
Zinc in Regulating Protein Kinases and Phosphatases in Neurodegenerative Diseases




Abstract
:1. Introduction
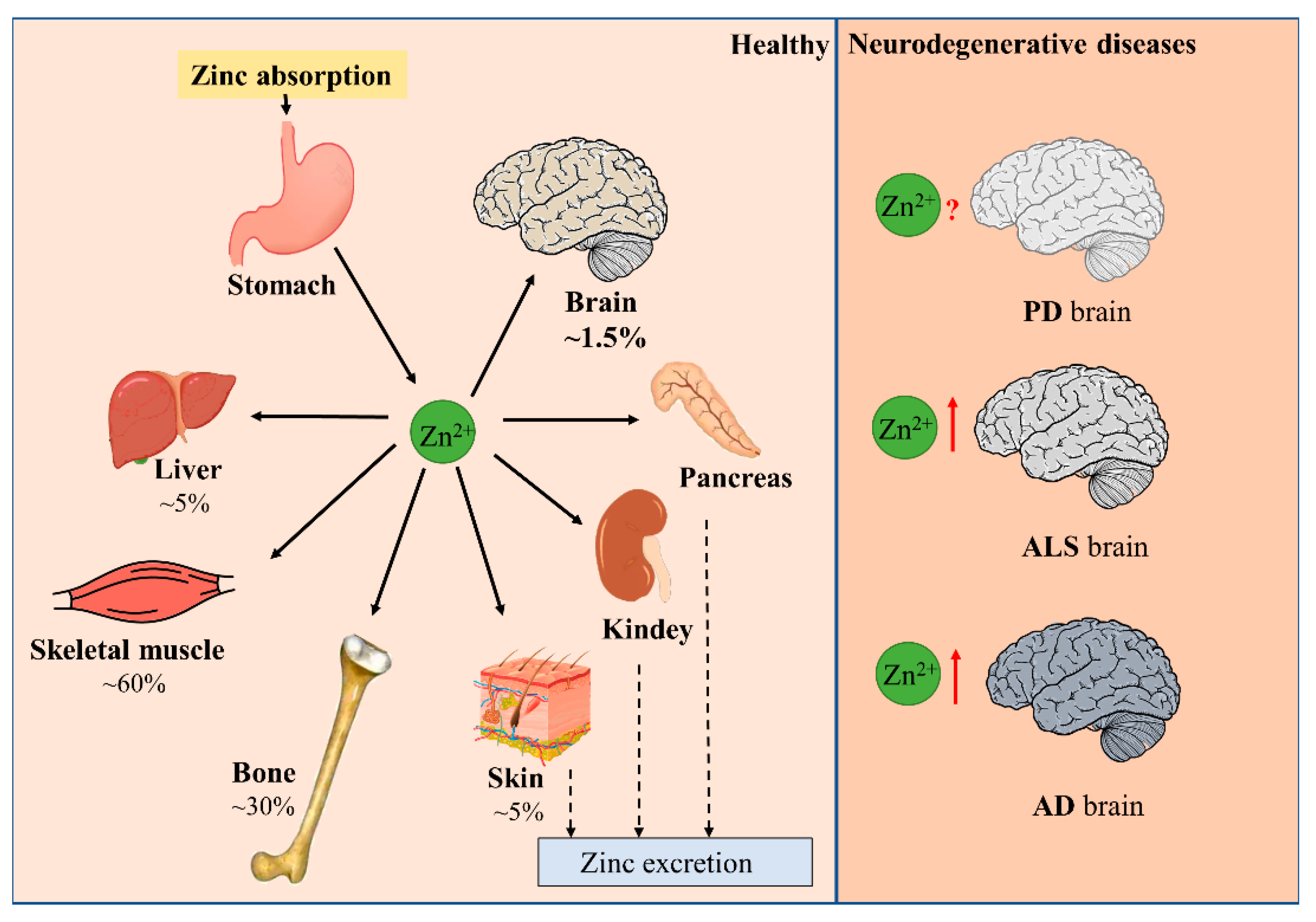
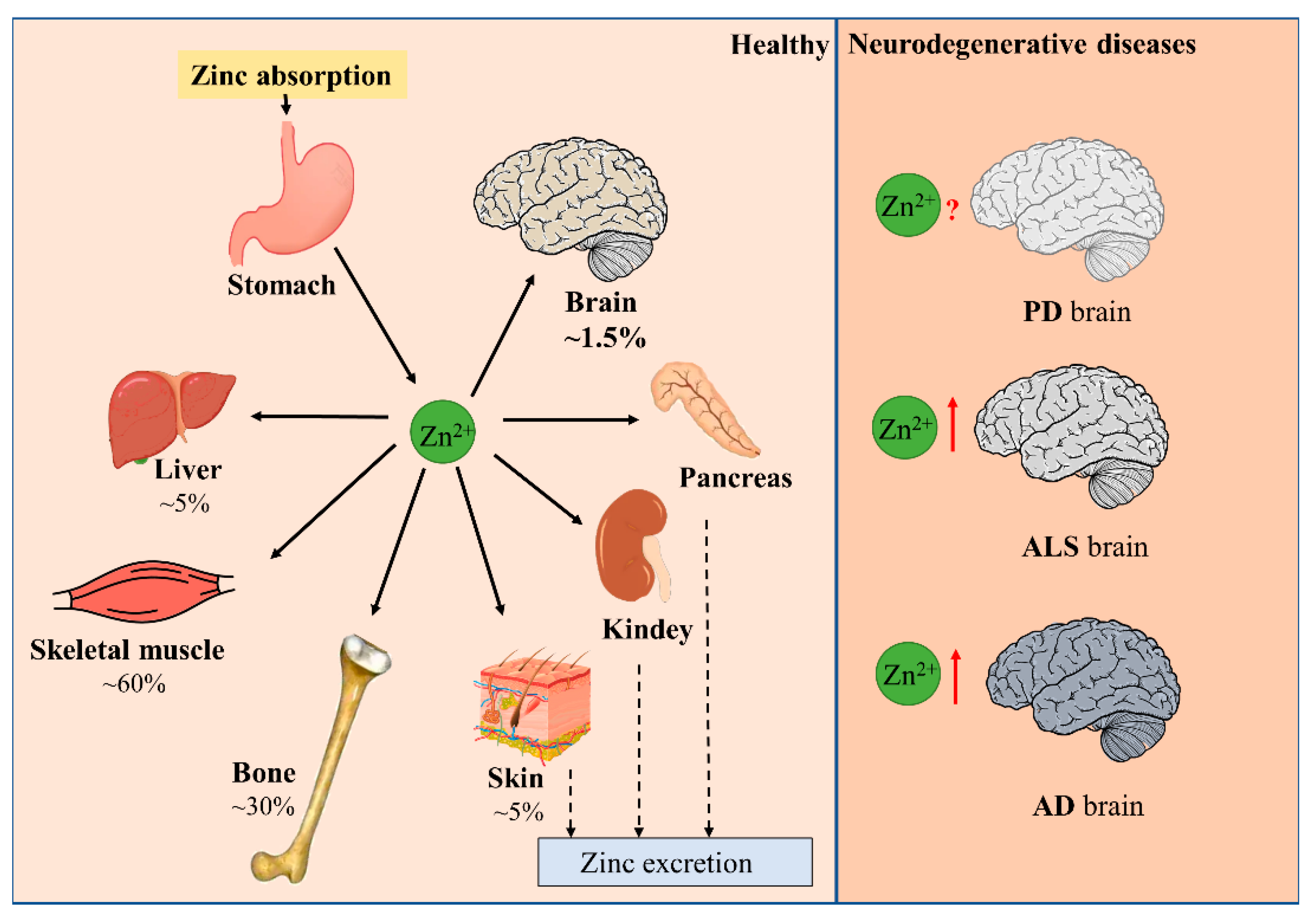
1.1. Zinc Balance in the Human Body
1.2. Distribution of Zinc and the Maintenance of Zinc Homeostasis in the Brain
1.3. Limited Effect of Peripheral Zinc Supplementation on Brain Zinc Level
2. Disturbance of Zinc Homeostasis in Neurodegenerative Diseases
2.1. Parkinson’s Disease (PD)
2.2. Amyotrophic Lateral Sclerosis (ALS)
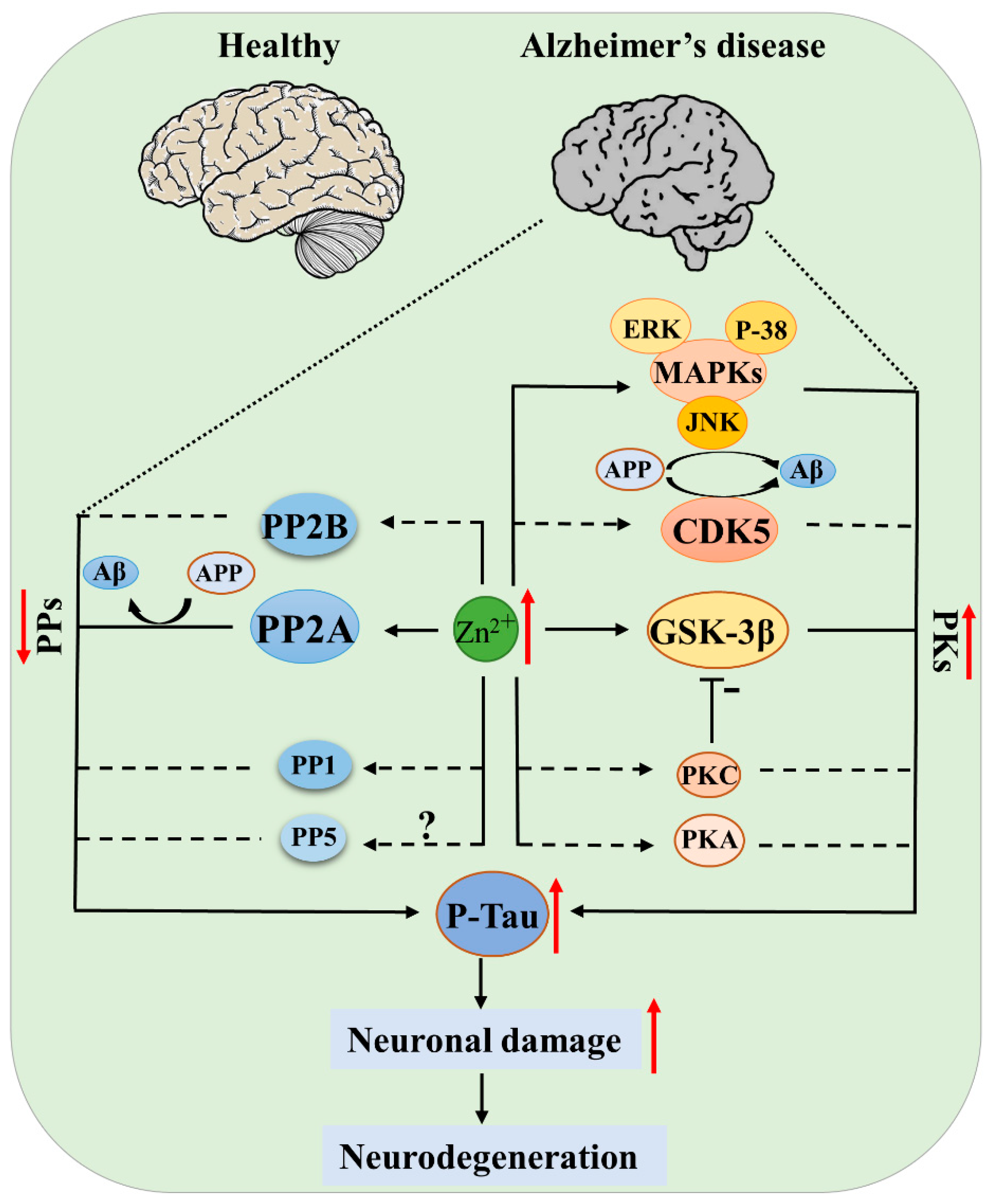
2.3. Alzheimer’s Disease (AD)
3. Zinc-Regulated Protein Kinases and Protein Phosphatases
3.1. Zinc and Protein Kinases
3.1.1. Proline-Directed Kinases
GSK-3β
CDK5
MAPKs (p38, ERK, JNK)
3.1.2. Non-Proline-Directed Kinases
PKA
PKC
3.2. Zinc and Protein Phosphatases
3.2.1. PP-1
3.2.2. PP-2A
3.2.3. Calcineurin/PP-2B
3.2.4. PP-5
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Dell, B.L.; Newberne, P.M.; Savage, J.E. Significance of dietary zinc for the growing chicken. J. Nutr. 1958, 65, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.L.; Lipman, C.B. Evidence on the indispensable nature of zinc and boron for higher green plants. Plant Physiol. 1926, 1, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M. Physiology of zinc: General aspects. In Zinc in Human Biology; Springer: Berlin/Heidelberg, Germany, 1989; pp. 1–14. [Google Scholar]
- Liuzzi, J.P.; Bobo, J.A.; Lichten, L.A.; Samuelson, D.A.; Cousins, R.J. Responsive transporter genes within the murine intestinal-pancreatic axis form a basis of zinc homeostasis. Proc. Natl. Acad. Sci. USA 2004, 101, 14355–14360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [CrossRef]
- Fukada, T.; Kambe, T. Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics 2011, 3, 662–674. [Google Scholar] [CrossRef]
- Maret, W.; Li, Y. Coordination dynamics of zinc in proteins. Chem. Rev. 2009, 109, 4682–4707. [Google Scholar] [CrossRef]
- Devirgiliis, C.; Zalewski, P.D.; Perozzi, G.; Murgia, C. Zinc fluxes and zinc transporter genes in chronic diseases. Mutat. Res. 2007, 622, 84–93. [Google Scholar] [CrossRef]
- Fraker, P.J.; King, L.E. Reprogramming of the immune system during zinc deficiency. Annu. Rev. Nutr. 2004, 24, 277–298. [Google Scholar] [CrossRef]
- Prasad, A.S. Discovery of human zinc deficiency and studies in an experimental human model. Am. J. Clin. Nutr. 1991, 53, 403–412. [Google Scholar] [CrossRef]
- Frederickson, C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989, 31, 145–238. [Google Scholar] [CrossRef]
- Ibata, Y.; Otsuka, N. Electron microscopic demonstration of zinc in the hippocampal formation using Timm’s sulfide silver technique. J. Histochem. Cytochem. 1969, 17, 171–175. [Google Scholar] [CrossRef]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Hashimoto, A.; Fujimoto, S. Current understanding of ZIP and ZnT zinc transporters in human health and diseases. Cell. Mol. Life Sci. 2014, 71, 3281–3295. [Google Scholar] [CrossRef] [PubMed]
- Lichten, L.A.; Cousins, R.J. Mammalian zinc transporters: Nutritional and physiologic regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xiao, G.; Liu, L.; Lang, M. Zinc transporters in Alzheimer’s disease. Mol. Brain 2019, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Bitanihirwe, B.K.; Cunningham, M.G. Zinc: The brain’s dark horse. Synapse 2009, 63, 1029–1049. [Google Scholar] [CrossRef]
- Choi, D.W.; Koh, J.Y. Zinc and brain injury. Annu. Rev. Neurosci. 1998, 21, 347–375. [Google Scholar] [CrossRef]
- Minami, A.; Takeda, A.; Yamaide, R.; Oku, N. Relationship between zinc and neurotransmitters released into the amygdalar extracellular space. Brain Res. 2002, 936, 91–94. [Google Scholar] [CrossRef]
- Assaf, S.Y.; Chung, S.H. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984, 308, 734–736. [Google Scholar] [CrossRef]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Huiliang, Z.; Mengzhe, Y.; Xiaochuan, W.; Hui, W.; Min, D.; Mengqi, W.; Jianzhi, W.; Zhongshan, C.; Caixia, P.; Rong, L. Zinc induces reactive astrogliosis through ERK-dependent activation of Stat3 and promotes synaptic degeneration. J. Neurochem. 2021, 159, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.W.; Yokoyama, M.; Koh, J. Zinc neurotoxicity in cortical cell culture. Neuroscience 1988, 24, 67–79. [Google Scholar] [CrossRef]
- Sensi, S.L.; Yin, H.Z.; Carriedo, S.G.; Rao, S.S.; Weiss, J.H. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. USA 1999, 96, 2414–2419. [Google Scholar] [CrossRef] [Green Version]
- Plum, L.M.; Rink, L.; Haase, H. The essential toxin: Impact of zinc on human health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jing, X.P.; Zhang, S.P.; Gu, R.X.; Tang, F.X.; Wang, X.L.; Xiong, Y.; Qiu, M.; Sun, X.Y.; Ke, D.; et al. High dose zinc supplementation induces hippocampal zinc deficiency and memory impairment with inhibition of BDNF signaling. PLoS ONE 2013, 8, e55384. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef] [Green Version]
- Calap-Quintana, P.; González-Fernández, J.; Sebastiá-Ortega, N.; Llorens, J.V.; Moltó, M.D. Drosophila melanogaster Models of Metal-Related Human Diseases and Metal Toxicity. Int. J. Mol. Sci. 2017, 18, 1456. [Google Scholar] [CrossRef] [Green Version]
- Shaw, B.F.; Valentine, J.S. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem. Sci. 2007, 32, 78–85. [Google Scholar] [CrossRef]
- Xiao, G.; Zhou, B. What can flies tell us about zinc homeostasis? Arch. Biochem. Biophys. 2016, 611, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, P.; Di Filippo, M.; Gallina, A.; Wang, Y.; Stankowski, J.N.; Picconi, B.; Dawson, V.L.; Dawson, T.M. New synaptic and molecular targets for neuroprotection in Parkinson’s disease. Mov. Disord. 2013, 28, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Sensi, S.L. Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 2015, 81, 25–37. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lu, L.; Wang, Q.; Liu, H.; Xue, W.; Zhou, T.; Xu, L.; Wang, K.; Wu, D.; Wei, F.; et al. Crocin Reverses Depression-Like Behavior in Parkinson Disease Mice via VTA-mPFC Pathway. Mol. Neurobiol. 2020, 57, 3158–3170. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Santosh, W. Metallomic profiling and linkage map analysis of early Parkinson’s disease: A new insight to aluminum marker for the possible diagnosis. PLoS ONE 2010, 5, e11252. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Shanmugavelu, P.; Vengamma, B.; Rao, T.S.; Menon, R.B.; Rao, R.V.; Rao, K.S. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson’s disease. J. Trace Elem. Med. Biol. 2004, 18, 163–171. [Google Scholar] [CrossRef]
- Alimonti, A.; Bocca, B.; Pino, A.; Ruggieri, F.; Forte, G.; Sancesario, G. Elemental profile of cerebrospinal fluid in patients with Parkinson’s disease. J. Trace Elem. Med. Biol. 2007, 21, 234–241. [Google Scholar] [CrossRef]
- Forte, G.; Bocca, B.; Senofonte, O.; Petrucci, F.; Brusa, L.; Stanzione, P.; Zannino, S.; Violante, N.; Alimonti, A.; Sancesario, G. Trace and major elements in whole blood, serum, cerebrospinal fluid and urine of patients with Parkinson’s disease. J. Neural Transm. 2004, 111, 1031–1040. [Google Scholar] [CrossRef]
- Ajjimaporn, A.; Phansuwan-Pujito, P.; Ebadi, M.; Govitrapong, P. Zinc protects SK-N-SH cells from methamphetamine-induced alpha-synuclein expression. Neurosci. Lett. 2007, 419, 59–63. [Google Scholar] [CrossRef]
- Ajjimaporn, A.; Shavali, S.; Ebadi, M.; Govitrapong, P. Zinc rescues dopaminergic SK-N-SH cell lines from methamphetamine-induced toxicity. Brain Res. Bull. 2008, 77, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Schaffner, W. Zinc supplement greatly improves the condition of parkin mutant Drosophila. Biol. Chem. 2010, 391, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pals, P.; Van Everbroeck, B.; Grubben, B.; Viaene, M.K.; Dom, R.; van der Linden, C.; Santens, P.; Martin, J.J.; Cras, P. Case-control study of environmental risk factors for Parkinson’s disease in Belgium. Eur. J. Epidemiol. 2003, 18, 1133–1142. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114 Pt 4, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Ali, S.F. Zinc potentiates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced dopamine depletion in caudate nucleus of mice brain. Neurosci. Lett. 2002, 335, 25–28. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, T.N.; Lim, N.K.; Grubman, A.; Li, Q.X.; Volitakis, I.; White, A.R.; Crouch, P.J. Increased metal content in the TDP-43(A315T) transgenic mouse model of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Front. Aging Neurosci. 2014, 6, 15. [Google Scholar] [CrossRef]
- Sillevis Smitt, P.A.; Mulder, T.P.; Verspaget, H.W.; Blaauwgeers, H.G.; Troost, D.; de Jong, J.M. Metallothionein in amyotrophic lateral sclerosis. Biol. Signals 1994, 3, 193–197. [Google Scholar] [CrossRef]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet. Neurology 2016, 15, 455–532. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, M. Dementia: A global health priority—Highlights from an ADI and World Health Organization report. Alzheimer’s Res. Ther. 2012, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Lashley, T.; Schott, J.M.; Weston, P.; Murray, C.E.; Wellington, H.; Keshavan, A.; Foti, S.C.; Foiani, M.; Toombs, J.; Rohrer, J.D.; et al. Molecular biomarkers of Alzheimer’s disease: Progress and prospects. Dis. Models Mech. 2018, 11, dmm031781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overk, C.R.; Masliah, E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem. Pharmacol. 2014, 88, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danscher, G.; Jensen, K.B.; Frederickson, C.J.; Kemp, K.; Andreasen, A.; Juhl, S.; Stoltenberg, M.; Ravid, R. Increased amount of zinc in the hippocampus and amygdala of Alzheimer’s diseased brains: A proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J. Neurosci. Methods 1997, 76, 53–59. [Google Scholar] [CrossRef]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Religa, D.; Strozyk, D.; Cherny, R.A.; Volitakis, I.; Haroutunian, V.; Winblad, B.; Naslund, J.; Bush, A.I. Elevated cortical zinc in Alzheimer disease. Neurology 2006, 67, 69–75. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Lovell, M.A. A potential role for alterations of zinc and zinc transport proteins in the progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 16, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Kim, J.; Mirica, L.M. The effect of Cu(2+) and Zn(2+) on the Aβ42 peptide aggregation and cellular toxicity. Metallomics 2013, 5, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Adlard, P.A.; West, A.K.; Vickers, J.C. Increased density of metallothionein I/II-immunopositive cortical glial cells in the early stages of Alzheimer’s disease. Neurobiol. Dis. 1998, 5, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Smith, J.L.; Xiong, S.; Markesbery, W.R. Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer’s disease. Neurotox. Res. 2005, 7, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Lyubartseva, G.; Smith, J.L.; Markesbery, W.R.; Lovell, M.A. Alterations of zinc transporter proteins ZnT-1, ZnT-4 and ZnT-6 in preclinical Alzheimer’s disease brain. Brain Pathol. 2010, 20, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, D.R.; Vallortigara, J.; Alghamdi, A.; Howlett, D.; Hortobágyi, T.; Johnson, M.; Attems, J.; Newhouse, S.; Ballard, C.; Thomas, A.J.; et al. Assessment of ZnT3 and PSD95 protein levels in Lewy body dementias and Alzheimer’s disease: Association with cognitive impairment. Neurobiol. Aging 2014, 35, 2836–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, A.I.; Pettingell, W.H., Jr.; Paradis, M.D.; Tanzi, R.E. Modulation of A beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 1994, 269, 12152–12158. [Google Scholar] [CrossRef]
- Sun, X.Y.; Wei, Y.P.; Xiong, Y.; Wang, X.C.; Xie, A.J.; Wang, X.L.; Yang, Y.; Wang, Q.; Lu, Y.M.; Liu, R.; et al. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J. Biol. Chem. 2012, 287, 11174–11182. [Google Scholar] [CrossRef] [Green Version]
- Boom, A.; Authelet, M.; Dedecker, R.; Frédérick, C.; Van Heurck, R.; Daubie, V.; Leroy, K.; Pochet, R.; Brion, J.P. Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn2+ in human-tau transfected cells. Biochim. Biophys. Acta 2009, 1793, 1058–1067. [Google Scholar] [CrossRef]
- Kim, I.; Park, E.J.; Seo, J.; Ko, S.J.; Lee, J.; Kim, C.H. Zinc stimulates tau S214 phosphorylation by the activation of Raf/mitogen-activated protein kinase-kinase/extracellular signal-regulated kinase pathway. Neuroreport 2011, 22, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Baudier, J.; Lee, S.H.; Cole, R.D. Separation of the different microtubule-associated tau protein species from bovine brain and their mode II phosphorylation by Ca2+/phospholipid-dependent protein kinase C. J. Biol. Chem. 1987, 262, 17584–17590. [Google Scholar] [CrossRef]
- Drewes, G.; Lichtenberg-Kraag, B.; Döring, F.; Mandelkow, E.M.; Biernat, J.; Goris, J.; Dorée, M.; Mandelkow, E. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992, 11, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Hasegawa, M.; Jakes, R.; Lawler, S.; Cuenda, A.; Cohen, P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997, 409, 57–62. [Google Scholar] [CrossRef]
- Imahori, K.; Uchida, T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J. Biochem. 1997, 121, 179–188. [Google Scholar]
- Scott, C.W.; Spreen, R.C.; Herman, J.L.; Chow, F.P.; Davison, M.D.; Young, J.; Caputo, C.B. Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J. Biol. Chem. 1993, 268, 1166–1173. [Google Scholar] [CrossRef]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; Tanaka, T.; Tung, Y.C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78. [Google Scholar] [CrossRef] [Green Version]
- An, W.L.; Bjorkdahl, C.; Liu, R.; Cowburn, R.F.; Winblad, B.; Pei, J.J. Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3beta in SH-SY5Y neuroblastoma cells. J. Neurochem. 2005, 92, 1104–1115. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Min, Y.K.; Lee, J.E.; Chung, K.C. Zinc induces cell death in immortalized embryonic hippocampal cells via activation of Akt-GSK-3beta signaling. Exp. Cell Res. 2007, 313, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arioka, M.; Tsukamoto, M.; Ishiguro, K.; Kato, R.; Sato, K.; Imahori, K.; Uchida, T. Tau protein kinase II is involved in the regulation of the normal phosphorylation state of tau protein. J. Neurochem. 1993, 60, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.T.; McKenna, R.; Evans, D.B.; Sharma, S.K.; Mathews, W.R. Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk5/p20) using mass spectrometry. J. Neurochem. 2001, 76, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Tuo, Q.Z.; Liuyang, Z.Y.; Lei, P.; Yan, X.; Shentu, Y.P.; Liang, J.W.; Zhou, H.; Pei, L.; Xiong, Y.; Hou, T.Y.; et al. Zinc induces CDK5 activation and neuronal death through CDK5-Tyr15 phosphorylation in ischemic stroke. Cell Death Dis. 2018, 9, 870. [Google Scholar] [CrossRef]
- Guo, C.; Wang, P.; Zhong, M.L.; Wang, T.; Huang, X.S.; Li, J.Y.; Wang, Z.Y. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef]
- Iijima, K.; Ando, K.; Takeda, S.; Satoh, Y.; Seki, T.; Itohara, S.; Greengard, P.; Kirino, Y.; Nairn, A.C.; Suzuki, T. Neuron-specific phosphorylation of Alzheimer’s beta-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 2000, 75, 1085–1091. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- McLaughlin, B.; Pal, S.; Tran, M.P.; Parsons, A.A.; Barone, F.C.; Erhardt, J.A.; Aizenman, E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J. Neurosci. 2001, 21, 3303–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.R.; Chong, S.A.; Lee, S.I.; Sung, J.Y.; Ahn, Y.S.; Chung, K.C.; Seo, J.T. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J. Neurochem. 2001, 78, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, M.; Du, Y.; Zhang, W.; Bai, M.; Zhang, Z.; Li, Z.; Miao, J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 2015, 77, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siano, G.; Caiazza, M.C.; Ollà, I.; Varisco, M.; Madaro, G.; Quercioli, V.; Calvello, M.; Cattaneo, A.; Di Primio, C. Identification of an ERK Inhibitor as a Therapeutic Drug Against Tau Aggregation in a New Cell-Based Assay. Front. Cell. Neurosci. 2019, 13, 386. [Google Scholar] [CrossRef]
- Carlyle, B.C.; Nairn, A.C.; Wang, M.; Yang, Y.; Jin, L.E.; Simen, A.A.; Ramos, B.P.; Bordner, K.A.; Craft, G.E.; Davies, P.; et al. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc. Natl. Acad. Sci. USA 2014, 111, 5036–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paspalas, C.D.; Carlyle, B.C.; Leslie, S.; Preuss, T.M.; Crimins, J.L.; Huttner, A.J.; van Dyck, C.H.; Rosene, D.L.; Nairn, A.C.; Arnsten, A.F.T. The aged rhesus macaque manifests Braak stage III/IV Alzheimer’s-like pathology. Alzheimer’s Dement. 2018, 14, 680–691. [Google Scholar] [CrossRef]
- Allouche-Fitoussi, D.; Breitbart, H. The Role of Zinc in Male Fertility. Int. J. Mol. Sci. 2020, 21, 7796. [Google Scholar] [CrossRef]
- Park, K.H.; Choi, Y.; Yoon, D.S.; Lee, K.M.; Kim, D.; Lee, J.W. Zinc Promotes Osteoblast Differentiation in Human Mesenchymal Stem Cells Via Activation of the cAMP-PKA-CREB Signaling Pathway. Stem Cells Dev. 2018, 27, 1125–1135. [Google Scholar] [CrossRef]
- Kang, J.H.; Toita, R.; Kim, C.W.; Katayama, Y. Protein kinase C (PKC) isozyme-specific substrates and their design. Biotechnol. Adv. 2012, 30, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isagawa, T.; Mukai, H.; Oishi, K.; Taniguchi, T.; Hasegawa, H.; Kawamata, T.; Tanaka, C.; Ono, Y. Dual effects of PKNalpha and protein kinase C on phosphorylation of tau protein by glycogen synthase kinase-3beta. Biochem. Biophys. Res. Commun. 2000, 273, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Correas, I.; Díaz-Nido, J.; Avila, J. Microtubule-associated protein tau is phosphorylated by protein kinase C on its tubulin binding domain. J. Biol. Chem. 1992, 267, 15721–15728. [Google Scholar] [CrossRef]
- Noh, K.M.; Koh, J.Y. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 2000, 20, Rc111. [Google Scholar] [CrossRef]
- Noh, K.M.; Kim, Y.H.; Koh, J.Y. Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J. Neurochem. 1999, 72, 1609–1616. [Google Scholar] [CrossRef]
- Sajan, M.P.; Hansen, B.C.; Higgs, M.G.; Kahn, C.R.; Braun, U.; Leitges, M.; Park, C.R.; Diamond, D.M.; Farese, R.V. Atypical PKC, PKCλ/ι, activates β-secretase and increases Aβ(1-40/42) and phospho-tau in mouse brain and isolated neuronal cells, and may link hyperinsulinemia and other aPKC activators to development of pathological and memory abnormalities in Alzheimer’s disease. Neurobiol. Aging 2018, 61, 225–237. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Kamada, R.; Kudoh, F.; Ito, S.; Tani, I.; Janairo, J.I.B.; Omichinski, J.G.; Sakaguchi, K. Metal-dependent Ser/Thr protein phosphatase PPM family: Evolution, structures, diseases and inhibitors. Pharmacol. Ther. 2020, 215, 107622. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Rahman, A.; Grundke-Iqbal, I.; Iqbal, K. Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem. Res. 2005, 30, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kelker, M.S.; Page, R.; Peti, W. Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: A novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J. Mol. Biol. 2009, 385, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.J.; Bai, G.; Deans-Zirattu, S.; Browner, M.F.; Lee, E.Y. Expression of the catalytic subunit of phosphorylase phosphatase (protein phosphatase-1) in Escherichia coli. J. Biol. Chem. 1992, 267, 1484–1490. [Google Scholar] [CrossRef]
- Chu, Y.; Lee, E.Y.; Schlender, K.K. Activation of protein phosphatase 1. Formation of a metalloenzyme. J. Biol. Chem. 1996, 271, 2574–2577. [Google Scholar] [CrossRef] [Green Version]
- Singla, N.; Dhawan, D.K. Regulatory role of zinc during aluminium-induced altered carbohydrate metabolism in rat brain. J. Neurosci. Res. 2012, 90, 698–705. [Google Scholar] [CrossRef]
- Gong, C.X.; Shaikh, S.; Wang, J.Z.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J. Neurochem. 1995, 65, 732–738. [Google Scholar] [CrossRef]
- Sun, L.; Liu, S.Y.; Zhou, X.W.; Wang, X.C.; Liu, R.; Wang, Q.; Wang, J.Z. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 2003, 118, 1175–1182. [Google Scholar] [CrossRef]
- Sontag, E.; Nunbhakdi-Craig, V.; Sontag, J.M.; Diaz-Arrastia, R.; Ogris, E.; Dayal, S.; Lentz, S.R.; Arning, E.; Bottiglieri, T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 2007, 27, 2751–2759. [Google Scholar] [CrossRef] [Green Version]
- Ingebritsen, T.S.; Cohen, P. The protein phosphatases involved in cellular regulation. 1. Classification and substrate specificities. Eur. J. Biochem. 1983, 132, 255–261. [Google Scholar] [CrossRef]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [Green Version]
- Nishito, Y.; Usui, H.; Tanabe, O.; Shimizu, M.; Takeda, M. Interconversion of Mn(2+)-dependent and -independent protein phosphatase 2A from human erythrocytes: Role of Zn(2+) and Fe(2+) in protein phosphatase 2A. J. Biochem. 1999, 126, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 1989, 58, 453–508. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Xiong, Y.; Luo, D.J.; Wang, X.L.; Qiu, M.; Yang, Y.; Yan, X.; Wang, J.Z.; Ye, Q.F.; Liu, R. Zinc binds to and directly inhibits protein phosphatase 2A in vitro. Neurosci. Bull. 2015, 31, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Martin, B.L.; Brautigan, D.L. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992, 257, 1261–1264. [Google Scholar] [CrossRef]
- Xiong, Y.; Jing, X.P.; Zhou, X.W.; Wang, X.L.; Yang, Y.; Sun, X.Y.; Qiu, M.; Cao, F.Y.; Lu, Y.M.; Liu, R.; et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol. Aging 2013, 34, 745–756. [Google Scholar] [CrossRef]
- Ferreira, A.; Kincaid, R.; Kosik, K.S. Calcineurin is associated with the cytoskeleton of cultured neurons and has a role in the acquisition of polarity. Mol. Biol. Cell 1993, 4, 1225–1238. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease abnormally phosphorylated tau is dephosphorylated by protein phosphatase-2B (calcineurin). J. Neurochem. 1994, 62, 803–806. [Google Scholar] [CrossRef]
- Wei, Q.; Holzer, M.; Brueckner, M.K.; Liu, Y.; Arendt, T. Dephosphorylation of tau protein by calcineurin triturated into neural living cells. Cell. Mol. Neurobiol. 2002, 22, 13–24. [Google Scholar] [CrossRef]
- Gratuze, M.; Noël, A.; Julien, C.; Cisbani, G.; Milot-Rousseau, P.; Morin, F.; Dickler, M.; Goupil, C.; Bezeau, F.; Poitras, I.; et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 86–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Akaishi, E.; Abe, Y.; Ishikawa, R.; Tanaka, S.; Hosaka, K.; Kubohara, Y. Zinc inhibits calcineurin activity in vitro by competing with nickel. Biochem. Biophys. Res. Commun. 2003, 307, 64–68. [Google Scholar] [CrossRef]
- Merten, K.E.; Jiang, Y.; Kang, Y.J. Zinc inhibits doxorubicin-activated calcineurin signal transduction pathway in H9c2 embryonic rat cardiac cells. Exp. Biol. Med. 2007, 232, 682–689. [Google Scholar]
- Park, K.H.; Park, B.; Yoon, D.S.; Kwon, S.H.; Shin, D.M.; Lee, J.W.; Lee, H.G.; Shim, J.H.; Park, J.H.; Lee, J.M. Zinc inhibits osteoclast differentiation by suppression of Ca2+-Calcineurin-NFATc1 signaling pathway. Cell Commun. Signal. 2013, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Liu, F.; Wu, G.; Rossie, S.; Wegiel, J.; Li, L.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of microtubule-associated protein tau by protein phosphatase 5. J. Neurochem. 2004, 88, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Lee, K.Y.; Clark, A.W.; Rosales, J.L.; Chapman, K.; Fung, T.; Johnston, R.N. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci. Res. 1999, 34, 21–29. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Larivière, R.C.; Julien, J.P. Deregulation of Cdk5 in a mouse model of ALS: Toxicity alleviated by perikaryal neurofilament inclusions. Neuron 2001, 30, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.D.; Crocker, S.J.; Jackson-Lewis, V.; Jordan-Sciutto, K.L.; Hayley, S.; Mount, M.P.; O’Hare, M.J.; Callaghan, S.; Slack, R.S.; Przedborski, S.; et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 13650–13655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Blanco, R.; Carmona, M.; Ribera, R.; Goutan, E.; Puig, B.; Rey, M.J.; Cardozo, A.; Viñals, F.; Ribalta, T. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001, 11, 144–158. [Google Scholar] [CrossRef]
- Kulich, S.M.; Chu, C.T. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J. Neurochem. 2001, 77, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, G.; Roder, H.; Nunomura, A.; Takeda, A.; Friedlich, A.L.; Zhu, X.; Raina, A.K.; Holbrook, N.; Siedlak, S.L.; Harris, P.L.; et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10, 2411–2415. [Google Scholar] [CrossRef]
- Zhu, X.; Castellani, R.J.; Takeda, A.; Nunomura, A.; Atwood, C.S.; Perry, G.; Smith, M.A. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: The ‘two hit’ hypothesis. Mech. Ageing Dev. 2001, 123, 39–46. [Google Scholar] [CrossRef]
- Atzori, C.; Ghetti, B.; Piva, R.; Srinivasan, A.N.; Zolo, P.; Delisle, M.B.; Mirra, S.S.; Migheli, A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Migheli, A.; Piva, R.; Atzori, C.; Troost, D.; Schiffer, D. c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1997, 56, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Dagda, R.K.; Das Banerjee, T. Role of protein kinase A in regulating mitochondrial function and neuronal development: Implications to neurodegenerative diseases. Rev. Neurosci. 2015, 26, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Govoni, S.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Bianchetti, A.; Trabucchi, M. Cytosol protein kinase C downregulation in fibroblasts from Alzheimer’s disease patients. Neurology 1993, 43, 2581–2586. [Google Scholar] [CrossRef]
- Wang, H.Y.; Pisano, M.R.; Friedman, E. Attenuated protein kinase C activity and translocation in Alzheimer’s disease brain. Neurobiol. Aging 1994, 15, 293–298. [Google Scholar] [CrossRef]
- Pei, J.J.; Gong, C.X.; Iqbal, K.; Grundke-Iqbal, I.; Wu, Q.L.; Winblad, B.; Cowburn, R.F. Subcellular distribution of protein phosphatases and abnormally phosphorylated tau in the temporal cortex from Alzheimer’s disease and control brains. J. Neural Transm. 1998, 105, 69–83. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Rossie, S.; Gong, C.X. Dephosphorylation of tau by protein phosphatase 5: Impairment in Alzheimer’s disease. J. Biol. Chem. 2005, 280, 1790–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Protein Kinase or Phosphatase | Expression Level or Activity in Disease Progression | Effect and Regulating Pathways of Zinc in Disease | Reference |
|---|---|---|---|
| GSK-3β | AD↑ | Activation zinc-MEK/ERK/GSK-3β | [81,140] |
| CDK5 | AD↑; ALS↑; PD ↑ | Activation, To be identified | [141,142,143] |
| ERK | AD↑; PD↑ | Activation zinc-Ras/Raf/MEK/ERK | [144,145,146,147] |
| JNK | AD↑; ALS↑ | Activation To be identified | [148,149,150] |
| p38 | AD↑ | Activation To be identified | [147,148] |
| PKA | AD↑; PD↑ | Not explored | [151,152] |
| PKC | AD↓ | Not explored | [153,154] |
| PP1 | AD↓ | Not explored | [114] |
| PP2A | AD↓ | Inhibition Direct effect, or through zinc-Src-PP2A(Y307) | [131,155] |
| PP2B | HD↓ | Inhibition To be identified | [135] |
| PP5 | AD↓ | Not explored | [156] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-L.; Wang, X.-C.; Liu, R. Zinc in Regulating Protein Kinases and Phosphatases in Neurodegenerative Diseases. Biomolecules 2022, 12, 785. https://doi.org/10.3390/biom12060785
Zhang H-L, Wang X-C, Liu R. Zinc in Regulating Protein Kinases and Phosphatases in Neurodegenerative Diseases. Biomolecules. 2022; 12(6):785. https://doi.org/10.3390/biom12060785
Chicago/Turabian StyleZhang, Hui-Liang, Xiao-Chuan Wang, and Rong Liu. 2022. "Zinc in Regulating Protein Kinases and Phosphatases in Neurodegenerative Diseases" Biomolecules 12, no. 6: 785. https://doi.org/10.3390/biom12060785
APA StyleZhang, H.-L., Wang, X.-C., & Liu, R. (2022). Zinc in Regulating Protein Kinases and Phosphatases in Neurodegenerative Diseases. Biomolecules, 12(6), 785. https://doi.org/10.3390/biom12060785