic. Biol. Med. 2021, 169, 382–396. [Google Scholar] [CrossRef] [PubMed]
- An, W.-L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Tang, Z.; Bereczki, E.; Zhang, H.; Wang, S.; Li, C.; Ji, X.; Branca, R.M.; Lehtiö, J.; Guan, Z.; Filipcik, P.; et al. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: Implication for Alzheimer disease. J. Biol. Chem. 2013, 288, 15556–15570. [Google Scholar] [CrossRef]
- Lai, C.; Chen, Z.; Ding, Y.; Chen, Q.; Su, S.; Liu, H.; Ni, R.; Tang, Z. Rapamycin Attenuated Zinc-Induced Tau Phosphorylation and Oxidative Stress in Rats: Involvement of Dual mTOR/p70S6K and Nrf2/HO-1 Pathways. Front. Immunol. 2022, 13, 782434. [Google Scholar] [CrossRef]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef]
- Grossman, E. Should melatonin be used to lower blood pressure? Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2013, 36, 682–683. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. = Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3 beta (GSK-3β) signaling: Implications for Parkinson’s disease. Pharmacol. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Han, J.; Jiang, J.; Shi, S.; Ma, X.; Liu, X.; Wang, C.; Nie, X.; He, Y.; Jiang, S.; et al. The downregulation of Wnt/β-catenin signaling pathway is associated with zinc deficiency-induced proliferative deficit of C17. 2 neural stem cells. Brain Res. 2015, 1615, 61–70. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Zinc Associated Protein | Function | Reference | |
|---|---|---|---|
| Zinc transporters | ZnT-1 |
| [104,110,112,113,114,115] |
| ZnT-3 |
| [117,118,119,120,123,124,125] | |
| ZnT-4 |
| [127,128,129,131,132,133,135] | |
| ZnT-6 |
| [114,136,137] | |
| Metallothionein | MT-I/MT-II |
| [143,144,145,146,149,151,152] |
| MT-III |
| [153,154,155,159] | |
| Zinc Finger Protein | Function | Reference |
|---|---|---|
| CIZ1 |
| [174,175,176] |
| Unkempt |
| [178,179,181,188,189] |
| ZFP804A |
| [193,195,200,201,202] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Wang, J.; Feng, J.; Cao, B. Zinc in Cognitive Impairment and Aging. Biomolecules 2022, 12, 1000. https://doi.org/10.3390/biom12071000
Sun R, Wang J, Feng J, Cao B. Zinc in Cognitive Impairment and Aging. Biomolecules. 2022; 12(7):1000. https://doi.org/10.3390/biom12071000
Chicago/Turabian StyleSun, Ruize, Jue Wang, Juan Feng, and Bin Cao. 2022. "Zinc in Cognitive Impairment and Aging" Biomolecules 12, no. 7: 1000. https://doi.org/10.3390/biom12071000
APA StyleSun, R., Wang, J., Feng, J., & Cao, B. (2022). Zinc in Cognitive Impairment and Aging. Biomolecules, 12(7), 1000. https://doi.org/10.3390/biom12071000