
Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present


Abstract
1. Introduction
2. Ribonucleotide Reductase
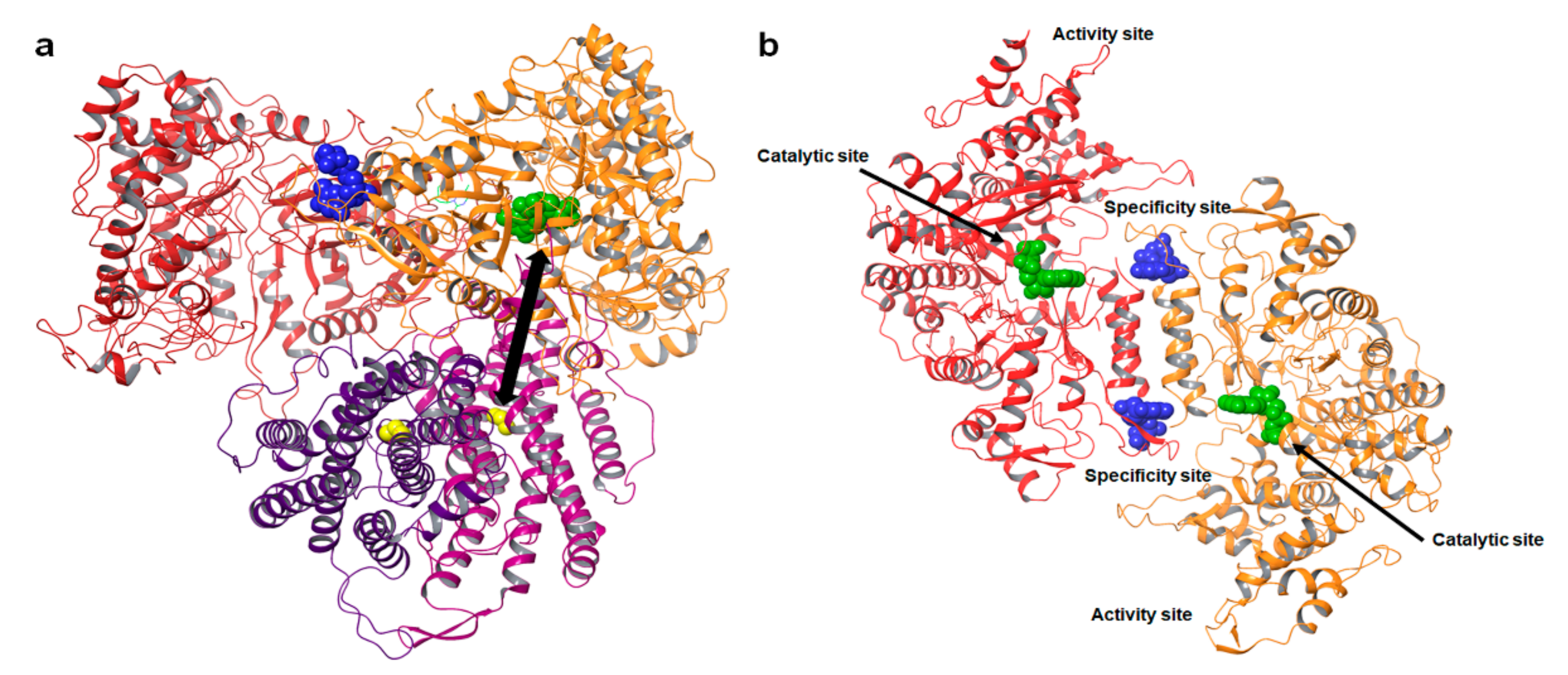
2.1. The Structure and Allosteric Regulation of Ribonucleotide Reductase
2.2. The Catalytic Mechanism of Class Ia RRs
3. Ribonucleotide Reductase as a Target for Cancer Therapeutics
3.1. The Role of RR in Cancer Biology
3.2. Cancer Chemotherapeutics Targeting hRRs
4. The Antimetabolite Class of hRRM1 Inhibitors
4.1. The Antimetabolite Class of RR Inhibitors: Cladribine (CLA) and Fludarabine (FLU)
4.2. The Antimetabolite Class of hRR Inhibitors: Clofarabine
4.3. The Antimetabolite Class of hRR Inhibitors: Gemcitabine
4.4. The Antimetabolite Class of hRR Inhibitors: Tezacitabine
4.5. Nucleoside Analog DMDC
4.6. hRRM1 Covalent Modifier Caracemide
5. Non-Nucleoside Inhibitors of hRRM2
5.1. The Radical Scavengers: Hydroxyurea
5.2. Metal Chelators: Triapine, Didox, Trimidox, Desferrioxamine, and Gallium Complexes
5.3. A Disruptor of hRRM1–hRRM2 Binding: COH29, Antisense Oligonucleotides, and siRNAs
6. Continuing Efforts to Identify Novel Inhibitors of hRR
6.1. Peptides Targeting the hRRM1 Subunit
6.2. Alkoxyphenols Targeting the hRR2 Subunit
6.3. Bivalent Inhibitors of hRR
6.4. S-Site Inhibition by the Non-Natural Nucleotide 5-NITP
6.5. Resveratrol Analog 4,4′-Trans-Dihydroxystiblene (DHS)
6.6. Modulation of the hRRM1 Oligomeric Equilibrium by OxoIsoIndoLys
6.7. NSAH and Other Acylhydrazones Targeting the C-Site of hRRM1
6.8. A Novel High Throughput PCR Assay Identifies Chemically Distinct RR Inhibitors
6.9. Transmetalative Iron Chelators That Inhibit hRRM2
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, N.C.; Canellakis, Z.N.; Lundin, B.; Reichard, P.; Thelander, L. Ribonucleoside diphosphate reductase. Purification of the two subunits, proteins B1 and B2. Eur. J. Biochem. 1969, 9, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.C.; Reichard, P. Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol. 1969, 46, 39–55. [Google Scholar] [CrossRef]
- Weinberg, G.; Ullman, B.; Martin, D.W., Jr. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. USA 1981, 78, 2447–2451. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Greene, B.L.; Kang, G.; Cui, C.; Bennati, M.; Nocera, D.G.; Drennan, C.L.; Stubbe, J. Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020, 89, 45–75. [Google Scholar] [CrossRef]
- Wijerathna, S.R.; Ahmad, M.F.; Xu, H.; Fairman, J.W.; Zhang, A.; Kaushal, P.S.; Wan, Q.; Kiser, J.; Dealwis, C.G. Targeting the Large Subunit of Human Ribonucleotide Reductase for Cancer Chemotherapy. Pharmaceuticals 2011, 4, 1328–1354. [Google Scholar] [CrossRef]
- Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Ribonucleotide reductase: A critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat. Anti-Cancer Drug Discov. 2007, 2, 11–29. [Google Scholar] [CrossRef]
- Gandhi, V.; Mineishi, S.; Huang, P.; Chapman, A.J.; Yang, Y.; Chen, F.; Nowak, B.; Chubb, S.; Hertel, L.W.; Plunkett, W. Cytotoxicity, metabolism, and mechanisms of action of 2′,2′-difluorodeoxyguanosine in Chinese hamster ovary cells. Cancer Res. 1995, 55, 1517–1524. [Google Scholar]
- Parker, W.B.; Shaddix, S.C.; Chang, C.H.; White, E.L.; Rose, L.M.; Brockman, R.W.; Shortnacy, A.T.; Montgomery, J.A.; Secrist, J.A., 3rd; Bennett, L.L., Jr. Effects of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5′-triphosphate. Cancer Res. 1991, 51, 2386–2394. [Google Scholar]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: A mechanism of self-potentiation. Cancer Res. 1992, 52, 533–539. [Google Scholar]
- Baker, C.H.; Banzon, J.; Bollinger, J.M.; Stubbe, J.; Samano, V.; Robins, M.J.; Lippert, B.; Jarvi, E.; Resvick, R. 2′-Deoxy-2′-methylenecytidine and 2′-deoxy-2′,2′-difluorocytidine 5′-diphosphates: Potent mechanism-based inhibitors of ribonucleotide reductase. J. Med. Chem. 1991, 34, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Motoi, F.; Kosuge, T.; Ueno, H.; Yamaue, H.; Satoi, S.; Sho, M.; Honda, G.; Matsumoto, I.; Wada, K.; Furuse, J.; et al. Randomized phase II/III trial of neoadjuvant chemotherapy with gemcitabine and S-1 versus upfront surgery for resectable pancreatic cancer (Prep-02/JSAP05). Jpn. J. Clin. Oncol. 2019, 49, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Sarvepalli, D.; Rashid, M.U.; Rahman, A.U.; Ullah, W.; Hussain, I.; Hasan, B.; Jehanzeb, S.; Khan, A.K.; Jain, A.G.; Khetpal, N.; et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit. Rev. Oncog. 2019, 24, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Honeywell, R.J.; Ruiz van Haperen, V.W.; Veerman, G.; Smid, K.; Peters, G.J. Inhibition of thymidylate synthase by 2′,2′-difluoro-2′-deoxycytidine (Gemcitabine) and its metabolite 2′,2′-difluoro-2′-deoxyuridine. Int. J. Biochem. Cell Biol. 2015, 60, 73–81. [Google Scholar] [CrossRef]
- Heinemann, V.; Plunkett, W. Modulation of deoxynucleotide metabolism by the deoxycytidylate deaminase inhibitor 3,4,5,6-tetrahydrodeoxyuridine. Biochem. Pharmacol. 1989, 38, 4115–4121. [Google Scholar] [CrossRef]
- Pourquier, P.; Gioffre, C.; Kohlhagen, G.; Urasaki, Y.; Goldwasser, F.; Hertel, L.W.; Yu, S.; Pon, R.T.; Gmeiner, W.H.; Pommier, Y. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin. Cancer Res.. 2002, 8, 2499–2504. [Google Scholar]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Tang, S.C.; Chen, Y.C. Novel therapeutic targets for pancreatic cancer. World J. Gastroenterol. 2014, 20, 10825–10844. [Google Scholar] [CrossRef]
- Mannargudi, M.B.; Deb, S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: Is it a viable cancer therapy? J. Cancer Res. Clin. Oncol. 2017, 143, 1499–1529. [Google Scholar] [CrossRef]
- Shao, J.; Zhou, B.; Chu, B.; Yen, Y. Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets 2006, 6, 409–431. [Google Scholar] [CrossRef]
- Pontarin, G.; Ferraro, P.; Rampazzo, C.; Kollberg, G.; Holme, E.; Reichard, P.; Bianchi, V. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J. Biol. Chem. 2011, 286, 11132–11140. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, J. Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 2003, 7, 183–188. [Google Scholar] [CrossRef]
- Srinivas, V.; Lebrette, H.; Lundin, D.; Kutin, Y.; Sahlin, M.; Lerche, M.; Eirich, J.; Branca, R.M.M.; Cox, N.; Sjöberg, B.M.; et al. Metal-free ribonucleotide reduction powered by a DOPA radical in Mycoplasma pathogens. Nature 2018, 563, 416–420. [Google Scholar] [CrossRef]
- Blaesi, E.J.; Palowitch, G.M.; Hu, K.; Kim, A.J.; Rose, H.R.; Alapati, R.; Lougee, M.G.; Kim, H.J.; Taguchi, A.T.; Tan, K.O.; et al. Metal-free class Ie ribonucleotide reductase from pathogens initiates catalysis with a tyrosine-derived dihydroxyphenylalanine radical. Proc. Natl. Acad. Sci. USA 2018, 115, 10022–10027. [Google Scholar] [CrossRef] [PubMed]
- Grāve, K.; Griese, J.J.; Berggren, G.; Bennett, M.D.; Högbom, M. The Bacillus anthracis class Ib ribonucleotide reductase subunit NrdF intrinsically selects manganese over iron. J. Biol. Inorg. Chem. 2020, 25, 571–582. [Google Scholar] [CrossRef]
- Hofer, A.; Crona, M.; Logan, D.T.; Sjöberg, B.M. DNA building blocks: Keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 50–63. [Google Scholar] [CrossRef]
- Kashlan, O.B.; Cooperman, B.S. Comprehensive model for allosteric regulation of mammalian ribonucleotide reductase: Refinements and consequences. Biochemistry 2003, 42, 1696–1706. [Google Scholar] [CrossRef]
- Rofougaran, R.; Vodnala, M.; Hofer, A. Enzymatically active mammalian ribonucleotide reductase exists primarily as an alpha6beta2 octamer. J. Biol. Chem. 2006, 281, 27705–27711. [Google Scholar] [CrossRef]
- Fairman, J.W.; Wijerathna, S.R.; Ahmad, M.F.; Xu, H.; Nakano, R.; Jha, S.; Prendergast, J.; Welin, R.M.; Flodin, S.; Roos, A.; et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011, 18, 316–322. [Google Scholar] [CrossRef]
- Ando, N.; Li, H.; Brignole, E.J.; Thompson, S.; McLaughlin, M.I.; Page, J.E.; Asturias, F.J.; Stubbe, J.; Drennan, C.L. Allosteric Inhibition of Human Ribonucleotide Reductase by dATP Entails the Stabilization of a Hexamer. Biochemistry 2016, 55, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Taguchi, A.T.; Stubbe, J.; Drennan, C.L. Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science 2020, 368, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Brignole, E.J.; Tsai, K.L.; Chittuluru, J.; Li, H.; Aye, Y.; Penczek, P.A.; Stubbe, J.; Drennan, C.L.; Asturias, F. 3.3-A resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. eLife 2018, 7, e31502. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Uhlin, U.; Ramaswamy, S.; Ekberg, M.; Regnstrom, K.; Sjoberg, B.M.; Eklund, H. Binding of allosteric effectors to ribonucleotide reductase protein R1: Reduction of active-site cysteines promotes substrate binding. Structure 1997, 5, 1077–1092. [Google Scholar] [CrossRef]
- Larsson, K.M.; Jordan, A.; Eliasson, R.; Reichard, P.; Logan, D.T.; Nordlund, P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat. Struct. Mol. Biol. 2004, 11, 1142–1149. [Google Scholar] [CrossRef]
- Zimanyi, C.M.; Chen, P.Y.; Kang, G.; Funk, M.A.; Drennan, C.L. Mol.ecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. eLife 2016, 5, e07141. [Google Scholar] [CrossRef]
- Xu, H.; Faber, C.; Uchiki, T.; Fairman, J.W.; Racca, J.; Dealwis, C. Structures of eukaryotic ribonucleotide reductase I provide insights into dNTP regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 4022–4027. [Google Scholar] [CrossRef]
- Rode, W.; Jarmuńa, A. [Thymidylate synthase-catalyzed reaction mechanism]. Postepy. Biochem. 2015, 61, 274–283. [Google Scholar]
- Ahmad, M.F.; Kaushal, P.S.; Wan, Q.; Wijerathna, S.R.; An, X.; Huang, M.; Dealwis, C.G. Role of arginine 293 and glutamine 288 in communication between catalytic and allosteric sites in yeast ribonucleotide reductase. J. Mol. Biol. 2012, 419, 315–329. [Google Scholar] [CrossRef]
- Minnihan, E.C.; Nocera, D.G.; Stubbe, J. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 2013, 46, 2524–2535. [Google Scholar] [CrossRef]
- Stubbe, J.; Riggs-Gelasco, P. Harnessing free radicals: Formation and function of the tyrosyl radical in ribonucleotide reductase. Trends Biochem. Sci. 1998, 23, 438–443. [Google Scholar] [CrossRef]
- Mao, S.S.; Holler, T.P.; Yu, G.X.; Bollinger, J.M., Jr.; Booker, S.; Johnston, M.I.; Stubbe, J. A model for the role of multiple cysteine residues involved in ribonucleotide reduction: Amazing and still confusing. Biochemistry 1992, 31, 9733–9743. [Google Scholar] [CrossRef] [PubMed]
- Licht, S.; Gerfen, G.J.; Stubbe, J. Thiyl radicals in ribonucleotide reductases. Science 1996, 271, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Reece, S.Y.; Hodgkiss, J.M.; Stubbe, J.; Nocera, D.G. Proton-coupled electron transfer: The mechanistic underpinning for radical transport and catalysis in biology. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, J.; Nocera, D.G.; Yee, C.S.; Chang, M.C. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem. Rev. 2003, 103, 2167–2201. [Google Scholar] [CrossRef]
- Stubbe, J. Ribonucleotide reductases: Amazing and confusing. J. Biol. Chem. 1990, 265, 5329–5332. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Elford, H.L.; Freese, M.; Passamani, E.; Morris, H.P. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J. Biol. Chem. 1970, 245, 5228–5233. [Google Scholar] [CrossRef]
- Morikawa, T.; Maeda, D.; Kume, H.; Homma, Y.; Fukayama, M. Ribonucleotide reductase M2 subunit is a novel diagnostic marker and a potential therapeutic target in bladder cancer. Histopathology 2010, 57, 885–892. [Google Scholar] [CrossRef]
- Wang, L.M.; Lu, F.F.; Zhang, S.Y.; Yao, R.Y.; Xing, X.M.; Wei, Z.M. Overexpression of catalytic subunit M2 in patients with ovarian cancer. Chin. Med. J. 2012, 125, 2151–2156. [Google Scholar]
- Morikawa, T.; Hino, R.; Uozaki, H.; Maeda, D.; Ushiku, T.; Shinozaki, A.; Sakatani, T.; Fukayama, M. Expression of ribonucleotide reductase M2 subunit in gastric cancer and effects of RRM2 inhibition in vitro. Hum. Pathol. 2010, 41, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.G.; Feng, H.; Wang, P.X.; Han, D.P.; Chen, X.H.; Zheng, M.H. Emerging roles of the ribonucleotide reductase M2 in colorectal cancer and ultraviolet-induced DNA damage repair. World J. Gastroenterol. 2012, 18, 4704–4713. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, H.; Lai, L.; Wang, X.; Loera, S.; Xue, L.; He, H.; Zhang, K.; Hu, S.; Huang, Y.; et al. Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clin. Sci. 2013, 124, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.; Salunga, R.; Tuggle, J.T.; Gaudet, J.; Enright, E.; McQuary, P.; Payette, T.; Pistone, M.; Stecker, K.; Zhang, B.M.; et al. Gene expression profiles of human breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 5974–5979. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef]
- Aird, K.M.; Li, H.; Xin, F.; Konstantinopoulos, P.A.; Zhang, R. Identification of ribonucleotide reductase M2 as a potential target for pro-senescence therapy in epithelial ovarian cancer. Cell Cycle 2014, 13, 199–207. [Google Scholar] [CrossRef]
- Uramoto, H.; Sugio, K.; Oyama, T.; Hanagiri, T.; Yasumoto, K. P53R2, p53 inducible ribonucleotide reductase gene, correlated with tumor progression of non-small cell lung cancer. AntiCancer Res. 2006, 26, 983–988. [Google Scholar] [PubMed]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene 2004, 23, 1539–1548. [Google Scholar] [CrossRef]
- Zhang, K.; Hu, S.; Wu, J.; Chen, L.; Lu, J.; Wang, X.; Liu, X.; Zhou, B.; Yen, Y. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: Implication of RRM2 in angiogenesis. Mol. Cancer 2009, 8, 11. [Google Scholar] [CrossRef]
- Gong, W.; Zhang, X.; Wu, J.; Chen, L.; Li, L.; Sun, J.; Lv, Y.; Wei, X.; Du, Y.; Jin, H.; et al. RRM1 expression and clinical outcome of gemcitabine-containing chemotherapy for advanced non-small-cell lung cancer: A meta-analysis. Lung Cancer 2012, 75, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, S.; Nakamori, S.; Tsujie, M.; Takahashi, Y.; Okami, J.; Yoshioka, S.; Yamasaki, M.; Marubashi, S.; Takemasa, I.; Miyamoto, A.; et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 2007, 120, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka, K.; Kohya, N.; Sato, K.; Kitajima, Y.; Ide, T.; Mitsuno, M.; Miyazaki, K. Ribonucleotide reductase subunit M1 is a possible chemoresistance marker to gemcitabine in biliary tract carcinoma. Oncol. Rep. 2008, 20, 279–286. [Google Scholar]
- Reynolds, C.; Obasaju, C.; Schell, M.J.; Li, X.; Zheng, Z.; Boulware, D.; Caton, J.R.; Demarco, L.C.; O’Rourke, M.A.; Shaw Wright, G.; et al. Randomized phase III trial of gemcitabine-based chemotherapy with in situ RRM1 and ERCC1 protein levels for response prediction in non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 5808–5815. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Zheng, Z.; Takeda, Y.; Kim, C.; Kittaka, N.; Kobayashi, S.; Marubashi, S.; Takemasa, I.; Nagano, H.; Dono, K.; et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene 2009, 28, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Boni, V.; Hernandez, A.; Bitarte, N.; Zarate, R.; Ponz-Sarvise, M.; Chopitea, A.; Bandres, E.; Garcia-Foncillas, J. Association of RRM1 -37A>C polymorphism with clinical outcome in colorectal cancer patients treated with gemcitabine-based chemotherapy. Eur. J. Cancer 2011, 47, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Seve, P.; Tredan, O.; Dumontet, C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol. 2011, 12, 693–702. [Google Scholar] [CrossRef]
- Ceppi, P.; Volante, M.; Novello, S.; Rapa, I.; Danenberg, K.D.; Danenberg, P.V.; Cambieri, A.; Selvaggi, G.; Saviozzi, S.; Calogero, R.; et al. ERCC1 and RRM1 gene expressions but not EGFR are predictive of shorter survival in advanced non-small-cell lung cancer treated with cisplatin and gemcitabine. Ann. Oncol. 2006, 17, 1818–1825. [Google Scholar] [CrossRef]
- Dong, X.; Hao, Y.; Wei, Y.; Yin, Q.; Du, J.; Zhao, X. Response to first-line chemotherapy in patients with non-small cell lung cancer according to RRM1 expression. PLoS ONE 2014, 9, e92320. [Google Scholar] [CrossRef]
- Bepler, G.; Sommers, K.E.; Cantor, A.; Li, X.; Sharma, A.; Williams, C.; Chiappori, A.; Haura, E.; Antonia, S.; Tanvetyanon, T.; et al. Clinical efficacy and predictive Mol.ecular markers of neoadjuvant gemcitabine and pemetrexed in resectable non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 1112–1118. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Guittet, O.; Lepoivre, M.; Galmarini, C.M.; Dumontet, C. Increased expression of the large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol. Cancer Ther. 2005, 4, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.D.; Ma, L.; Flagella, M.; Geeganage, S.; Gelbert, L.M.; Slapak, C.A. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004, 64, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.M.; Eijk, P.P.; Ruiz van Haperen, V.W.; Smid, K.; Veerman, G.; Hubeek, I.; van den Ijssel, P.; Ylstra, B.; Peters, G.J. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005, 65, 9510–9516. [Google Scholar] [CrossRef] [PubMed]
- Bepler, G.; Kusmartseva, I.; Sharma, S.; Gautam, A.; Cantor, A.; Sharma, A.; Simon, G. RRM1 modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4731–4737. [Google Scholar] [CrossRef]
- Ferrandina, G.; Mey, V.; Nannizzi, S.; Ricciardi, S.; Petrillo, M.; Ferlini, C.; Danesi, R.; Scambia, G.; Del Tacca, M. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother. Pharmacol. 2010, 65, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Itoi, T.; Sofuni, A.; Fukushima, N.; Itokawa, F.; Tsuchiya, T.; Kurihara, T.; Moriyasu, F.; Tsuchida, A.; Kasuya, K. Ribonucleotide reductase subunit M2 mRNA expression in pretreatment biopsies obtained from unresectable pancreatic carcinomas. J. Gastroenterol. 2007, 42, 389–394. [Google Scholar] [CrossRef]
- Avolio, T.M.; Lee, Y.; Feng, N.; Xiong, K.; Jin, H.; Wang, M.; Vassilakos, A.; Wright, J.; Young, A. RNA interference targeting the R2 subunit of ribonucleotide reductase inhibits growth of tumor cells in vitro and in vivo. Anticancer Drugs 2007, 18, 377–388. [Google Scholar] [CrossRef]
- Xu, X.; Page, J.L.; Surtees, J.A.; Liu, H.; Lagedrost, S.; Lu, Y.; Bronson, R.; Alani, E.; Nikitin, A.Y.; Weiss, R.S. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008, 68, 2652–2660. [Google Scholar] [CrossRef]
- Martin, K.R.; Barrett, J.C. Reactive oxygen species as double-edged swords in cellular processes: Low-dose cell signaling versus high-dose toxicity. Hum. Exp. Toxicol. 2002, 21, 71–75. [Google Scholar] [CrossRef]
- Feig, D.I.; Reid, T.M.; Loeb, L.A. Reactive oxygen species in tumorigenesis. Cancer Res. 1994, 54, 1890s–1894s. [Google Scholar]
- Lee, H.S.; Lee, N.C.; Kouprina, N.; Kim, J.H.; Kagansky, A.; Bates, S.; Trepel, J.B.; Pommier, Y.; Sackett, D.; Larionov, V. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res. 2016, 76, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.C.; Kearsey, S.E. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Stubbe, J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. USA 2011, 108, 9815–9820. [Google Scholar] [CrossRef]
- Griffig, J.; Koob, R.; Blakley, R.L. Mechanisms of inhibition of DNA synthesis by 2-chlorodeoxyadenosine in human lymphoblastic cells. Cancer Res. 1989, 49, 6923–6928. [Google Scholar]
- Gandhi, V.; Plunkett, W. Modulatory activity of 2′,2′-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res. 1990, 50, 3675–3680. [Google Scholar]
- Stubbe, J.; van der Donk, W.A. Ribonucleotide reductases: Radical enzymes with suicidal tendencies. Chem. Biol. 1995, 2, 793–801. [Google Scholar] [CrossRef]
- Plunkett, W.; Huang, P.; Gandhi, V. Preclinical characteristics of gemcitabine. Anticancer Drugs 1995, 6 (Suppl. 6), 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, G.; Thelander, L.; Graslund, A. EPR stopped-flow studies of the reaction of the tyrosyl radical of protein R2 from ribonucleotide reductase with hydroxyurea. Biochem. Biophys. Res. Commun. 1992, 188, 879–887. [Google Scholar] [CrossRef]
- Nigovic, B.; Kujundzic, N.; Sankovic, K. Electron transfer in N-hydroxyurea complexes with iron(III). Eur. J. Med. Chem. 2005, 40, 51–55. [Google Scholar] [CrossRef]
- Agrawal, K.C.; Sartorelli, A.C. The chemistry and biological activity of alpha-(N)-heterocyclic carboxaldehyde thiosemicarbazones. Prog. Med. Chem. 1978, 15, 321–356. [Google Scholar] [CrossRef]
- Cory, J.G.; Cory, A.H.; Rappa, G.; Lorico, A.; Liu, M.C.; Lin, T.S.; Sartorelli, A.C. Structure-function relationships for a new series of pyridine-2-carboxaldehyde thiosemicarbazones on ribonucleotide reductase activity and tumor cell growth in culture and in vivo. Adv. Enzyme Regul. 1995, 35, 55–68. [Google Scholar] [CrossRef]
- Cory, J.G.; Cory, A.H.; Rappa, G.; Lorico, A.; Liu, M.C.; Lin, T.S.; Sartorelli, A.C. Inhibitors of ribonucleotide reductase. Comparative effects of amino- and hydroxy-substituted pyridine-2-carboxaldehyde thiosemicarbazones. Biochem. Pharmacol. 1994, 48, 335–344. [Google Scholar] [CrossRef]
- Fritscher, J.; Artin, E.; Wnuk, S.; Bar, G.; Robblee, J.H.; Kacprzak, S.; Kaupp, M.; Griffin, R.G.; Bennati, M.; Stubbe, J. Structure of the nitrogen-centered radical formed during inactivation of E. coli ribonucleotide reductase by 2′-azido-2′-deoxyuridine-5′-diphosphate: Trapping of the 3′-ketonucleotide. J. Am. Chem. Soc. 2005, 127, 7729–7738. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.J.; Estey, E.; O’Brien, S.; Kantarjian, H.; Robertson, L.E.; Plunkett, W. Clinical experience with fludarabine in leukaemia. Drugs 1994, 47 (Suppl. 6), 39–49. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.J.; O’Brien, S.; Plunkett, W.; Robertson, L.E.; Gandhi, V.; Estey, E.; Dimopoulos, M.; Cabanillas, F.; Kemena, A.; Kantarjian, H. Fludarabine phosphate: A new active agent in hematologic malignancies. Semin. Hematol. 1994, 31, 28–39. [Google Scholar]
- Wright, S.J.; Robertson, L.E.; O’Brien, S.; Plunkett, W.; Keating, M.J. The role of fludarabine in hematological malignancies. Blood Rev. 1994, 8, 125–134. [Google Scholar] [CrossRef]
- Montefusco, E.; Fazi, F.; Cordone, I.; Ariola, C.; Nanni, M.; Spadea, A.; Spiriti, M.A.; Fenu, S.; Mandelli, F.; Petti, M.C. Mol.ecular remission following high-dose hydroxyurea and fludarabine plus cytarabine in a patient with simultaneous acute myeloid leukemia and low-grade lymphoma. Leuk. Lymphoma 2001, 40, 671–674. [Google Scholar] [CrossRef]
- Huang, P.; Chubb, S.; Plunkett, W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 1990, 265, 16617–16625. [Google Scholar] [CrossRef]
- Yang, S.W.; Huang, P.; Plunkett, W.; Becker, F.F.; Chan, J.Y. Dual mode of inhibition of purified DNA ligase I from human cells by 9-beta-D-arabinofuranosyl-2-fluoroadenine triphosphate. J. Biol. Chem. 1992, 267, 2345–2349. [Google Scholar] [CrossRef]
- Gandhi, V.; Huang, P.; Plunkett, W. Fludarabine inhibits DNA replication: A rationale for its use in the treatment of acute leukemias. Leuk. Lymphoma 1994, 14 (Suppl. 2), 3–9. [Google Scholar] [CrossRef]
- Chihara, D.; Arons, E.; Stetler-Stevenson, M.; Yuan, C.M.; Wang, H.W.; Zhou, H.; Raffeld, M.; Xi, L.; Steinberg, S.M.; Feurtado, J.; et al. Randomized Phase II Study of First-Line Cladribine With Concurrent or Delayed Rituximab in Patients With Hairy Cell Leukemia. J. Clin. Oncol. 2020, 38, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Qasrawi, A.; Bahaj, W.; Qasrawi, L.; Abughanimeh, O.; Foxworth, J.; Gaur, R. Cladribine in the remission induction of adult acute myeloid leukemia: Where do we stand? Ann. Hematol. 2019, 98, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Fidias, P.; Chabner, B.A.; Grossbard, M.L. Purine Analogs for the Treatment of Low-Grade Lymphoproliferative Disorders. Oncologist 1996, 1, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.C.; Gill, D.M. Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA Mol.ecules containing different types of strand breaks. J. Biol. Chem. 1980, 255, 10502–10508. [Google Scholar] [CrossRef]
- Wisitpitthaya, S.; Zhao, Y.; Long, M.J.; Li, M.; Fletcher, E.A.; Blessing, W.A.; Weiss, R.S.; Aye, Y. Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition. ACS Chem. Biol. 2016, 11, 2021–2032. [Google Scholar] [CrossRef]
- Faderl, S.; Gandhi, V.; Keating, M.J.; Jeha, S.; Plunkett, W.; Kantarjian, H.M. The role of clofarabine in hematologic and solid malignancies--development of a next-generation nucleoside analog. Cancer 2005, 103, 1985–1995. [Google Scholar] [CrossRef]
- Pui, C.H.; Jeha, S. Clofarabine. Nat. Rev. Drug Discov. 2005, 4, 369–370. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Jeha, S.; Gandhi, V.; Wess, M.; Faderl, S. Clofarabine: Past, present, and future. Leuk. Lymphoma 2007, 48, 1922–1930. [Google Scholar] [CrossRef]
- Salzer, W.L.; Burke, M.J.; Devidas, M.; Chen, S.; Gore, L.; Larsen, E.C.; Borowitz, M.; Wood, B.; Heerema, N.A.; Carroll, A.J.; et al. Toxicity associated with intensive postinduction therapy incorporating clofarabine in the very high-risk stratum of patients with newly diagnosed high-risk B-lymphoblastic leukemia: A report from the Children’s Oncology Group study AALL1131. Cancer 2018, 124, 1150–1159. [Google Scholar] [CrossRef]
- Aye, Y.; Brignole, E.J.; Long, M.J.; Chittuluru, J.; Drennan, C.L.; Asturias, F.J.; Stubbe, J. Clofarabine targets the large subunit (α) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol. 2012, 19, 799–805. [Google Scholar] [CrossRef]
- Genini, D.; Adachi, S.; Chao, Q.; Rose, D.W.; Carrera, C.J.; Cottam, H.B.; Carson, D.A.; Leoni, L.M. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood 2000, 96, 3537–3543. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Nowak, B.J.; Keating, M.J.; Plunkett, W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin. Cancer Res. 2001, 7, 3580–3589. [Google Scholar] [PubMed]
- Storniolo, A.M.; Enas, N.H.; Brown, C.A.; Voi, M.; Rothenberg, M.L.; Schilsky, R. An investigational new drug treatment program for patients with gemcitabine: Results for over 3000 patients with pancreatic carcinoma. Cancer 1999, 85, 1261–1268. [Google Scholar] [CrossRef]
- Vallo, S.; Michaelis, M.; Rothweiler, F.; Bartsch, G.; Gust, K.M.; Limbart, D.M.; Rodel, F.; Wezel, F.; Haferkamp, A.; Cinatl, J., Jr. Drug-Resistant Urothelial Cancer Cell Lines Display Diverse Sensitivity Profiles to Potential Second-Line Therapeutics. Transl. Oncol. 2015, 8, 210–216. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Parikh, P.; von Pawel, J.; Biesma, B.; Vansteenkiste, J.; Manegold, C.; Serwatowski, P.; Gatzemeier, U.; Digumarti, R.; Zukin, M.; et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 3543–3551. [Google Scholar] [CrossRef]
- Pfisterer, J.; Vergote, I.; Du Bois, A.; Eisenhauer, E.; Ago, O.; Ncic, C.T.G.; Eortc, G.C.G. Combination therapy with gemcitabine and carboplatin in recurrent ovarian cancer. Int. J. Gynecol. Cancer 2005, 15 (Suppl. 1), 36–41. [Google Scholar] [CrossRef]
- Roy, V.; LaPlant, B.R.; Gross, G.G.; Bane, C.L.; Palmieri, F.M.; North Central Cancer Treatment, G. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel) (Abraxane) in combination with gemcitabine in patients with metastatic breast cancer (N0531). Ann. Oncol. 2009, 20, 449–453. [Google Scholar] [CrossRef]
- Xu, H.; Faber, C.; Uchiki, T.; Racca, J.; Dealwis, C. Structures of eukaryotic ribonucleotide reductase I define gemcitabine diphosphate binding and subunit assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 4028–4033. [Google Scholar] [CrossRef]
- Wang, J.; Lohman, G.J.; Stubbe, J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. USA 2007, 104, 14324–14329. [Google Scholar] [CrossRef]
- Lohman, G.J.; Gerfen, G.J.; Stubbe, J. Inactivation of Lactobacillus leichmannii ribonucleotide reductase by 2′,2′-difluoro-2′-deoxycytidine 5′-triphosphate: Adenosylcobalamin destruction and formation of a nucleotide-based radical. Biochemistry 2010, 49, 1396–1403. [Google Scholar] [CrossRef]
- Ono, H.; Basson, M.D.; Ito, H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301–51310. [Google Scholar] [CrossRef]
- Plunkett, W.; Huang, P.; Searcy, C.E.; Gandhi, V. Gemcitabine: Preclinical pharmacology and mechanisms of action. Semin. Oncol. 1996, 23, 3–15. [Google Scholar]
- Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar] [PubMed]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates 2015, 23, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wen, J.Y.; Chen, J.; Ma, X.K.; Wu, D.H.; Chen, Z.H.; Huang, J.L. Oncogenic ADAM28 induces gemcitabine resistance and predicts a poor prognosis in pancreatic cancer. World J. Gastroenterol. 2019, 25, 5590–5603. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Guan, W.; Cao, Z.; Guo, W.; Xiong, G.; Zhao, F.; Feng, M.; Qiu, J.; Liu, Y.; Zhang, M.Q.; et al. Integrative Genomic Analysis of Gemcitabine Resistance in Pancreatic Cancer by Patient-derived Xenograft Models. Clin. Cancer Res. 2021, 27, 3383–3396. [Google Scholar] [CrossRef] [PubMed]
- Palam, L.R.; Gore, J.; Craven, K.E.; Wilson, J.L.; Korc, M. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis. 2015, 6, e1913. [Google Scholar] [CrossRef]
- Morimoto, M.; Matsuo, Y.; Koide, S.; Tsuboi, K.; Shamoto, T.; Sato, T.; Saito, K.; Takahashi, H.; Takeyama, H. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: Effect of CXCR4 antagonists. BMC Cancer 2016, 16, 305. [Google Scholar] [CrossRef]
- Long, J.; Zhang, Y.; Yu, X.; Yang, J.; LeBrun, D.G.; Chen, C.; Yao, Q.; Li, M. Overcoming drug resistance in pancreatic cancer. Expert Opin. Ther. Targets 2011, 15, 817–828. [Google Scholar] [CrossRef]
- Gu, J.; Huang, W.; Wang, X.; Zhang, J.; Tao, T.; Zheng, Y.; Liu, S.; Yang, J.; Chen, Z.S.; Cai, C.Y.; et al. Hsa-miR-3178/RhoB/PI3K/Akt, a novel signaling pathway regulates ABC transporters to reverse gemcitabine resistance in pancreatic cancer. Mol. Cancer 2022, 21, 112. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, R.K.; Leon, F.; Rachagani, S.; Rauth, S.; Nallasamy, P.; Marimuthu, S.; Shailendra, G.K.; Chhonker, Y.S.; Chugh, S.; Chirravuri, R.; et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Chen, J.; Cao, J.; Diao, F.; Huang, P. Low-intensity low-frequency ultrasound enhances the chemosensitivity of gemcitabine-resistant ASPC-1 cells via PI3K/AKT/NF-κB pathway-mediated ABC transporters. Oncol. Rep. 2020, 44, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Takahashi, N.; Takayama, T.; Goel, A. LAMC2 promotes cancer progression and gemcitabine resistance through modulation of EMT and ATP-binding cassette transporters in pancreatic ductal adenocarcinoma. Carcinogenesis 2021, 42, 546–556. [Google Scholar] [CrossRef]
- Yang, J.; Sontag, D.; Gong, Y.; Minuk, G.Y. Enhanced gemcitabine cytotoxicity with knockdown of multidrug resistance protein genes in human cholangiocarcinoma cell lines. J. Gastroenterol. Hepatol. 2021, 36, 1103–1109. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Springfeld, C.; Jäger, D.; Büchler, M.W.; Strobel, O.; Hackert, T.; Palmer, D.H.; Neoptolemos, J.P. Chemotherapy for pancreatic cancer. La Presse Med. 2019, 48, e159–e174. [Google Scholar] [CrossRef]
- Bao, K.; Li, X.; He, X.; Jian, L. Pharmacoeconomic Evaluation of Erlotinib for the Treatment of Pancreatic Cancer. Clin. Ther. 2021, 43, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine with or without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef]
- Raymond, E.; Faivre, S.; Armand, J.P. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs discussion 41-12. 2000, 60 (Suppl. 1), 15–23. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nakashima, A.; Kanazawa, J.; Yamaguchi, K.; Akinaga, S.; Tamaoki, T.; Okabe, M. Metabolism and ribonucleotide reductase inhibition of (E)-2′-deoxy-2′-(fluoromethylene)cytidine, MDL 101,731, in human cervical carcinoma HeLa S3 cells. Cancer Chemother. Pharmacol. 1998, 41, 268–274. [Google Scholar] [CrossRef]
- Skierski, J.S.; Koronkiewicz, M.; Grieb, P. Effect of FMdC on the cell cycle of some leukemia cell lines. Cytometry 1999, 37, 302–307. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Stevenson, J.P.; Gallagher, M.; Giantonio, B.; Algazy, K.M.; Sun, W.; Haller, D.G.; O’Dwyer, P.J. Dose escalation study of tezacitabine in combination with cisplatin in patients with advanced cancer. Cancer 2003, 97, 1985–1990. [Google Scholar] [CrossRef]
- Fernandes, P.A.; Ramos, M.J. Theoretical studies on the mode of inhibition of ribonucleotide reductase by 2′-substituted substrate analogues. Chemistry 2003, 9, 5916–5925. [Google Scholar] [CrossRef]
- Bitonti, A.J.; Bush, T.L.; Lewis, M.T.; Sunkara, P.S. Response of human colon and prostate tumor xenografts to (E)-2′-deoxy-2′-(fluoromethylene) cytidine, an inhibitor of ribonucleotide reductase. Anticancer Res. 1995, 15, 1179–1182. [Google Scholar]
- Masuda, N.; Negoro, S.; Takeda, K.; Takifuji, N.; Hirashima, T.; Yana, T.; Kurata, N.; Kuwabara, T.; Kobayashi, S.; Kudoh, S.; et al. Phase I and pharmacologic study of oral (E)-2′-deoxy-2′-(fluoromethylene) cytidine: On a daily x 5-day schedule. Investig. New Drugs 1998, 16, 245–254. [Google Scholar] [CrossRef]
- Seley, K.L. Tezacitabine Hoechst Marion Roussel. Curr. Opin. Investig. Drugs 2000, 1, 135–140. [Google Scholar] [PubMed]
- Kanazawa, J.; Takahashi, T.; Akinaga, S.; Tamaoki, T.; Okabe, M. The relationship between the antitumor activity and the ribonucleotide reductase inhibitory activity of (E)-2′-deoxy-2′-(fluoromethylene) cytidine, MDL 101,731. Anticancer Drugs 1998, 9, 653–657. [Google Scholar] [CrossRef]
- Zhou, Y.; Achanta, G.; Pelicano, H.; Gandhi, V.; Plunkett, W.; Huang, P. Action of (E)-2′-deoxy-2′-(fluoromethylene)cytidine on DNA metabolism: Incorporation, excision, and cellular response. Mol. Pharmacol. 2002, 61, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Belker, M.; Stoltz, M.; Peccerillo, K.M.; Lamb, L.A.; Chmael, S.E.; McKeon, A.; Clark, M.B.; Winship, J.; Marsh, J.C.; et al. A phase I study of the antimetabolite (E)-2′-fluoromethylene-2′-deoxycytidine (MDL 101,731) administered as a twice-weekly infusion. Cancer J. 2000, 6, 309–315. [Google Scholar] [PubMed]
- Rodriguez, G.I.; Jones, R.E.; Orenberg, E.K.; Stoltz, M.L.; Brooks, D.J. Phase I clinical trials of tezacitabine [(E)-2′-deoxy-2′-(fluoromethylene)cytidine] in patients with refractory solid tumors. Clin. Cancer Res. 2002, 8, 2828–2834. [Google Scholar] [PubMed]
- Bendell, J.C.; Eder, J.P.; Clark, J.W.; Fidias, P.; Lynch, T.J.; Seiden, M.V.; Ryan, D.P. Phase I dose-escalation study of tezacitabine in combination with 5-fluorouracil in patients with advanced solid tumors. Cancer 2005, 103, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Takenuki, K.; Matsuda, A.; Ueda, T.; Sasaki, T.; Fujii, A.; Yamagami, K. Design, synthesis, and antineoplastic activity of 2′-deoxy-2′-methylidenecytidine. J. Med. Chem. 1988, 31, 1063–1064. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, A.; Takenuki, K.; Tanaka, M.; Sasaki, T.; Ueda, T. Nucleosides and nucleotides. 97. Synthesis of new broad spectrum antineoplastic nucleosides, 2′-deoxy-2′-methylidenecytidine (DMDC) and its derivatives. J. Med. Chem. 1991, 34, 812–819. [Google Scholar] [CrossRef]
- Miwa, M.; Eda, H.; Ura, M.; Ouchi, K.F.; Keith, D.D.; Foley, L.H.; Ishitsuka, H. High susceptibility of human cancer xenografts with higher levels of cytidine deaminase to a 2′-deoxycytidine antimetabolite, 2′-deoxy-2′-methylidenecytidine. Clin. Cancer Res. 1998, 4, 493–497. [Google Scholar]
- Brindley, C.J.; Morrison, R.; Gordon, R.J.; Devlin, A.J.; van der Gaast, A.; Verweij, L.; Funaki, T. Clinical pharmacokinetics of 2′-deoxy-2′-methylidenecytidine (DMDC), a deoxycytidine analogue antineoplastic agent. Clin. Pharm. 2000, 38, 475–491. [Google Scholar] [CrossRef]
- Eda, H.; Ura, M.; Kaori, F.O.; Tanaka, Y.; Miwa, M.; Ishitsuka, H. The antiproliferative activity of DMDC is modulated by inhibition of cytidine deaminase. Cancer Res. 1998, 58, 1165–1169. [Google Scholar] [PubMed]
- Gemma, A.; Kudoh, S.; Fukuoka, M.; Kurita, Y.; Hasegawa, K.; Harada, M.; Mori, K.; Ariyoshi, Y.; Kurihara, M.; Furuse, K.; et al. Phase I study on DMDC. Gan To Kagaku Ryoho 1996, 23, 1799–1811. [Google Scholar] [PubMed]
- Friberg, L.E.; Brindley, C.J.; Karlsson, M.O.; Devlin, A.J. Models of schedule dependent haematological toxicity of 2′-deoxy-2′-methylidenecytidine (DMDC). Eur. J. Clin. Pharmacol. 2000, 56, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Larsen, I.K.; Cornett, C.; Karlsson, M.; Sahlin, M.; Sjöberg, B.M. Caracemide, a site-specific irreversible inhibitor of protein R1 of Escherichia coli ribonucleotide reductase. J. Biol. Chem. 1992, 267, 12627–12631. [Google Scholar] [CrossRef]
- Satyamoorthy, K.; Chitnis, M.P.; Advani, S.H. In vitro cytotoxicity of caracemide alone and in combination with hydroxyurea or iron-chelating agents in human chronic myeloid leukemia cells and murine tumors. Neoplasma 1988, 35, 27–35. [Google Scholar]
- Newman, R.A.; Farquhar, D.; Lu, K.; Meyn, R.; Moore, E.C.; Massia, S.; Korp, J.D.; Wright, J.A.; McKinney, M. Biochemical pharmacology of N-acetyl-N-(methylcarbamoyloxy)-N’-methylurea (caracemide; NSC-253272). Biochem. Pharmacol. 1986, 35, 2781–2787. [Google Scholar] [CrossRef]
- Buccafusco, J.J.; Smith, M.D. In vivo and in vitro cholinesterase inhibitor property of the antitumor agent caracemide. Res. Commun. Chem. Pathol. Pharmacol. 1990, 67, 219–227. [Google Scholar]
- Pazdur, R.; Chabot, G.G.; Baker, L.H. Phase I study and pharmacokinetics of caracemide (NSC-253272) administered as a short infusion. Investig. New Drugs 1987, 5, 365–371. [Google Scholar] [CrossRef]
- Raber, M.N.; Adams, F.; Kavanagh, J.; Legha, S.; Dimery, I.; Krakoff, I. Phase I trial of caracemide using bolus and infusion schedules. Cancer Treat. Rep. 1987, 71, 349–352. [Google Scholar]
- Belani, C.P.; Eisenberger, M.; Van Echo, D.; Hiponia, D.; Aisner, J. Phase II study of caracemide in advanced or recurrent non-small cell lung cancer. Cancer Treat. Rep. 1987, 71, 1099–1100. [Google Scholar]
- Lad, T.; Schor, J.; Mullane, M.; Carroll, R.; Chernicoff, D.; Blough, R.; Weidner, L. Phase II trial of caracemide (NSC 253272) in advanced unresectable non-small cell bronchogenic carcinoma. An Illinois Cancer Council study. Investig. New Drugs 1992, 10, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Witte, R.S.; Hsieh, P.; Elson, P.; Oken, M.M.; Trump, D.L. A phase II trial of amonafide, caracemide, and homoharringtonine in the treatment of patients with advanced renal cell cancer. Investig. New Drugs 1996, 14, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Witte, R.S.; Lipsitz, S.; Goodman, T.L.; Asbury, R.F.; Wilding, G.; Strnad, C.M.; Smith, T.J.; Haller, D.G. A phase II trial of homoharringtonine and caracemide in the treatment of patients with advanced large bowel cancer. Investig. New Drugs 1999, 17, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Donehower, R.C. An overview of the clinical experience with hydroxyurea. Semin. Oncol. 1992, 19, 11–19. [Google Scholar]
- Kennedy, B.J. The evolution of hydroxyurea therapy in chronic myelogenous leukemia. Semin. Oncol. 1992, 19, 21–26. [Google Scholar]
- Yarbro, J.W. Mechanism of action of hydroxyurea. Semin. Oncol. 1992, 19, 1–10. [Google Scholar]
- King, S.B. Nitric oxide production from hydroxyurea. Free Radic. Biol. Med. 2004, 37, 737–744. [Google Scholar] [CrossRef]
- Ho, J.A.; Pickens, C.V.; Gamcsik, M.P.; Colvin, O.M.; Ware, R.E. In vitro induction of fetal hemoglobin in human erythroid progenitor cells. Exp. Hematol. 2003, 31, 586–591. [Google Scholar] [CrossRef]
- Baliga, B.S.; Pace, B.S.; Chen, H.H.; Shah, A.K.; Yang, Y.M. Mechanism for fetal hemoglobin induction by hydroxyurea in sickle cell erythroid progenitors. Am. J. Hematol. 2000, 65, 227–233. [Google Scholar] [CrossRef]
- Zhou, B.S.; Hsu, N.Y.; Pan, B.C.; Doroshow, J.H.; Yen, Y. Overexpression of ribonucleotide reductase in transfected human KB cells increases their resistance to hydroxyurea: M2 but not M1 is sufficient to increase resistance to hydroxyurea in transfected cells. Cancer Res. 1995, 55, 1328–1333. [Google Scholar]
- Zhou, B.; Mo, X.; Liu, X.; Qiu, W.; Yen, Y. Human ribonucleotide reductase M2 subunit gene amplification and transcriptional regulation in a homogeneous staining chromosome region responsible for the mechanism of drug resistance. Cytogenet. Genome Res. 2001, 95, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Quinn, J.A.; Vredenburgh, J.J.; Sathornsumetee, S.; Friedman, A.H.; Herndon, J.E.; McLendon, R.E.; Provenzale, J.M.; Rich, J.N.; Sampson, J.H.; et al. Phase II study of imatinib mesylate and hydroxyurea for recurrent grade III malignant gliomas. J. Neurooncol. 2007, 83, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, L.J.; Rankin, C.; Carraway, H.; Albain, K.S.; Townsend, J.J.; Budd, G.T.; Kish, J.A.; Rivkin, S.E.; Blumenthal, D.T. A phase II study of cisplatin preceded by a 12-h continuous infusion of concurrent hydroxyurea and cytosine arabinoside (Ara-C) for adult patients with malignant gliomas (Southwest Oncology Group S9149). J. Neurooncol. 2008, 86, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Dresemann, G.; Weller, M.; Rosenthal, M.A.; Wedding, U.; Wagner, W.; Engel, E.; Heinrich, B.; Mayer-Steinacker, R.; Karup-Hansen, A.; Fluge, O.; et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J. Neurooncol. 2010, 96, 393–402. [Google Scholar] [CrossRef]
- Kao, J.; Genden, E.M.; Gupta, V.; Policarpio, E.L.; Burri, R.J.; Rivera, M.; Gurudutt, V.; Som, P.M.; Teng, M.; Packer, S.H. Phase 2 trial of concurrent 5-fluorouracil, hydroxyurea, cetuximab, and hyperfractionated intensity-modulated radiation therapy for locally advanced head and neck cancer. Cancer 2011, 117, 318–326. [Google Scholar] [CrossRef]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; McMahon, R.P.; Barton, F.B.; Waclawiw, M.; Eckert, S.V. Design of the multicenter study of hydroxyurea in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea. Control. Clin. Trials 1995, 16, 432–446. [Google Scholar] [CrossRef]
- Ma, B.; Goh, B.C.; Tan, E.H.; Lam, K.C.; Soo, R.; Leong, S.S.; Wang, L.Z.; Mo, F.; Chan, A.T.; Zee, B.; et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Investig. New Drugs 2008, 26, 169–173. [Google Scholar] [CrossRef]
- Shao, J.; Zhou, B.; Di Bilio, A.J.; Zhu, L.; Wang, T.; Qi, C.; Shih, J.; Yen, Y. A Ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol. Cancer Ther. 2006, 5, 586–592. [Google Scholar] [CrossRef]
- Aye, Y.; Long, M.J.C.; Stubbe, J. Mechanistic studies of semicarbazone triapine targeting human ribonucleotide reductase in vitro and in mammalian cells: Tyrosyl radical quenching not involving reactive oxygen species. J. Biol. Chem. 2012, 287, 35768–35778. [Google Scholar] [CrossRef]
- Karp, J.E.; Giles, F.J.; Gojo, I.; Morris, L.; Greer, J.; Johnson, B.; Thein, M.; Sznol, M.; Low, J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk. Res. 2008, 32, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Fracasso, P.M.; Kantarjian, H.M.; Cortes, J.E.; Brown, R.A.; Verstovsek, S.; Alvarado, Y.; Thomas, D.A.; Faderl, S.; Garcia-Manero, G.; et al. Phase I and pharmacodynamic study of Triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk. Res. 2003, 27, 1077–1083. [Google Scholar] [CrossRef]
- Gojo, I.; Tidwell, M.L.; Greer, J.; Takebe, N.; Seiter, K.; Pochron, M.F.; Johnson, B.; Sznol, M.; Karp, J.E. Phase I and pharmacokinetic study of Triapine, a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leuk. Res. 2007, 31, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Nutting, C.M.; van Herpen, C.M.; Miah, A.B.; Bhide, S.A.; Machiels, J.P.; Buter, J.; Kelly, C.; de Raucourt, D.; Harrington, K.J. Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2009, 20, 1275–1279. [Google Scholar] [CrossRef]
- Knox, J.J.; Hotte, S.J.; Kollmannsberger, C.; Winquist, E.; Fisher, B.; Eisenhauer, E.A. Phase II study of Triapine in patients with metastatic renal cell carcinoma: A trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). Investig. New Drugs 2007, 25, 471–477. [Google Scholar] [CrossRef]
- Schelman, W.R.; Morgan-Meadows, S.; Marnocha, R.; Lee, F.; Eickhoff, J.; Huang, W.; Pomplun, M.; Jiang, Z.; Alberti, D.; Kolesar, J.M.; et al. A phase I study of Triapine in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2009, 63, 1147–1156. [Google Scholar] [CrossRef]
- Yen, Y.; Margolin, K.; Doroshow, J.; Fishman, M.; Johnson, B.; Clairmont, C.; Sullivan, D.; Sznol, M. A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother. Pharmacol. 2004, 54, 331–342. [Google Scholar] [CrossRef]
- Wadler, S.; Makower, D.; Clairmont, C.; Lambert, P.; Fehn, K.; Sznol, M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J. Clin. Oncol. 2004, 22, 1553–1563. [Google Scholar] [CrossRef]
- Li, J.; Zheng, L.M.; King, I.; Doyle, T.W.; Chen, S.H. Syntheses and antitumor activities of potent inhibitors of ribonucleotide reductase: 3-amino-4-methylpyridine-2-carboxaldehyde-thiosemicarba-zone (3-AMP), 3-amino-pyridine-2-carboxaldehyde-thiosemicarbazone (3-AP) and its water-soluble prodrugs. Curr. Med. Chem. 2001, 8, 121–133. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Karp, J.E.; Blackford, A.L.; Smith, B.D.; Gojo, I.; Gore, S.D.; Levis, M.J.; Carraway, H.E.; Greer, J.M.; Ivy, S.P.; et al. A phase II trial of sequential ribonucleotide reductase inhibition in aggressive myeloproliferative neoplasms. Haematologica 2014, 99, 672–678. [Google Scholar] [CrossRef]
- Elford, H.L.; Wampler, G.L.; van’t Riet, B. New ribonucleotide reductase inhibitors with antineoplastic activity. Cancer Res. 1979, 39, 844–851. [Google Scholar]
- Elford, H.L.; Van’t Riet, B.; Wampler, G.L.; Lin, A.L.; Elford, R.M. Regulation of ribonucleotide reductase in mammalian cells by chemotherapeutic agents. Adv. Enzym. Regul. 1980, 19, 151–168. [Google Scholar] [CrossRef]
- Tihan, T.; Elford, H.L.; Cory, J.G. Studies on the mechanisms of inhibition of L1210 cell growth by 3,4-dihydroxybenzohydroxamic acid and 3,4-dihydroxybenzamidoxime. Adv. Enzym. Regul. 1991, 31, 71–83. [Google Scholar] [CrossRef]
- Szekeres, T.; Gharehbaghi, K.; Fritzer, M.; Woody, M.; Srivastava, A.; van’t Riet, B.; Jayaram, H.N.; Elford, H.L. Biochemical and antitumor activity of trimidox, a new inhibitor of ribonucleotide reductase. Cancer Chemother. Pharmacol. 1994, 34, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, T.; Vielnascher, E.; Novotny, L.; Vachalkova, A.; Fritzer, M.; Findenig, G.; Gobl, R.; Elford, H.L.; Goldenberg, H. Iron binding capacity of trimidox (3,4,5-trihydroxybenzamidoxime), a new inhibitor of the enzyme ribonucleotide reductase. Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 785–789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Szekeres, T.; Fritzer, M.; Strobl, H.; Gharehbaghi, K.; Findenig, G.; Elford, H.L.; Lhotka, C.; Schoen, H.J.; Jayaram, H.N. Synergistic growth inhibitory and differentiating effects of trimidox and tiazofurin in human promyelocytic leukemia HL-60 cells. Blood 1994, 84, 4316–4321. [Google Scholar] [CrossRef]
- Iyamu, W.E.; Adunyah, S.E.; Fasold, H.; Horiuchi, K.; Elford, H.L.; Asakura, T.; Turner, E.A. Enhancement of hemoglobin and F-cell production by targeting growth inhibition and differentiation of K562 cells with ribonucleotide reductase inhibitors (didox and trimidox) in combination with streptozotocin. Am. J. Hematol. 2000, 63, 176–183. [Google Scholar] [CrossRef]
- Horvath, Z.; Bauer, W.; Hoechtl, T.; Saiko, P.; Fritzer-Szekeres, M.; Tihan, T.; Szekeres, T. Combination chemotherapy of BCNU and Didox acts synergystically in 9L glioma cells. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1531–1535. [Google Scholar] [CrossRef]
- Horvath, Z.; Hochtl, T.; Bauer, W.; Fritzer-Szekeres, M.; Elford, H.L.; Szekeres, T.; Tihan, T. Synergistic cytotoxicity of the ribonucleotide reductase inhibitor didox (3,4-dihydroxy-benzohydroxamic acid) and the alkylating agent carmustine (BCNU) in 9L rat gliosarcoma cells and DAOY human medulloblastoma cells. Cancer Chemother. Pharmacol. 2004, 54, 139–145. [Google Scholar] [CrossRef]
- Figul, M.; Soling, A.; Dong, H.J.; Chou, T.C.; Rainov, N.G. Combined effects of temozolomide and the ribonucleotide reductase inhibitors didox and trimidox in malignant brain tumor cells. Cancer Chemother. Pharmacol. 2003, 52, 41–46. [Google Scholar] [CrossRef]
- Wakisaka, N.; Yoshizaki, T.; Raab-Traub, N.; Pagano, J.S. Ribonucleotide reductase inhibitors enhance cidofovir-induced apoptosis in EBV-positive nasopharyngeal carcinoma xenografts. Int. J. Cancer 2005, 116, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.; Carmichael, J.; Cantwell, B.M.; Elford, H.L.; Blackie, R.; Kerr, D.J.; Kaye, S.B.; Harris, A.L. A phase 1 and pharmacokinetic study of didox: A ribonucleotide reductase inhibitor. Br. J. Cancer 1988, 58, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; Cantwell, B.M.; Mannix, K.A.; Veale, D.; Elford, H.L.; Blackie, R.; Kerr, D.J.; Kaye, S.B.; Harris, A.L. A phase I and pharmacokinetic study of didox administered by 36 hour infusion. The Cancer Research Campaign Phase I/II Clinical Trials Committee. Br. J. Cancer 1990, 61, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Rubens, R.D.; Kaye, S.B.; Soukop, M.; Williams, C.J.; Brampton, M.H.; Harris, A.L. Phase II trial of didox in advanced breast cancer. Cancer Research Campaign Phase I/II Clinical Trials Committee. Br. J. Cancer 1991, 64, 1187–1188. [Google Scholar] [CrossRef][Green Version]
- Inayat, M.S.; Chendil, D.; Mohiuddin, M.; Elford, H.L.; Gallicchio, V.S.; Ahmed, M.M. Didox (a novel ribonucleotide reductase inhibitor) overcomes Bcl-2 mediated radiation resistance in prostate cancer cell line PC-3. Cancer Biol. Ther. 2002, 1, 539–545. [Google Scholar] [CrossRef]
- Fritzer-Szekeres, M.; Salamon, A.; Grusch, M.; Horvath, Z.; Hochtl, T.; Steinbrugger, R.; Jager, W.; Krupitza, G.; Elford, H.L.; Szekeres, T. Trimidox, an inhibitor of ribonucleotide reductase, synergistically enhances the inhibition of colony formation by Ara-C in HL-60 human promyelocytic leukemia cells. Biochem. Pharmacol. 2002, 64, 481–485. [Google Scholar] [CrossRef]
- Novotny, L.; Rauko, P.; Liska, J.; Elford, H.L.; Szekeres, T. Potentiation of the activity of cisplatin and cyclophosphamide by trimidox, a novel ribonucleotide reductase inhibitor, in leukemia-bearing mice. Cancer Lett. 2006, 233, 178–184. [Google Scholar] [CrossRef]
- Cooper, C.E.; Lynagh, G.R.; Hoyes, K.P.; Hider, R.C.; Cammack, R.; Porter, J.B. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J. Biol. Chem. 1996, 271, 20291–20299. [Google Scholar] [CrossRef]
- Komoto, K.; Nomoto, T.; El Muttaqien, S.; Takemoto, H.; Matsui, M.; Miura, Y.; Nishiyama, N. Iron chelation cancer therapy using hydrophilic block copolymers conjugated with deferoxamine. Cancer Sci. 2021, 112, 410–421. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.; Lin, T.M.; Chanana, S.; Xiong, M.P. Nanogel-DFO conjugates as a model to investigate pharmacokinetics, biodistribution, and iron chelation in vivo. Int. J. Pharm. 2018, 538, 79–86. [Google Scholar] [CrossRef]
- Dayani, P.N.; Bishop, M.C.; Black, K.; Zeltzer, P.M. Desferoxamine (DFO)—mediated iron chelation: Rationale for a novel approach to therapy for brain cancer. J. Neurooncol. 2004, 67, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, D.S.; Richardson, D.R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005, 57, 547–583. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Novel chelators for cancer treatment: Where are we now? Antioxid. Redox Signal. 2013, 18, 973–1006. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Kalinowski, D.S.; Lau, S.; Jansson, P.J.; Lovejoy, D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta 2009, 1790, 702–717. [Google Scholar] [CrossRef]
- Fan, L.; Iyer, J.; Zhu, S.; Frick, K.K.; Wada, R.K.; Eskenazi, A.E.; Berg, P.E.; Ikegaki, N.; Kennett, R.H.; Frantz, C.N. Inhibition of N-myc expression and induction of apoptosis by iron chelation in human neuroblastoma cells. Cancer Res. 2001, 61, 1073–1079. [Google Scholar] [PubMed]
- Zhou, T.; Ma, Y.; Kong, X.; Hider, R.C. Design of iron chelators with therapeutic application. Coord. Chem. Rev. 2012, 41, 6371–6389. [Google Scholar] [CrossRef] [PubMed]
- Donfrancesco, A.; Deb, G.; Dominici, C.; Pileggi, D.; Castello, M.A.; Helson, L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990, 50, 4929–4930. [Google Scholar] [PubMed]
- Blatt, J. Deferoxamine in children with recurrent neuroblastoma. Anticancer Res. 1994, 14, 2109–2112. [Google Scholar]
- Lang, J.; Zhao, X.; Wang, X.; Zhao, Y.; Li, Y.; Zhao, R.; Cheng, K.; Li, Y.; Han, X.; Zheng, X.; et al. Targeted Co-delivery of the Iron Chelator Deferoxamine and a HIF1alpha Inhibitor Impairs Pancreatic Tumor Growth. ACS Nano 2019, 13, 2176–2189. [Google Scholar] [CrossRef]
- Krakoff, I.H. Gallium nitrate in the treatment of cancer-related hypercalcemia. Semin. Oncol. 1991, 18, 3. [Google Scholar]
- Higashi, T.; Wakao, H.; Yamaguchi, M.; Suga, K. The relationship between Ga-67 accumulation and cell cycle in malignant tumor cells in vitro. Eur. J. Nucl. Med. 1988, 14, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Chitambar, C.R.; Narasimhan, J.; Guy, J.; Sem, D.S.; O’Brien, W.J. Inhibition of ribonucleotide reductase by gallium in murine leukemic L1210 cells. Cancer Res. 1991, 51, 6199–6201. [Google Scholar] [PubMed]
- Bernstein, L.R. Mechanisms of therapeutic activity for gallium. Pharmacol. Rev. 1998, 50, 665–682. [Google Scholar] [PubMed]
- Chitambar, C.R.; Purpi, D.P.; Woodliff, J.; Yang, M.; Wereley, J.P. Development of gallium compounds for treatment of lymphoma: Gallium maltolate, a novel hydroxypyrone gallium compound, induces apoptosis and circumvents lymphoma cell resistance to gallium nitrate. J. Pharmacol. Exp. Ther. 2007, 322, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Hashemy, S.I.; Ungerstedt, J.S.; Zahedi Avval, F.; Holmgren, A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J. Biol. Chem. 2006, 281, 10691–10697. [Google Scholar] [CrossRef]
- William, W.N., Jr.; Zinner, R.G.; Karp, D.D.; Oh, Y.W.; Glisson, B.S.; Phan, S.C.; Stewart, D.J. Phase I trial of motexafin gadolinium in combination with docetaxel and cisplatin for the treatment of non-small cell lung cancer. J. Thorac. Oncol. 2007, 2, 745–750. [Google Scholar] [CrossRef]
- Edelman, M.J.; Otterson, G.; Leach, J.; Malpass, T.; Salgia, R.; Jones, D.; Mody, T.D.; Govindan, R. Multicenter phase II trial of Motexafin gadolinium and pemetrexed for second-line treatment in patients with non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 786–789. [Google Scholar] [CrossRef]
- Evens, A.M.; Spies, W.G.; Helenowski, I.B.; Patton, D.; Spies, S.; Jovanovic, B.D.; Miyata, S.; Hamilton, E.; Variakojis, D.; Chen, J.; et al. The novel expanded porphyrin, motexafin gadolinium, combined with [90Y]ibritumomab tiuxetan for relapsed/refractory non-Hodgkin’s lymphoma: Preclinical findings and results of a phase I trial. Clin. Cancer Res. 2009, 15, 6462–6471. [Google Scholar] [CrossRef]
- Brachman, D.G.; Pugh, S.L.; Ashby, L.S.; Thomas, T.A.; Dunbar, E.M.; Narayan, S.; Robins, H.I.; Bovi, J.A.; Rockhill, J.K.; Won, M.; et al. Phase 1/2 trials of Temozolomide, Motexafin Gadolinium, and 60-Gy fractionated radiation for newly diagnosed supratentorial glioblastoma multiforme: Final results of RTOG 0513. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 961–967. [Google Scholar] [CrossRef]
- McHaffie, D.R.; Chabot, P.; Dagnault, A.; Suh, J.H.; Fortin, M.A.; Chang, E.; Timmerman, R.; Souhami, L.; Grecula, J.; Nabid, A.; et al. Safety and feasibility of motexafin gadolinium administration with whole brain radiation therapy and stereotactic radiosurgery boost in the treatment of </= 6 brain metastases: A multi-institutional phase II trial. J. Neurooncol. 2011, 105, 301–308. [Google Scholar] [CrossRef]
- Ford, J.M.; Seiferheld, W.; Alger, J.R.; Wu, G.; Endicott, T.J.; Mehta, M.; Curran, W.; Phan, S.C. Results of the phase I dose-escalating study of motexafin gadolinium with standard radiotherapy in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Pollack, I.F.; Reid, J.M.; Adamson, P.C.; Ames, M.M.; Vezina, G.; Blaney, S.; Ivy, P.; Zhou, T.; Krailo, M.; et al. Motexafin gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: A Children’s Oncology Group phase I study. Neuro-Oncology 2008, 10, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.P.; Shapiro, W.R.; Phan, S.C.; Gervais, R.; Carrie, C.; Chabot, P.; Patchell, R.A.; Glantz, M.J.; Recht, L.; Langer, C.; et al. Motexafin gadolinium combined with prompt whole brain radiotherapy prolongs time to neurologic progression in non-small-cell lung cancer patients with brain metastases: Results of a phase III trial. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.A.; Smith, J.A.; Bezjak, A.; Mehta, M.P.; Liebmann, J.; Illidge, T.; Kunkler, I.; Caudrelier, J.M.; Eisenberg, P.D.; Meerwaldt, J.; et al. Neurocognitive function and progression in patients with brain metastases treated with whole-brain radiation and motexafin gadolinium: Results of a randomized phase III trial. J. Clin. Oncol. 2004, 22, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Naumovski, L.; Lecane, P.S.; Lucas, M.S.; Moran, M.E.; Cheney, C.; Lucas, D.M.; Phan, S.C.; Miller, R.A.; Byrd, J.C. Effects of motexafin gadolinium in a phase II trial in refractory chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 1977–1982. [Google Scholar] [CrossRef]
- Amato, R.J.; Jac, J.; Hernandez-McClain, J. Motexafin gadolinium for the treatment of metastatic renal cell carcinoma: Phase II study results. Clin. Genitourin. Cancer 2008, 6, 73–78. [Google Scholar] [CrossRef]
- Zhou, B.; Su, L.; Hu, S.; Hu, W.; Yip, M.L.; Wu, J.; Gaur, S.; Smith, D.L.; Yuan, Y.C.; Synold, T.W.; et al. A small-Mol.ecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res. 2013, 73, 6484–6493. [Google Scholar] [CrossRef]
- Chen, M.C.; Zhou, B.; Zhang, K.; Yuan, Y.C.; Un, F.; Hu, S.; Chou, C.M.; Chen, C.H.; Wu, J.; Wang, Y.; et al. The Novel Ribonucleotide Reductase Inhibitor COH29 Inhibits DNA Repair In Vitro. Mol. Pharmacol. 2015, 87, 996–1005. [Google Scholar] [CrossRef]
- Orr, R.M. GTI-2040. Lorus Therapeutics. Curr. Opin. Investig. Drugs 2001, 2, 1462–1466. [Google Scholar]
- Lee, Y.; Vassilakos, A.; Feng, N.; Lam, V.; Xie, H.; Wang, M.; Jin, H.; Xiong, K.; Liu, C.; Wright, J.; et al. GTI-2040, an antisense agent targeting the small subunit component (R2) of human ribonucleotide reductase, shows potent antitumor activity against a variety of tumors. Cancer Res. 2003, 63, 2802–2811. [Google Scholar]
- Tu, G.C.; Tu, X. GTI-2501. Lorus Therapeutics. Curr. Opin. Investig. Drugs 2001, 2, 1467–1470. [Google Scholar] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.A.; Gaudreau, P.; Brazeau, P.; Langelier, Y. Specific inhibition of herpesvirus ribonucleotide reductase by a nonapeptide derived from the carboxy terminus of subunit 2. Nature 1986, 321, 441–443. [Google Scholar] [CrossRef]
- Dutia, B.M.; Frame, M.C.; Subak-Sharpe, J.H.; Clark, W.N.; Marsden, H.S. Specific inhibition of herpesvirus ribonucleotide reductase by synthetic peptides. Nature 1986, 321, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Climent, I.; Sjöberg, B.M.; Huang, C.Y. Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: Kinetic analysis of inhibition studies. Biochemistry 1991, 30, 5164–5171. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, G.; Lavallee, P.; Rakhit, S.; Plante, R.; Gaudette, Y.; Lawetz, C.; Whitehead, P.W.; Duceppe, J.S.; Lepine-Frenette, C.; Dansereau, N.; et al. Specific inhibition of ribonucleotide reductases by peptides corresponding to the C-terminal of their second subunit. Biochem. Cell Biol. 1991, 69, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Lycksell, P.O.; Ingemarson, R.; Davis, R.; Graslund, A.; Thelander, L. 1H NMR studies of mouse ribonucleotide reductase: The R2 protein carboxyl-terminal tail, essential for subunit interaction, is highly flexible but becomes rigid in the presence of protein R1. Biochemistry 1994, 33, 2838–2842. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.D.; Spanevello, R.A.; Celiker, I.; Hirschmann, R.; Rubin, H.; Cooperman, B.S. The carboxyl terminus heptapeptide of the R2 subunit of mammalian ribonucleotide reductase inhibits enzyme activity and can be used to purify the R1 subunit. FEBS Lett. 1990, 272, 61–64. [Google Scholar] [CrossRef]
- Wnuk, S.F.; Robins, M.J. Ribonucleotide reductase inhibitors as anti-herpes agents. Antiviral. Res. 2006, 71, 122–126. [Google Scholar] [CrossRef]
- Xu, H.; Fairman, J.W.; Wijerathna, S.R.; Kreischer, N.R.; LaMacchia, J.; Helmbrecht, E.; Cooperman, B.S.; Dealwis, C. The structural basis for peptidomimetic inhibition of eukaryotic ribonucleotide reductase: A conformationally flexible pharmacophore. J. Med. Chem. 2008, 51, 4653–4659. [Google Scholar] [CrossRef]
- Lassmann, G.; Potsch, S. Structure of transient radicals from cytostatic-active p-alkoxyphenols by continuous-flow EPR. Free Radic. Biol. Med. 1995, 19, 533–539. [Google Scholar] [CrossRef]
- Potsch, S.; Drechsler, H.; Liermann, B.; Graslund, A.; Lassmann, G. p-Alkoxyphenols, a new class of inhibitors of mammalian R2 ribonucleotide reductase: Possible candidates for antimelanotic drugs. Mol. Pharmacol. 1994, 45, 792–796. [Google Scholar] [PubMed]
- Potsch, S.; Sahlin, M.; Langelier, Y.; Graslund, A.; Lassmann, G. Reduction of the tyrosyl radical and the iron center in protein R2 of ribonucleotide reductase from mouse, herpes simplex virus and E. coli by p-alkoxyphenols. FEBS Lett. 1995, 374, 95–99. [Google Scholar] [CrossRef]
- Wu, X.; Cooperman, B.S. Synthesis and biological activity of a bivalent nucleotide inhibitor of ribonucleotide reductase. Bioorg. Med. Chem. Lett. 2000, 10, 2387–2389. [Google Scholar] [CrossRef]
- Petrelli, R.; Meli, M.; Vita, P.; Torquati, I.; Ferro, A.; Vodnala, M.; D’Alessandro, N.; Tolomeo, M.; Del Bello, F.; Kusumanchi, P.; et al. From the covalent linkage of drugs to novel inhibitors of ribonucleotide reductase: Synthesis and biological evaluation of valproic esters of 3′-C-methyladenosine. Bioorg. Med. Chem. Lett. 2014, 24, 5304–5309. [Google Scholar] [CrossRef]
- Ahmad, M.F.; Wan, Q.; Jha, S.; Motea, E.; Berdis, A.; Dealwis, C. Evaluating the therapeutic potential of a non-natural nucleotide that inhibits human ribonucleotide reductase. Mol. Cancer Ther. 2012, 11, 2077–2086. [Google Scholar] [CrossRef]
- Fan, G.J.; Liu, X.D.; Qian, Y.P.; Shang, Y.J.; Li, X.Z.; Dai, F.; Fang, J.G.; Jin, X.L.; Zhou, B. 4,4′-Dihydroxy-trans-stilbene, a resveratrol analogue, exhibited enhanced antioxidant activity and cytotoxicity. Bioorg. Med. Chem. 2009, 17, 2360–2365. [Google Scholar] [CrossRef]
- Maccario, C.; Savio, M.; Ferraro, D.; Bianchi, L.; Pizzala, R.; Pretali, L.; Forti, L.; Stivala, L.A. The resveratrol analog 4,4′-dihydroxy-trans-stilbene suppresses transformation in normal mouse fibroblasts and inhibits proliferation and invasion of human breast cancer cells. Carcinogenesis 2012, 33, 2172–2180. [Google Scholar] [CrossRef]
- Balan, K.V.; Wang, Y.; Chen, S.W.; Chen, J.C.; Zheng, L.F.; Yang, L.; Liu, Z.L.; Pantazis, P.; Wyche, J.H.; Han, Z. Proteasome-independent down-regulation of estrogen receptor-alpha (ERalpha) in breast cancer cells treated with 4,4′-dihydroxy-trans-stilbene. Biochem. Pharmacol. 2006, 72, 573–581. [Google Scholar] [CrossRef]
- Kimura, Y.; Sumiyoshi, M.; Baba, K. Antitumor activities of synthetic and natural stilbenes through antiangiogenic action. Cancer Sci. 2008, 99, 2083–2096. [Google Scholar] [CrossRef]
- Saha, B.; Patro, B.S.; Koli, M.; Pai, G.; Ray, J.; Bandyopadhyay, S.K.; Chattopadhyay, S. trans-4,4′-Dihydroxystilbene (DHS) inhibits human neuroblastoma tumor growth and induces mitochondrial and lysosomal damages in neuroblastoma cell lines. Oncotarget 2017, 8, 73905–73924. [Google Scholar] [CrossRef] [PubMed]
- Savio, M.; Ferraro, D.; Maccario, C.; Vaccarone, R.; Jensen, L.D.; Corana, F.; Mannucci, B.; Bianchi, L.; Cao, Y.; Stivala, L.A. Resveratrol analogue 4,4′-dihydroxy-trans-stilbene potently inhibits cancer invasion and metastasis. Sci. Rep. 2016, 6, 19973. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Li, Y.; Hu, S.; Zhou, W.; Meng, Y.; Li, Z.; Zhang, Y.; Sun, J.; Bo, Z.; DePamphilis, M.L.; et al. DHS (trans-4,4′-dihydroxystilbene) suppresses DNA replication and tumor growth by inhibiting RRM2 (ribonucleotide reductase regulatory subunit M2). Oncogene 2019, 38, 2364–2379. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.F.; Huff, S.E.; Pink, J.; Alam, I.; Zhang, A.; Perry, K.; Harris, M.E.; Misko, T.; Porwal, S.K.; Oleinick, N.L.; et al. Identification of Non-nucleoside Human Ribonucleotide Reductase Modulators. J. Med. Chem. 2015, 58, 9498–9509. [Google Scholar] [CrossRef]
- Ahmad, M.F.; Alam, I.; Huff, S.E.; Pink, J.; Flanagan, S.A.; Shewach, D.; Misko, T.A.; Oleinick, N.L.; Harte, W.E.; Viswanathan, R.; et al. Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small Mol.ecule. Proc. Natl. Acad. Sci. USA 2017, 114, 8241–8246. [Google Scholar] [CrossRef]
- Huff, S.E.; Mohammed, F.A.; Yang, M.; Agrawal, P.; Pink, J.; Harris, M.E.; Dealwis, C.G.; Viswanathan, R. Structure-Guided Synthesis and Mechanistic Studies Reveal Sweetspots on Naphthyl Salicyl Hydrazone Scaffold as Non-Nucleosidic Competitive, Reversible Inhibitors of Human Ribonucleotide Reductase. J. Med. Chem. 2018, 61, 666–680. [Google Scholar] [CrossRef]
- Misko, T.A.; Liu, Y.T.; Harris, M.E.; Oleinick, N.L.; Pink, J.; Lee, H.Y.; Dealwis, C.G. Structure-guided design of anti-cancer ribonucleotide reductase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 438–450. [Google Scholar] [CrossRef]
- Tholander, F.; Sjöberg, B.M. Discovery of antimicrobial ribonucleotide reductase inhibitors by screening in microwell format. Proc. Natl. Acad. Sci. USA 2012, 109, 9798–9803. [Google Scholar] [CrossRef]
- Berggren, G.; Sahlin, M.; Crona, M.; Tholander, F.; Sjoberg, B.M. Compounds with capacity to quench the tyrosyl radical in Pseudomonas aeruginosa ribonucleotide reductase. J. Biol. Inorg. Chem. 2019, 24, 841–848. [Google Scholar] [CrossRef]
- Crona, M.; Codo, P.; Jonna, V.R.; Hofer, A.; Fernandes, A.P.; Tholander, F. A ribonucleotide reductase inhibitor with deoxyribonucleoside-reversible cytotoxicity. Mol. Oncol. 2016, 10, 1375–1386. [Google Scholar] [CrossRef]
- Cleaveland, E.S.; Monks, A.; Vaigro-Wolff, A.; Zaharevitz, D.W.; Paull, K.; Ardalan, K.; Cooney, D.A.; Ford, H., Jr. Site of action of two novel pyrimidine biosynthesis inhibitors accurately predicted by the compare program. Biochem. Pharmacol. 1995, 49, 947–954. [Google Scholar] [CrossRef]
- Knecht, W.; Loffler, M. Redoxal as a new lead structure for dihydroorotate dehydrogenase inhibitors: A kinetic study of the inhibition mechanism. FEBS Lett. 2000, 467, 27–30. [Google Scholar] [CrossRef]
- Gaur, K.; Perez Otero, S.C.; Benjamin-Rivera, J.A.; Rodriguez, I.; Loza-Rosas, S.A.; Vazquez Salgado, A.M.; Akam, E.A.; Hernandez-Matias, L.; Sharma, R.K.; Alicea, N.; et al. Iron Chelator Transmetalative Approach to Inhibit Human Ribonucleotide Reductase. JACS Au 2021, 1, 865–878. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Name | Cellular Mechanism | RR Subunit Targeted | Mechanism of RR Inhibition | Indications: Single Agent | Indications: Combination |
|---|---|---|---|---|---|---|
 | Fludarabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Induces formation of inactive a6 hexamers | Chronic lymphocytic leukemia | With cyclophosphamide, mitoxantrone, dexamethasone, and rituximab: non-Hodgkin’s lymphoma With cyclophosphamide, mitoxantrone, dexamethasone, and granulocyte colony-stimulating factor: acute myeloid leukemia |
 | Cladribine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Induces formation of inactive a6 hexamers | Hairy cell leukemia B-cell chronic lymphocytic leukemic | |
 | Gemcitabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Alkylates the C-site irreversibly | Pancreatic cancer Off label: cholangiocarcinoma and other biliary tract cancers | With nab-paclitaxel: pancreatic cancer With cisplatin: advanced or metastatic bladder cancer; advanced or metastatic non-small cell lung cancer With carboplatin: ovarian cancer With paclitaxel: metastatic breast cancer |
 | Clofarabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Induces formation of inactive a6 hexamers | Relapsed or refractory acute lymphoblastic leukemia | |
 | Hydroxyurea | Radical Scavenger | hRRM2 | Quenches the radical center | Chronic myelogenous leukemia |
| Structure | Name | Cellular Mechanism | RR Subunit Targeted | Mechanism of RR Inhibition |
|---|---|---|---|---|
 | Tezacitabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Irreversibly modifies the C-site |
 | 2′-deoxy-2′-methylenecytidine (DMDC) | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Inhibits the C-site |
 | Triapine | Metal chelation | hRRM2 | Chelates FeIII from the radical center |
 | Caracemide | Radical Scavenger | hRRM1 | Quenches the radical center |
 | COH29 | Interferes with DNA synthesis and repair mechanisms | hRRM2 | Binds hRRM2 near the C-terminal tail to prevent association with hRRM1 |
 | Didox | Radical Scavenger | hRRM2 | Chelates FeIII from the radical center |
 | Trimidox | Radical Scavenger | hRRM2 | Chelates FeIII from the radical center |
 | Gallium nitrate | FeIII mimic | hRRM2 | Interferes with FeIII radical center |
 | Gallium maltolate | FeIII mimic | hRRM2 | Interferes with FeIII radical center |
 | Motexafin gadolinium | FeIII mimic | hRRM2 | Interferes with FeIII radical center |
| GTI-2040 | antisense ologonucleotide | hRRM2 | Inhibits hRRM2 | |
| GTI-2501 | antisense ologonucleotide | hRRM2 | Inhibits hRRM2 | |
| CALAA-01 | siRNA-containing nanoparticle | hRRM2 | Inhibits hRRM2 |
| Structure | Name | RR Subunit Targeted | Mechanism of RR Inhibition |
|---|---|---|---|
 | P6 | RR1 | Prevents binding of the RR2 subunit |
 | P7 | RR1 | Prevents binding of the RR2 subunit |
 | p-Alkoxyphenols | RR2 | Quenches the tyrosyl radical |
 | ADP-S-HBES-S-dGTP | RR1 | Targets the S-site |
 | A167 | RR1 | Compeites with ATP to bind the A-site |
 | 5-NITP | RR1 | S-site inhibitor that prevents RR1 hexamerization |
 | DHS | RR2 | Induces degradation of RR2 proteosome degredation pathway |
 | OxolsolndoLys | RR1 | Induces formation of inactive a6 hexamers |
 | NSAH | RR1 | Reversible C-site inhibitor |
 | TP6 | RR1 | Reversible C-site inhibitor |
 | NSC73735 | RR1 and RR2 | Prevents alpha subunit hexamerization and quenches the tyrosyl radical of RR2 |
 | Ti(HBED) | RR2 | Depletes intracellular labile iron pools by transmetalation |
 | Ti(Deferasirox)2 | RR2 | Depletes intracellular labile iron pools by transmetalation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huff, S.E.; Winter, J.M.; Dealwis, C.G. Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules 2022, 12, 815. https://doi.org/10.3390/biom12060815
Huff SE, Winter JM, Dealwis CG. Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules. 2022; 12(6):815. https://doi.org/10.3390/biom12060815
Chicago/Turabian StyleHuff, Sarah E., Jordan M. Winter, and Chris G. Dealwis. 2022. "Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present" Biomolecules 12, no. 6: 815. https://doi.org/10.3390/biom12060815
APA StyleHuff, S. E., Winter, J. M., & Dealwis, C. G. (2022). Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules, 12(6), 815. https://doi.org/10.3390/biom12060815