
PPARα Signaling: A Candidate Target in Psychiatric Disorder Management

 ,
,  ,
,  ,
,
Abstract
1. Introduction
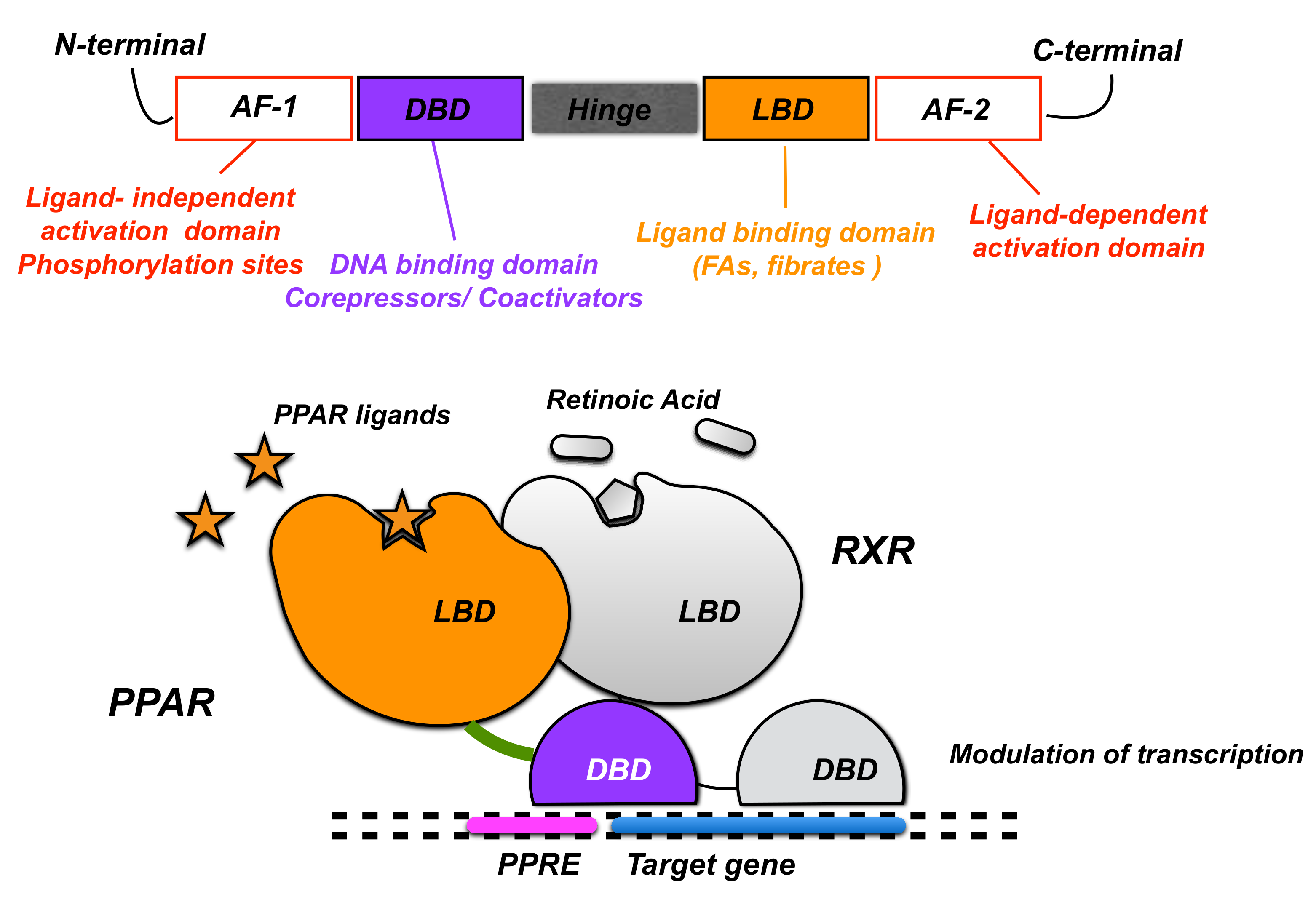
2. PPARα Expression and Physiological Role in the CNS
2.1. Modulation of Neuroinflammation
2.2. Neuroprotection
2.3. Regulation of Neurotransmission
3. PPARα in CNS Disorders
3.1. Role of PPARα in Depression and Anhedonia
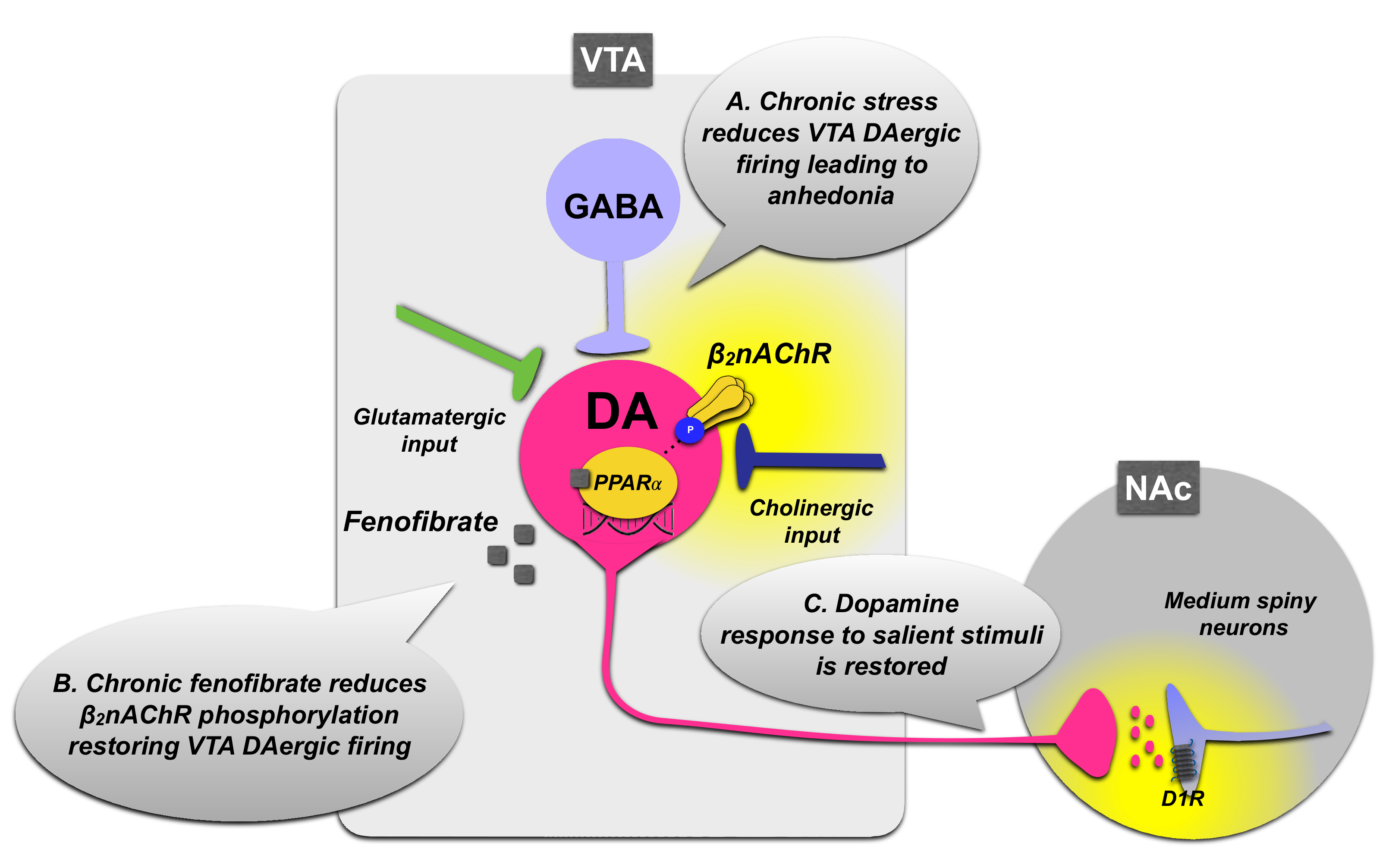
3.1.1. PPARα Regulation of VTA Dopamine Neurons
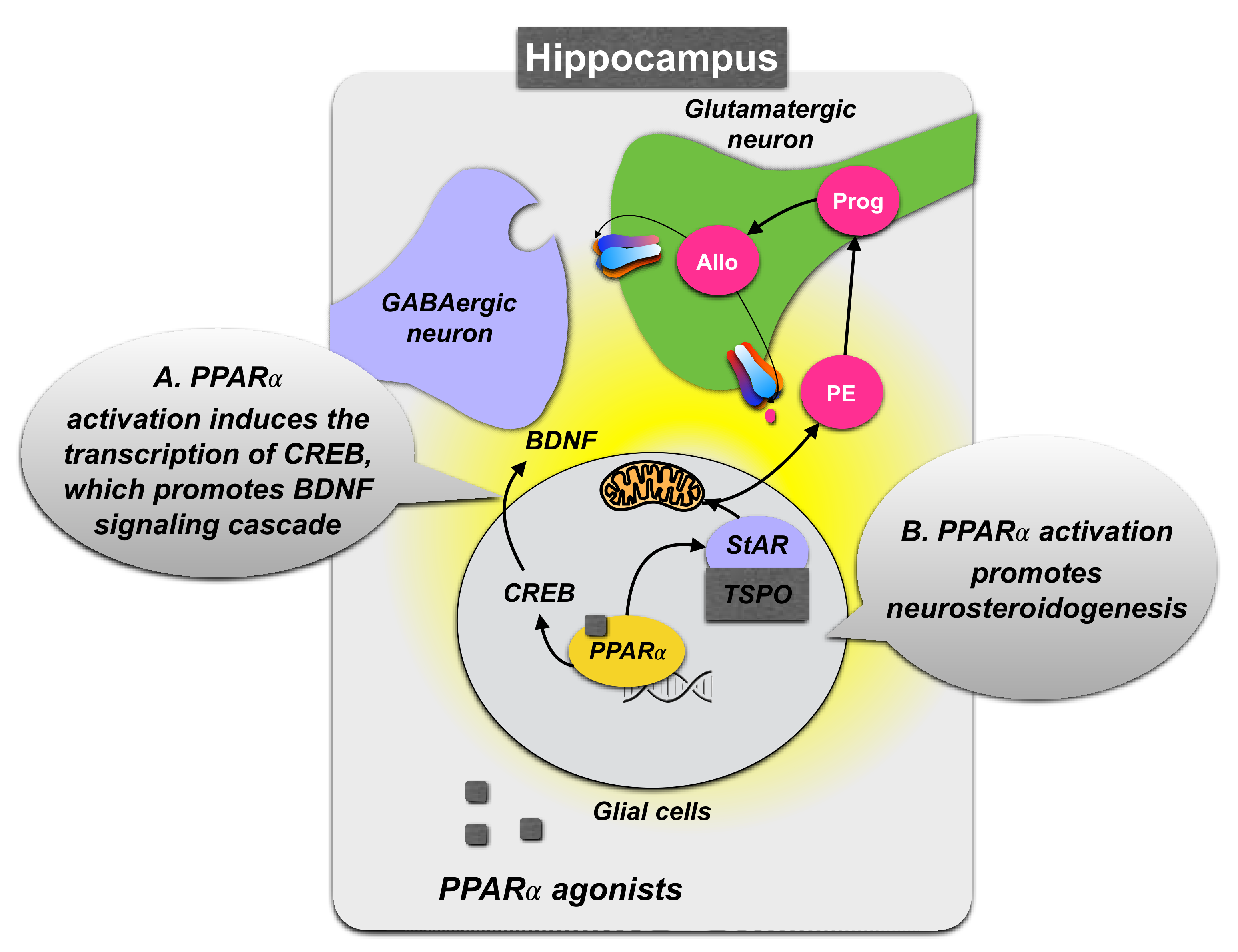
3.1.2. Role of PPARα Expression in the Hippocampus
3.1.3. PPARα Neurosteroidogenic Effects
3.2. Role of PPARα in Autism Spectrum Disorder (ASD)
3.3. Role of PPARα in Schizophrenia
3.4. Neurodegenerative Disorders
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duval, C.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Staels, B. The Role of PPARs in Atherosclerosis. Trends Mol. Med. 2002, 8, 422–430. [Google Scholar] [CrossRef]
- Skerrett, R.; Malm, T.; Landreth, G. Nuclear Receptors in Neurodegenerative Diseases. Neurobiol. Dis. 2014, 72, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome Proliferator-Activated Receptors: Nuclear Control of Metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Borel, V.; Gallot, D.; Marceau, G.; Sapin, V.; Blanchon, L. Placental Implications of Peroxisome Proliferator-Activated Receptors in Gestation and Parturition. PPAR Res. 2008, 2008, 758562. [Google Scholar] [CrossRef] [PubMed]
- Fournier, T.; Tsatsaris, V.; Handschuh, K.; Evain-Brion, D. PPARs and the Placenta. Placenta 2007, 28, 65–76. [Google Scholar] [CrossRef]
- Green, S. PPAR: A Mediator of Peroxisome Proliferator Action. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 1995, 333, 101–109. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-Cis Retinoic Acid and Peroxisome Proliferator Signalling Pathways through Heterodimer Formation of Their Receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef]
- Pineda Torra, I.; Jamshidi, Y.; Flavell, D.M.; Fruchart, J.-C.; Staels, B. Characterization of the Human PPARα Promoter: Identification of a Functional Nuclear Receptor Response Element. Mol. Endocrinol. 2002, 16, 1013–1028. [Google Scholar] [CrossRef]
- Kota, B.P.; Huang, T.H.W.; Roufogalis, B.D. An Overview on Biological Mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef]
- Anbalagan, M.; Huderson, B.; Murphy, L.; Rowan, B.G. Post-Translational Modifications of Nuclear Receptors and Human Disease. Nucl. Recept. Signal. 2012, 10, e001. [Google Scholar] [CrossRef]
- Poulsen, L.L.C.; Siersbæk, M.; Mandrup, S. PPARs: Fatty Acid Sensors Controlling Metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and Lipid Ligands in Inflammation and Metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.; Sharma, B. Benefits of Fenofibrate in Prenatal Valproic Acid-Induced Autism Spectrum Disorder Related Phenotype in Rats. Brain Res. Bull. 2019, 147, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Toobian, D.; Ghosh, P.; Katkar, G.D. Parsing the Role of PPARs in Macrophage Processes. Front. Immunol. 2021, 12, 783780. [Google Scholar] [CrossRef]
- Antonosante, A.; D’Angelo, M.; Castelli, V.; Catanesi, M.; Iannotta, D.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells. Int. J. Mol. Sci. 2018, 19, 1907. [Google Scholar] [CrossRef]
- Gervois, P.; Torra, I.P.; Fruchart, J.C.; Staels, B. Regulation of Lipid and Lipoprotein Metabolism by PPAR Activators. Clin. Chem. Lab. Med. 2000, 38, 3–11. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of Energy Metabolism by Long-Chain Fatty Acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Tsuboi, K.; Ikematsu, N.; Uyama, T.; Deutsch, D.G.; Tokumura, A.; Ueda, N. Biosynthetic Pathways of Bioactive N-Acylethanolamines in Brain. CNS Neurol. Disord.-Drug Targets 2013, 12, 7–16. [Google Scholar] [CrossRef]
- Sun, Y.; Alexander, S.P.H.; Garle, M.J.; Gibson, C.L.; Hewitt, K.; Murphy, S.P.; Kendall, D.A.; Bennett, A.J. Cannabinoid Activation of PPARα; a Novel Neuroprotective Mechanism. Br. J. Pharmacol. 2007, 152, 734–743. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An Update on PPAR Activation by Cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- Hostetler, H.A.; Petrescu, A.D.; Kier, A.B.; Schroeder, F. Peroxisome Proliferator-Activated Receptor α Interacts with High Affinity and Is Conformationally Responsive to Endogenous Ligands. J. Biol. Chem. 2005, 280, 18667–18682. [Google Scholar] [CrossRef] [PubMed]
- Balenga, N.A.B.; Aflaki, E.; Kargl, J.; Platzer, W.; Schröder, R.; Blättermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 Regulates Cannabinoid 2 Receptor-Mediated Responses in Human Neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Sahebkar, A.; Maffioli, P. The Role of Various Peroxisome Proliferator-Activated Receptors and Their Ligands in Clinical Practice. J. Cell. Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.; Antonosante, A.; Castelli, V.; Catanesi, M.; Moorthy, N.; Iannotta, D.; Cimini, A.; Benedetti, E. PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation. Int. J. Mol. Sci. 2018, 19, 1869. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Fanelli, F.; Ceru, M.; Moreno, S. Neuroprotective Properties of Peroxisome Proliferator-Activated Receptor Alpha (PPARa) and Its Lipid Ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Target in Neurodegenerative Disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- Tufano, M.; Pinna, G. Is There a Future for PPARs in the Treatment of Neuropsychiatric Disorders? Molecules 2020, 25, 1062. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef]
- Matheson, J.; Le Foll, B. Therapeutic Potential of Peroxisome Proliferator-Activated Receptor (PPAR) Agonists in Substance Use Disorders: A Synthesis of Preclinical and Human Evidence. Cells 2020, 9, 1196. [Google Scholar] [CrossRef]
- Cullingford, T.E.; Bhakoo, K.; Peuchen, S.; Dolphin, C.T.; Patel, R.; Clark, J.B. Distribution of MRNAs Encoding the Peroxisome Proliferator-Activated Receptor α, β, and γ and the Retinoid X Receptor α, β, and γ in Rat Central Nervous System. J. Neurochem. 1998, 70, 1366–1375. [Google Scholar] [CrossRef]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR Isotypes in the Adult Mouse and Human Brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Farioli-vecchioli, S.; Cerù, M.P. Immunolocalization of Peroxisome Proliferator-Activated Receptors and Retinoid X Receptors in the Adult Rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J. Recent Update on the PPARα-Null Mouse. Biochimie 1997, 79, 139–144. [Google Scholar] [CrossRef]
- Sayd, A.; Anton, M.; Alen, F.; Caso, J.R.; Pavon, J.; Leza, J.C.; Rodriguez de Fonseca, F.; Garcia-Bueno, B.; Orio, L. Systemic Administration of Oleoylethanolamide Protects from Neuroinflammation and Anhedonia Induced by LPS in Rats. Int. J. Neuropsychopharmacol. 2015, 18, pyu111. [Google Scholar] [CrossRef]
- Roy, A.; Jana, M.; Corbett, G.T.; Ramaswamy, S.; Kordower, J.H.; Gonzalez, F.J.; Pahan, K. Regulation of Cyclic AMP Response Element Binding and Hippocampal Plasticity-Related Genes by Peroxisome Proliferator-Activated Receptor α. Cell Rep. 2013, 4, 724–737. [Google Scholar] [CrossRef]
- Huang, H.T.; Liao, C.K.; Chiu, W.T.; Tzeng, S.F. Ligands of Peroxisome Proliferator-Activated Receptor-Alpha Promote Glutamate Transporter-1 Endocytosis in Astrocytes. Int. J. Biochem. Cell Biol. 2017, 86, 42–53. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, J.H.; Liu, L.; Liu, Y.; Tang, W.Q.; Ji, C.H.; Guan, W.; Zhao, X.Y.; Sun, Y.F.; Xu, D.W.; et al. Hippocampal PPARα Plays a Role in the Pharmacological Mechanism of Vortioxetine, a Multimodal-Acting Antidepressant. Front. Pharmacol. 2021, 12, 673221. [Google Scholar] [CrossRef]
- Roy, A.; Kundu, M.; Jana, M.; Mishra, R.K.; Yung, Y.; Luan, C.H.; Gonzalez, F.J.; Pahan, K. Identification and Characterization of PPARα Ligands in the Hippocampus. Nat. Chem. Biol. 2016, 12, 1075–1083. [Google Scholar] [CrossRef]
- Patel, D.; Roy, A.; Raha, S.; Kundu, M.; Gonzalez, F.J.; Pahan, K. Upregulation of BDNF and Hippocampal Functions by a Hippocampal Ligand of PPARα. JCI Insight 2020, 5, e136654. [Google Scholar] [CrossRef]
- Melis, M.; Carta, S.; Fattore, L.; Tolu, S.; Yasar, S.; Goldberg, S.R.; Fratta, W.; Maskos, U.; Pistis, M. Peroxisome Proliferator-Activated Receptors-Alpha Modulate Dopamine Cell Activity through Nicotinic Receptors. Biol. Psychiatry 2010, 68, 256–264. [Google Scholar] [CrossRef]
- Melis, M.; Scheggi, S.; Carta, G.; Madeddu, C.; Lecca, S.; Luchicchi, A.; Cadeddu, F.; Frau, R.; Fattore, L.; Fadda, P.; et al. PPARα Regulates Cholinergic-Driven Activity of Midbrain Dopamine Neurons via a Novel Mechanism Involving A7 Nicotinic Acetylcholine Receptors. J. Neurosci. 2013, 33, 6203–6211. [Google Scholar] [CrossRef] [PubMed]
- Nisbett, K.E.; Pinna, G. Emerging Therapeutic Role of PPAR-α in Cognition and Emotions. Front. Pharmacol. 2018, 9, 998. [Google Scholar] [CrossRef] [PubMed]
- Locci, A.; Pinna, G. Stimulation of Peroxisome Proliferator-Activated Receptor-α by N-Palmitoylethanolamine Engages Allopregnanolone Biosynthesis to Modulate Emotional Behavior. Biol. Psychiatry 2019, 85, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Galan-Rodriguez, B.; Suarez, J.; Gonzalez-Aparicio, R.; Bermudez-Silva, F.J.; Maldonado, R.; Robledo, P.; Rodriguez de Fonseca, F.; Fernandez-Espejo, E. Oleoylethanolamide Exerts Partial and Dose-Dependent Neuroprotection of Substantia Nigra Dopamine Neurons. Neuropharmacology 2009, 56, 653–664. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple Sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective Activities of Palmitoylethanolamide in an Animal Model of Parkinson’s Disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Yadav, S.; Gupta, R.K.; Waggoner, G.R.; Deloach, A.; Calingasan, N.Y.; Flint Beal, M.; Kiaei, M. Preferential PPAR-α Activation Reduces Neuroinflammation, and Blocks Neurodegeneration in Vivo. Hum. Mol. Genet. 2016, 25, 317–327. [Google Scholar] [CrossRef]
- Locci, A.; Geoffroy, P.; Miesch, M.; Mensah-Nyagan, A.G.; Pinna, G. Social Isolation in Early versus Late Adolescent Mice Is Associated with Persistent Behavioral Deficits That Can Be Improved by Neurosteroid-Based Treatment. Front. Cell. Neurosci. 2017, 11, 208. [Google Scholar] [CrossRef]
- Wada, Y.; Maekawa, M.; Ohnishi, T.; Balan, S.; Matsuoka, S.; Iwamoto, K.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Hisano, Y.; et al. Peroxisome Proliferator-Activated Receptor α as a Novel Therapeutic Target for Schizophrenia. eBioMedicine 2020, 62, 103130. [Google Scholar] [CrossRef]
- Lee, D.; Tomita, Y.; Allen, W.; Tsubota, K.; Negishi, K.; Kurihara, T. PPARα Modulation-Based Therapy in Central Nervous System Diseases. Life 2021, 11, 1168. [Google Scholar] [CrossRef]
- Matrisciano, F.; Pinna, G. PPAR-α Hypermethylation in the Hippocampus of Mice Exposed to Social Isolation Stress Is Associated with Enhanced Neuroinflammation and Aggressive Behavior. Int. J. Mol. Sci. 2021, 22, 10678. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Mazzon, E.; Impellizzeri, D.; Esposito, E.; Cuzzocrea, S. N-palmitoylethanolamide Treatment Exhibits Antidepressant Effects In A Mouse Model Of Anxiety/Depressive Like Behavior. FASEB J. 2012, 26, 1042.6. [Google Scholar] [CrossRef]
- Hasler, G.; Drevets, W.C.; Manji, H.K.; Charney, D.S. Discovering Endophenotypes for Major Depression. Neuropsychopharmacology 2004, 29, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- McCabe, C. Neural Correlates of Anhedonia as a Trait Marker for Depression. In Anhedonia: A Comprehensive Handbook Volume II: Neuropsychiatric and Physical Disorders; Springer: Dordrecht, The Netherlands, 2014; pp. 159–174. ISBN 9789401786102. [Google Scholar]
- Gong, L.; He, C.; Zhang, H.; Zhang, H.; Zhang, Z.; Xie, C. Disrupted Reward and Cognitive Control Networks Contribute to Anhedonia in Depression. J. Psychiatr. Res. 2018, 103, 61–68. [Google Scholar] [CrossRef]
- McMakin, D.L.; Olino, T.M.; Porta, G.; Dietz, L.J.; Emslie, G.; Clarke, G.; Wagner, K.D.; Asarnow, J.R.; Ryan, N.D.; Birmaher, B.; et al. Anhedonia Predicts Poorer Recovery among Youth with Selective Serotonin Reuptake Inhibitor Treatmentresistant Depression. J. Am. Acad. Child Adolesc. Psychiatry 2012, 51, 404–411. [Google Scholar] [CrossRef]
- Vinckier, F.; Gourion, D.; Mouchabac, S. Anhedonia Predicts Poor Psychosocial Functioning: Results from a Large Cohort of Patients Treated for Major Depressive Disorder by General Practitioners. Eur. Psychiatry 2017, 44, 1–8. [Google Scholar] [CrossRef]
- Yu, H.L.; Deng, X.Q.; Li, Y.J.; Li, Y.C.; Quan, Z.S.; Sun, X.Y. N-Palmitoylethanolamide, an Endocannabinoid, Exhibits Antidepressant Effects in the Forced Swim Test and the Tail Suspension Test in Mice. Pharmacol. Rep. 2011, 63, 834–839. [Google Scholar] [CrossRef]
- Cordaro, M.; Cuzzocrea, S.; Crupi, R. An Update of Palmitoylethanolamide and Luteolin Effects in Preclinical and Clinical Studies of Neuroinflammatory Events. Antioxidants 2020, 9, 216. [Google Scholar] [CrossRef]
- Yang, R.; Wang, P.; Chen, Z.; Hu, W.; Gong, Y.; Zhang, W.; Huang, C. WY-14643, a Selective Agonist of Peroxisome Proliferator-Activated Receptor-α, Ameliorates Lipopolysaccharide-Induced Depressive-like Behaviors by Preventing Neuroinflammation and Oxido-Nitrosative Stress in Mice. Pharmacol. Biochem. Behav. 2017, 153, 97–104. [Google Scholar] [CrossRef]
- Ghazizadeh-Hashemi, M.; Ghajar, A.; Shalbafan, M.R.; Ghazizadeh-Hashemi, F.; Afarideh, M.; Malekpour, F.; Ghaleiha, A.; Ardebili, M.E.; Akhondzadeh, S. Palmitoylethanolamide as Adjunctive Therapy in Major Depressive Disorder: A Double-Blind, Randomized and Placebo-Controlled Trial. J. Affect. Disord. 2018, 232, 127–133. [Google Scholar] [CrossRef]
- McNab, F.; Varrone, A.; Farde, L.; Jucaite, A.; Bystritsky, P.; Forssberg, H.; Klingberg, T. Changes in Cortical Dopamine D1 Receptor Binding Associated with Cognitive Training. Science 2009, 323, 800–802. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.; Axelsson, J.; Riklund, K.; Nyberg, L.; Dayan, P.; Bäckman, L.; Guitart-Masip, M. Attenuation of Dopamine-Modulated Prefrontal Value Signals Underlies Probabilistic Reward Learning Deficits in Old Age. Elife 2017, 6, e26424. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.L.; Ishibashi, K.; Mandelkern, M.A.; Brown, A.K.; Ghahremani, D.G.; Sabb, F.; Bilder, R.; Cannon, T.; Borg, J.; London, E.D. Striatal D1- and D2-Type Dopamine Receptors Are Linked to Motor Response Inhibition in Human Subjects. J. Neurosci. 2015, 35, 5990–5997. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.J.; Nestler, E.J. The Brain Reward Circuitry in Mood Disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Pizzagalli, D.A.; Iosifescu, D.; Hallett, L.A.; Ratner, K.G.; Fava, M. Reduced Hedonic Capacity in Major Depressive Disorder: Evidence from a Probabilistic Reward Task. J. Psychiatr. Res. 2008, 43, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Green, I.W.; Pizzagalli, D.A.; Admon, R.; Kumar, P. Anhedonia Modulates the Effects of Positive Mood Induction on Reward-Related Brain Activation. Neuroimage 2019, 193, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Young, C.B.; Chen, T.; Nusslock, R.; Keller, J.; Schatzberg, A.F.; Menon, V. Anhedonia and General Distress Show Dissociable Ventromedial Prefrontal Cortex Connectivity in Major Depressive Disorder. Transl. Psychiatry 2016, 6, e810. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Chen, X.; Zhang, Z.; Xiao, L.; Zhou, Y. Anhedonia Correlates with Functional Connectivity of the Nucleus Accumbens Subregions in Patients with Major Depressive Disorder. NeuroImage Clin. 2021, 30, 102599. [Google Scholar] [CrossRef]
- Marchese, G.; Scheggi, S.; Secci, M.E.; De Montis, M.G.; Gambarana, C. Anti-Anhedonic Activity of Long-Term Lithium Treatment in Rats Exposed to Repeated Unavoidable Stress. Int. J. Neuropsychopharmacol. 2013, 16, 1611–1621. [Google Scholar] [CrossRef][Green Version]
- Scheggi, S.; Pelliccia, T.; Ferrari, A.; De Montis, M.G.; Gambarana, C. Impramine, Fluoxetine and Clozapine Differently Affected Reactivity to Positive and Negative Stimuli in a Model of Motivational Anhedonia in Rats. Neuroscience 2015, 291, 189–202. [Google Scholar] [CrossRef]
- Scheggi, S.; Melis, M.; De Felice, M.; Aroni, S.; Muntoni, A.L.; Pelliccia, T.; Gambarana, C.; De Montis, M.G.; Pistis, M. PPARα Modulation of Mesolimbic Dopamine Transmission Rescues Depression-Related Behaviors. Neuropharmacology 2016, 110, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Scheggi, S.; Pelliccia, T.; Gambarana, C.; De Montis, M.G. Aripiprazole Relieves Motivational Anhedonia in Rats. J. Affect. Disord. 2018, 227, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Van Zessen, R.; Phillips, J.L.; Budygin, E.A.; Stuber, G.D. Activation of VTA GABA Neurons Disrupts Reward Consumption. Neuron 2012, 73, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Derst, C.; Veh, R.W.; Zahm, D.S. Glutamatergic Afferents of the Ventral Tegmental Area in the Rat. J. Neurosci. 2007, 27, 5730–5743. [Google Scholar] [CrossRef]
- Zell, V.; Steinkellner, T.; Hollon, N.G.; Warlow, S.M.; Souter, E.; Faget, L.; Hunker, A.C.; Jin, X.; Zweifel, L.S.; Hnasko, T.S. VTA Glutamate Neuron Activity Drives Positive Reinforcement Absent Dopamine Co-Release. Neuron 2020, 107, 864–873.e4. [Google Scholar] [CrossRef]
- Changeux, J.P. Nicotine Addiction and Nicotinic Receptors: Lessons from Genetically Modified Mice. Nat. Rev. Neurosci. 2010, 11, 389–401. [Google Scholar] [CrossRef]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of Native Nicotinic Receptor Subtypes in Mammalian Brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef]
- Grady, S.R.; Salminen, O.; Laverty, D.C.; Whiteaker, P.; McIntosh, J.M.; Collins, A.C.; Marks, M.J. The Subtypes of Nicotinic Acetylcholine Receptors on Dopaminergic Terminals of Mouse Striatum. Biochem. Pharmacol. 2007, 74, 1235–1246. [Google Scholar] [CrossRef]
- Mameli-Engvall, M.; Evrard, A.; Pons, S.; Maskos, U.; Svensson, T.H.; Changeux, J.P.; Faure, P. Hierarchical Control of Dopamine Neuron-Firing Patterns by Nicotinic Receptors. Neuron 2006, 50, 911–921. [Google Scholar] [CrossRef]
- Tye, K.M.; Mirzabekov, J.J.; Warden, M.R.; Ferenczi, E.A.; Tsai, H.C.; Finkelstein, J.; Kim, S.Y.; Adhikari, A.; Thompson, K.R.; Andalman, A.S.; et al. Dopamine Neurons Modulate Neural Encoding and Expression of Depression-Related Behaviour. Nature 2013, 493, 537–541. [Google Scholar] [CrossRef]
- Chang, C.H.; Grace, A.A. Amygdala-Ventral Pallidum Pathway Decreases Dopamine Activity after Chronic Mild Stress in Rats. Biol. Psychiatry 2014, 76, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Rauggi, R.; Scheggi, S.; Cassanelli, A.; De Montis, M.G.; Tagliamonte, A.; Gambarana, C. The Mesolimbic Dopaminergic Response to Novel Palatable Food Consumption Increases Dopamine-D1 Receptor-Mediated Signalling with Complex Modifications of the DARPP-32 Phosphorylation Pattern. J. Neurochem. 2005, 92, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Scheggi, S.; Guzzi, F.; Braccagni, G.; De Montis, M.G.; Parenti, M.; Gambarana, C. Targeting PPARα in the Rat Valproic Acid Model of Autism: Focus on Social Motivational Impairment and Sex-Related Differences. Mol. Autism 2020, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Scheggi, S.; De Montis, M.G.; Gambarana, C. DARPP-32 in the Orchestration of Responses to Positive Natural Stimuli. J. Neurochem. 2018, 147, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Francis, T.C.; Chandra, R.; Friend, D.M.; Finkel, E.; Dayrit, G.; Miranda, J.; Brooks, J.M.; Iñiguez, S.D.; O’Donnell, P.; Kravitz, A.; et al. Nucleus Accumbens Medium Spiny Neuron Subtypes Mediate Depression-Related Outcomes to Social Defeat Stress. Biol. Psychiatry 2015, 77, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chikahisa, S.; Chida, D.; Shiuchi, T.; Harada, S.; Shimizu, N.; Otsuka, A.; Tanioka, D.; Séi, H. Enhancement of Fear Learning in PPARα Knockout Mice. Behav. Brain Res. 2019, 359, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, H.; Wang, Y.J.; Wang, J.L.; Zhu, Q.; Wu, F.; Zhang, W.; Jiang, B. Hippocampal PPARα Is a Novel Therapeutic Target for Depression and Mediates the Antidepressant Actions of Fluoxetine in Mice. Br. J. Pharmacol. 2018, 175, 2968–2987. [Google Scholar] [CrossRef]
- Chen, C.; Shen, J.H.; Xu, H.; Chen, P.; Chen, F.; Guan, Y.X.; Jiang, B.; Wu, Z.H. Hippocampal PPARα Is Involved in the Antidepressant-like Effects of Venlafaxine in Mice. Brain Res. Bull. 2019, 153, 171–180. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The Molecular Neurobiology of Depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, Y.-J.; Wang, H.; Song, L.; Huang, C.; Zhu, Q.; Wu, F.; Zhang, W. Antidepressant-like Effects of Fenofibrate in Mice via the Hippocampal Brain-Derived Neurotrophic Factor Signalling Pathway. Br. J. Pharmacol. 2017, 174, 177–194. [Google Scholar] [CrossRef]
- Ni, Y.F.; Wang, H.; Gu, Q.Y.; Wang, F.Y.; Wang, Y.J.; Wang, J.L.; Jiang, B. Gemfibrozil Has Antidepressant Effects in Mice: Involvement of the Hippocampal Brain-Derived Neurotrophic Factor System. J. Psychopharmacol. 2018, 32, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-M.; Wang, D.; Bi, W.-P.; Jiang, Z.-E.; Piao, R.-L.; Yu, H.-L. N-Palmitoylethanolamide Exerts Antidepressant-like Effects in Rats: Involvement of PPARa Pathway in the Hippocampus. J. Pharmacol. Exp. Ther. 2019, 369, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Pahan, K. PPARα Signaling in the Hippocampus: Crosstalk Between Fat and Memory. J. Neuroimmune Pharmacol. 2015, 10, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Raso, G.M.; Esposito, E.; Vitiello, S.; Iacono, A.; Santoro, A.; D’Agostino, G.; Sasso, O.; Russo, R.; Piazza, P.V.; Calignano, A.; et al. Palmitoylethanolamide Stimulation Induces Allopregnanolone Synthesis in C6 Cells and Primary Astrocytes: Involvement of Peroxisome-Proliferator Activated Receptor-α. J. Neuroendocrinol. 2011, 23, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Sasso, O.; Moreno-Sanz, G.; Martucci, C.; Realini, N.; Dionisi, M.; Mengatto, L.; Duranti, A.; Tarozzo, G.; Tarzia, G.; Mor, M.; et al. Antinociceptive Effects of the N-Acylethanolamine Acid Amidase Inhibitor ARN077 in Rodent Pain Models. Pain 2013, 154, 350–360. [Google Scholar] [CrossRef][Green Version]
- American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5). In Diagnostic and Statistical Manual of Mental Disorders DMS V, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [CrossRef]
- Masi, A.; Quintana, D.S.; Glozier, N.; Lloyd, A.R.; Hickie, I.B.; Guastella, A.J. Cytokine Aberrations in Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2015, 20, 440–446. [Google Scholar] [CrossRef]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem Brain Abnormalities of the Glutamate Neurotransmitter System in Autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef]
- Shinohe, A.; Hashimoto, K.; Nakamura, K.; Tsujii, M.; Iwata, Y.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Increased Serum Levels of Glutamate in Adult Patients with Autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1472–1477. [Google Scholar] [CrossRef]
- D’Agostino, G.; Cristiano, C.; Lyons, D.J.; Citraro, R.; Russo, E.; Avagliano, C.; Russo, R.; Raso, G.M.; Meli, R.; De Sarro, G.; et al. Peroxisome Proliferator-Activated Receptor Alpha Plays a Crucial Role in Behavioral Repetition and Cognitive Flexibility in Mice. Mol. Metab. 2015, 4, 528–536. [Google Scholar] [CrossRef]
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide Counteracts Autistic-like Behaviours in BTBR T+tf/J Mice: Contribution of Central and Peripheral Mechanisms. Brain. Behav. Immun. 2018, 74, 166–175. [Google Scholar] [CrossRef]
- Mirza, R.; Sharma, B. Selective Modulator of Peroxisome Proliferator-Activated Receptor-α Protects Propionic Acid Induced Autism-like Phenotypes in Rats. Life Sci. 2018, 214, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Elnahas, E.M.; Abuelezz, S.A.; Mohamad, M.I.; Nabil, M.M.; Abdelraouf, S.M.; Bahaa, N.; Hassan, G.A.M.; Aboul-Fotouh, S. Novel Role of Peroxisome Proliferator Activated Receptor-α in Valproic Acid Rat Model of Autism: Mechanistic Study of Risperidone and Metformin Monotherapy versus Combination. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2022, 116, 110522. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, B.; Crupi, R.; Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Beneficial Effects of Co-Ultramicronized Palmitoylethanolamide/Luteolin in a Mouse Model of Autism and in a Case Report of Autism. CNS Neurosci. Ther. 2017, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and Related N-Acylethanolamines: Biological Activities and Metabolism Makoto Murakami. Inflamm. Regen. 2018, 38, 28. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, M.; Bortoletto, R.; Costa, R.; Zoccante, L. Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence. Nutrients 2021, 13, 1346. [Google Scholar] [CrossRef]
- Aran, A.; Eylon, M.; Harel, M.; Polianski, L.; Nemirovski, A.; Tepper, S.; Schnapp, A.; Cassuto, H.; Wattad, N.; Tam, J. Lower Circulating Endocannabinoid Levels in Children with Autism Spectrum Disorder. Mol. Autism 2019, 10, 2. [Google Scholar] [CrossRef]
- Antonucci, N.; Cirillo, A.; Siniscalco, D. Beneficial Effects of Palmitoylethanolamide on Expressive Language, Cognition, and Behaviors in Autism: A Report of Two Cases. Case Rep. Psychiatry 2015, 2015, 325061. [Google Scholar] [CrossRef]
- Khalaj, M.; Saghazadeh, A.; Shirazi, E.; Shalbafan, M.-R.; Alavi, K.; Shooshtari, M.H.; Laksari, F.Y.; Hosseini, M.; Mohammadi, M.-R.; Akhondzadeh, S. Palmitoylethanolamide as Adjunctive Therapy for Autism: Efficacy and Safety Results from a Randomized Controlled Trial. J. Psychiatr. Res. 2018, 103, 104–111. [Google Scholar] [CrossRef]
- Chevallier, C.; Kohls, G.; Troiani, V.; Brodkin, E.S.; Schultz, R.T. The Social Motivation Theory of Autism. Trends Cogn. Sci. 2012, 16, 231–239. [Google Scholar] [CrossRef]
- Kohls, G.; Chevallier, C.; Troiani, V.; Schultz, R.T. Social ‘Wanting’ Dysfunction in Autism: Neurobiological Underpinnings and Treatment Implications. J. Neurodev. Disord. 2012, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Burnside, K.; Wright, K.; Poulin-Dubois, D. Social Motivation and Implicit Theory of Mind in Children with Autism Spectrum Disorder. Autism Res. 2017, 10, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Done, D.J.; Johnstone, E.C.; Frith, C.D.; Golding, J.; Shepherd, P.M.; Crow, T.J. Complications of Pregnancy and Delivery in Relation to Psychosis in Adult Life: Data from the British Perinatal Mortality Survey Sample. Br. Med. J. 1991, 302, 1576–1580. [Google Scholar] [CrossRef]
- Buka, S.L.; Tsuang, M.T.; Lipsitt, L.P. Pregnancy/Delivery Complications and Psychiatric Diagnosis: A Prospective Study. Arch. Gen. Psychiatry 1993, 50, 151–156. [Google Scholar] [CrossRef]
- Jones, P.; Murray, R.; Rodgers, B.; Marmot, M. Child Developmental Risk Factors for Adult Schizophrenia in the British 1946 Birth Cohort. Lancet 1994, 344, 1398–1402. [Google Scholar] [CrossRef]
- Jones, P.B.; Rantakallio, P.; Hartikainen, A.L.; Isohanni, M.; Sipila, P. Schizophrenia as a Long-Term Outcome of Pregnancy, Delivery, and Perinatal Complications: A 28-Year Follow-up of the 1966 North Finland General Population Birth Cohort. Am. J. Psychiatry 1998, 155, 355–364. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Chiang, M.C.; Huang, Y.C.; Yeh, T.H.; Shih, H.Y.; Liu, H.F.; Chen, H.Y.; Wang, C.P.; Cheng, Y.C. Pparα Deficiency Inhibits the Proliferation of Neuronal and Glial Precursors in the Zebrafish Central Nervous System. Dev. Dyn. 2018, 247, 1264–1275. [Google Scholar] [CrossRef]
- Cimini, A.; Benedetti, E.; Cristiano, L.; Sebastiani, P.; D’Amico, M.A.; D’Angelo, B.; Di Loreto, S. Expression of Peroxisome Proliferator-Activated Receptors (PPARs) and Retinoic Acid Receptors (RXRs) in Rat Cortical Neurons. Neuroscience 2005, 130, 325–337. [Google Scholar] [CrossRef]
- Sagheddu, C.; Melis, M.; Muntoni, A.L.; Pistis, M. Repurposing Peroxisome Proliferator-Activated Receptor Agonists in Neurological and Psychiatric Disorders. Pharmaceuticals 2021, 14, 1025. [Google Scholar] [CrossRef]
- Reddy, R.; Sahebarao, M.P.; Mukherjee, S.; Murthy, J.N. Enzymes of the Antioxidant Defense System in Chronic Schizophrenic Patients. Biol. Psychiatry 1991, 30, 409–412. [Google Scholar] [CrossRef]
- Wang, A.M.; Pradhan, S.; Coughlin, J.M.; Trivedi, A.; Dubois, S.L.; Crawford, J.L.; Sedlak, T.W.; Nucifora, F.C.; Nestadt, G.; Nucifora, L.G.; et al. Assessing Brain Metabolism with 7-T Proton Magnetic Resonance Spectroscopy in Patients with First-Episode Psychosis. JAMA Psychiatry 2019, 76, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione Deficit in Cerebrospinal Fluid and Prefrontal Cortex in Vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, J.M.; Ishizuka, K.; Kano, S.I.; Edwards, J.A.; Seifuddin, F.T.; Shimano, M.A.; Daley, E.L.; Zandi, P.P.; Leweke, F.M.; Cascella, N.G.; et al. Marked Reduction of Soluble Superoxide Dismutase-1 (SOD1) in Cerebrospinal Fluid of Patients with Recent-Onset Schizophrenia. Mol. Psychiatry 2013, 18, 10–11. [Google Scholar] [CrossRef]
- Do, K.Q.; Cabungcal, J.H.; Frank, A.; Steullet, P.; Cuenod, M. Redox Dysregulation, Neurodevelopment, and Schizophrenia. Curr. Opin. Neurobiol. 2009, 19, 220–230. [Google Scholar] [CrossRef]
- Tosic, M.; Ott, J.; Barral, S.; Bovet, P.; Deppen, P.; Gheorghita, F.; Matthey, M.L.; Parnas, J.; Preisig, M.; Saraga, M.; et al. Schizophrenia and Oxidative Stress: Glutamate Cysteine Ligase Modifier as a Susceptibility Gene. Am. J. Hum. Genet. 2006, 79, 586–592. [Google Scholar] [CrossRef]
- Gysin, R.; Kraftsik, R.; Sandell, J.; Bovet, P.; Chappuis, C.; Conus, P.; Deppen, P.; Preisig, M.; Ruiz, V.; Steullet, P.; et al. Impaired Glutathione Synthesis in Schizophrenia: Convergent Genetic and Functional Evidence. Proc. Natl. Acad. Sci. USA 2007, 104, 16621–16626. [Google Scholar] [CrossRef]
- Powell, S.B. Models of Neurodevelopmental Abnormalities in Schizophrenia. Curr. Top. Behav. Neurosci. 2010, 4, 435–481. [Google Scholar] [CrossRef]
- Rolland, B.; Marche, K.; Cottencin, O.; Bordet, R. The PPAR α Agonist Fenofibrate Reduces Prepulse Inhibition Disruption in a Neurodevelopmental Model of Schizophrenia. Schizophr. Res. Treat. 2012, 2012, 839853. [Google Scholar] [CrossRef]
- De Felice, M.; Melis, M.; Aroni, S.; Muntoni, A.L.; Fanni, S.; Frau, R.; Devoto, P.; Pistis, M. The PPARα Agonist Fenofibrate Attenuates Disruption of Dopamine Function in a Maternal Immune Activation Rat Model of Schizophrenia. CNS Neurosci. Ther. 2019, 25, 549–561. [Google Scholar] [CrossRef]
- Zuckerman, L.; Rehavi, M.; Nachman, R.; Weiner, I. Immune Activation during Pregnancy in Rats Leads to a Postpubertal Emergence of Disrupted Latent Inhibition, Dopaminergic Hyperfunction, and Altered Limbic Morphology in the Offspring: A Novel Neurodevelopmental Model of Schizophrenia. Neuropsychopharmacology 2003, 28, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Robbins, M.E. PPARα Ligands Inhibit Radiation-Induced Microglial Inflammatory Responses by Negatively Regulating NF-ΚB and AP-1 Pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Squassina, A.; Congiu, D.; Chillotti, C.; Niola, P.; Galderisi, S.; Pistis, M.; Del Zompo, M. Investigation of Endocannabinoid System Genes Suggests Association between Peroxisome Proliferator Activator Receptor-α Gene (PPARA) and Schizophrenia. Eur. Neuropsychopharmacol. 2013, 23, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Okubo, Y.; Ohtsuki, T.; Yamakawa-Kobayashi, K.; Arinami, T. Mutation Analysis of the Retinoid X Receptor Beta, Nuclear-Related Receptor 1, and Peroxisome Proliferator-Activated Receptor Alpha Genes in Schizophrenia and Alcohol Dependence: Possible Haplotype Association of Nuclear-Related Receptor 1 Gene to Alcohol. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2002, 114, 15–23. [Google Scholar] [CrossRef]
- Doherty, J.L.; O’Donovan, M.C.; Owen, M.J. Recent Genomic Advances in Schizophrenia. Clin. Genet. 2012, 81, 103–109. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Y.; Keaton, M.; Baranova, A.; Liu, S.; Hu, X.; Li, Q.; Cheng, L.; Zhou, P.; Cao, H.; et al. Variants and Expression Changes in PPAR-Encoding Genes Display No Significant Association with Schizophrenia. Biosci. Rep. 2020, 40, BSR20201083. [Google Scholar] [CrossRef]
- Maulik, M.; Westaway, D.; Jhamandas, J.H.; Kar, S. Role of Cholesterol in APP Metabolism and Its Significance in Alzheimer’s Disease Pathogenesis. Mol. Neurobiol. 2013, 47, 37–63. [Google Scholar] [CrossRef]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of Peroxisome Proliferator-Activated Receptor α Stimulates ADAM10-Mediated Proteolysis of APP. Proc. Natl. Acad. Sci. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Qiao, P.; Zhao, F.; Yan, Y. Fenofibrate Reduces Amyloidogenic Processing of APP in APP/PS1 Transgenic Mice via PPAR-α/PI3-K Pathway. Int. J. Dev. Neurosci. 2014, 38, 223–231. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Qiao, P.-F.; Zhao, F.-L.; Yan, Y. PPAR-α Agonist Regulates Amyloid-β Generation via Inhibiting BACE-1 Activity in Human Neuroblastoma SH-SY5Y Cells Transfected with APPswe Gene. Mol. Cell. Biochem. 2015, 408, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Walker, A.W.; Hosken, I.T.; Chua, B.E.; Zhang, C.; Haidar, M.; Gundlach, A.L. Relaxin-3/RXFP3 Networks: An Emerging Target for the Treatment of Depression and Other Neuropsychiatric Diseases? Front. Pharmacol. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Deng, Y.; Luo, Y.; Zhang, S.; Zou, H.; Cai, F.; Wada, K.; Song, W. Control of BACE1 Degradaton and APP Processing by Ubiquitin Carboxyl-Terminal Hydrolase L1. J. Neurochem. 2012, 120, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.X.; He, J.H.; Cui, Z.Q.; Yang, T.; Sun, X.H. PPAR-α Agonist GW7647 Protects Against Oxidative Stress and Iron Deposit via GPx4 in a Transgenic Mouse Model of Alzheimer’s Diseases. ACS Chem. Neurosci. 2022, 13, 207–216. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C.; et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front. Aging Neurosci. 2021, 13, 713726. [Google Scholar] [CrossRef]
- Chandra, S.; Pahan, K. Gemfibrozil, a Lipid-Lowering Drug, Lowers Amyloid Plaque Pathology and Enhances Memory in a Mouse Model of Alzheimer’s Disease via Peroxisome Proliferator-Activated Receptor α. J. Alzheimer’s Dis. Rep. 2019, 3, 149–168. [Google Scholar] [CrossRef]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-Mediated Autophagy Reduces Alzheimer Disease-like Pathology and Cognitive Decline in a Murine Model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Raha, S.; Ghosh, A.; Dutta, D.; Patel, D.R.; Pahan, K. Activation of PPARα Enhances Astroglial Uptake and Degradation of β-Amyloid. Sci. Signal. 2021, 14, eabg4747. [Google Scholar] [CrossRef]
- Brune, S.; Kölsch, H.; Ptok, U.; Majores, M.; Schulz, A.; Schlosser, R.; Rao, M.L.; Maier, W.; Heun, R. Polymorphism in the Peroxisome Proliferator-Activated Receptor α Gene Influences the Risk for Alzheimer’s Disease. J. Neural Transm. 2003, 110, 1041–1050. [Google Scholar] [CrossRef]
- Sjölander, A.; Minthon, L.; Bogdanovic, N.; Wallin, A.; Zetterberg, H.; Blennow, K. The PPAR-α Gene in Alzheimer’s Disease: Lack of Replication of Earlier Association. Neurobiol. Aging 2009, 30, 666–668. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M.; Wands, J.R. Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Orellana, F.; Leroy, T.; Ribeiro, F.; Kreis, A.; Leroy, K.; Lalloyer, F.; Baugé, E.; Staels, B.; Duyckaerts, C.; Brion, J.P.; et al. Regulation of PPAR α by APP in Alzheimer Disease Affects the Pharmacological Modulation of Synaptic Activity. JCI Insight 2021, 6, e150099. [Google Scholar] [CrossRef] [PubMed]
- Kreisler, A.; Gelé, P.; Wiart, J.-F.; Lhermitte, M.; Destée, A.; Bordet, R. Lipid-Lowering Drugs in the MPTP Mouse Model of Parkinson’s Disease: Fenofibrate Has a Neuroprotective Effect, Whereas Bezafibrate and HMG-CoA Reductase Inhibitors Do Not. Brain Res. 2007, 1135, 77–84. [Google Scholar] [CrossRef]
- Barbiero, J.K.; Santiago, R.; Tonin, F.S.; Boschen, S.; Da Silva, L.M.; De Paula Werner, M.F.; Da Cunha, C.; Lima, M.M.S.; Vital, M.A.B.F. PPAR-α Agonist Fenofibrate Protects against the Damaging Effects of MPTP in a Rat Model of Parkinson’s Disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 53, 35–44. [Google Scholar] [CrossRef]
- Gottschalk, C.G.; Jana, M.; Roy, A.; Patel, D.R.; Pahan, K. Gemfibrozil Protects Dopaminergic Neurons in a Mouse Model of Parkinson’s Disease via Ppara-Dependent Astrocytic Gdnf Pathway. J. Neurosci. 2021, 41, 2287–2300. [Google Scholar] [CrossRef]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic Acid: A Mitochondrial Toxin to Uncover Physiopathological Mechanisms Underlying Striatal Degeneration in Huntington’s Disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef]
- Bhateja, D.K.; Dhull, D.K.; Gill, A.; Sidhu, A.; Sharma, S.; Reddy, B.V.K.; Padi, S.S.V. Peroxisome Proliferator-Activated Receptor-α Activation Attenuates 3-Nitropropionic Acid Induced Behavioral and Biochemical Alterations in Rats: Possible Neuroprotective Mechanisms. Eur. J. Pharmacol. 2011, 674, 33–43. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| PPARα Agonist | Disease | Preclinical Model | Output Features | Ref. |
|---|---|---|---|---|
| PEA −/+ luteolin | Depression | Corticosterone-induced depression | Increased neurogenesis | [52] |
| Fenofibrate | Chronic unavoidable stress in rat | Regulation of VTA dopamine neurons | [72] | |
| Fenofibrate | Chronic social defeat in mice | Normalization of BDNF signaling in hippocampus | [91] | |
| Gemfibrozil | Chronic mild stress in mice | Normalization of BDNF signaling in hippocampus | [92] | |
| PEA | Social isolation | Increase in steroidogenesis in limbic areas | [48] | |
| WY-14643 (synthetic ligand) | (LPS)-induced depressive-like behaviors | Prevention of neuroinflammation and oxidative stress | [60] | |
| Fenofibrate | Autism Spectrum Disorder | Propionic acid model | Reduction in oxidative stress and neuroinflammation | [103] |
| PEA + luteolin | Valproic acid model | Reduction in inflammation and apoptosis | [106] | |
| PEA | BTBR T+tf/J mice | Modulation of neuroprotection, inflammation, and gut–brain axis. | [102] | |
| Fenofibrate | Valproic acid model | Reinstatement of dopaminergic response to social stimuli | [84] | |
| Fenofibrate | Schizophrenia | Kainic acid model | Reduction in behavioral impairment | [131] |
| Fenofibrate | Maternal immune activation | Modulation of pathways underlying oxidative stress and neuroinflammation | [132] | |
| Fenofibrate | Alzheimer Disease | APP/PS1 transgenic mice | Reduction in Ab deposition and levels of b-secretase | [142] |
| GW7647 | APP/PS1 transgenic mice | Reduction in Ab deposition, improved cognition, and decrease in oxidative stress | [146] | |
| Gemfibrozil | 5XFAD mice | Proteolysis of APP by stimulating a-secretase | [140] | |
| Gemfibrozil | 5XFAD mice | Reduction in a microgliosis and astrogliosis in hippocampus and cortex | [149] | |
| Gemfibrozil | APP-PSEN1DE9 | Reduction in Ab accumulation and improved cognitive impairment by modulation of autophagy | [150] | |
| Gemfibrozil + retinoic acid | 5XFAD mice | Astroglial uptake and degradation of Ab | [151] | |
| Fenofibrate | Parkinson’s Disease | MPTP | Prevention of MPTP-induced dopaminergic loss in SNpc | [156] |
| Fenofibrate | MPTP | Reduced hypolocomotion, oxidative stress, and degeneration of dopamine neurons in SNpc | [157] | |
| PEA | MPTP | Reduction in MPTP-induced microglia activation and motor deficits | [46] | |
| Gemfibrozil | MPTP | Neuroprotection via GDNF pathway | [158] |
| Drug | Disease | Clinical Study | Output | Ref. |
|---|---|---|---|---|
| Ultramicronized PEA + Citalopram | Depression | Randomized, double-blind placebo-controlled trial | As add-on therapy to antidepressant treatment, PEA increased antidepressant response rate | [61] |
| Ultramicronized PEA | Autism Spectrum Disorder | Two cases report | Beneficial effects on expressive language and cognition | [110] |
| Ultramicronized PEA + luteolin | A case report | Improvement of ASD symptoms | [106] | |
| Ultramicronized PEA + Risperidone | Randomized, double-blind placebo-controlled trial | As add-on therapy to antipsychotic treatment, PEA reduced autism-related irritability and hyperactivity | [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheggi, S.; Pinna, G.; Braccagni, G.; De Montis, M.G.; Gambarana, C. PPARα Signaling: A Candidate Target in Psychiatric Disorder Management. Biomolecules 2022, 12, 723. https://doi.org/10.3390/biom12050723
Scheggi S, Pinna G, Braccagni G, De Montis MG, Gambarana C. PPARα Signaling: A Candidate Target in Psychiatric Disorder Management. Biomolecules. 2022; 12(5):723. https://doi.org/10.3390/biom12050723
Chicago/Turabian StyleScheggi, Simona, Graziano Pinna, Giulia Braccagni, Maria Graziella De Montis, and Carla Gambarana. 2022. "PPARα Signaling: A Candidate Target in Psychiatric Disorder Management" Biomolecules 12, no. 5: 723. https://doi.org/10.3390/biom12050723
APA StyleScheggi, S., Pinna, G., Braccagni, G., De Montis, M. G., & Gambarana, C. (2022). PPARα Signaling: A Candidate Target in Psychiatric Disorder Management. Biomolecules, 12(5), 723. https://doi.org/10.3390/biom12050723