
Feature Reduction for Molecular Similarity Searching Based on Autoencoder Deep Learning

 ,
, 
 ,
,  and
and 
Abstract
:1. Introduction
- Proposing a novel ligand-based virtual screening dimensionality reduction method based on Autoencoder deep learning offers low-dimensional representations of molecular features while removing irrelevant and redundant features that affect similarity searching.
- Enhancing the effectiveness of the similarity searching by applying the proposed low dimensional representation of molecules.
- The proposed method has demonstrated superior results in terms of overall performances than the benchmark methods, e.g., TAN, ASMTP, and SQB.
2. Related Work
3. Materials and Methods
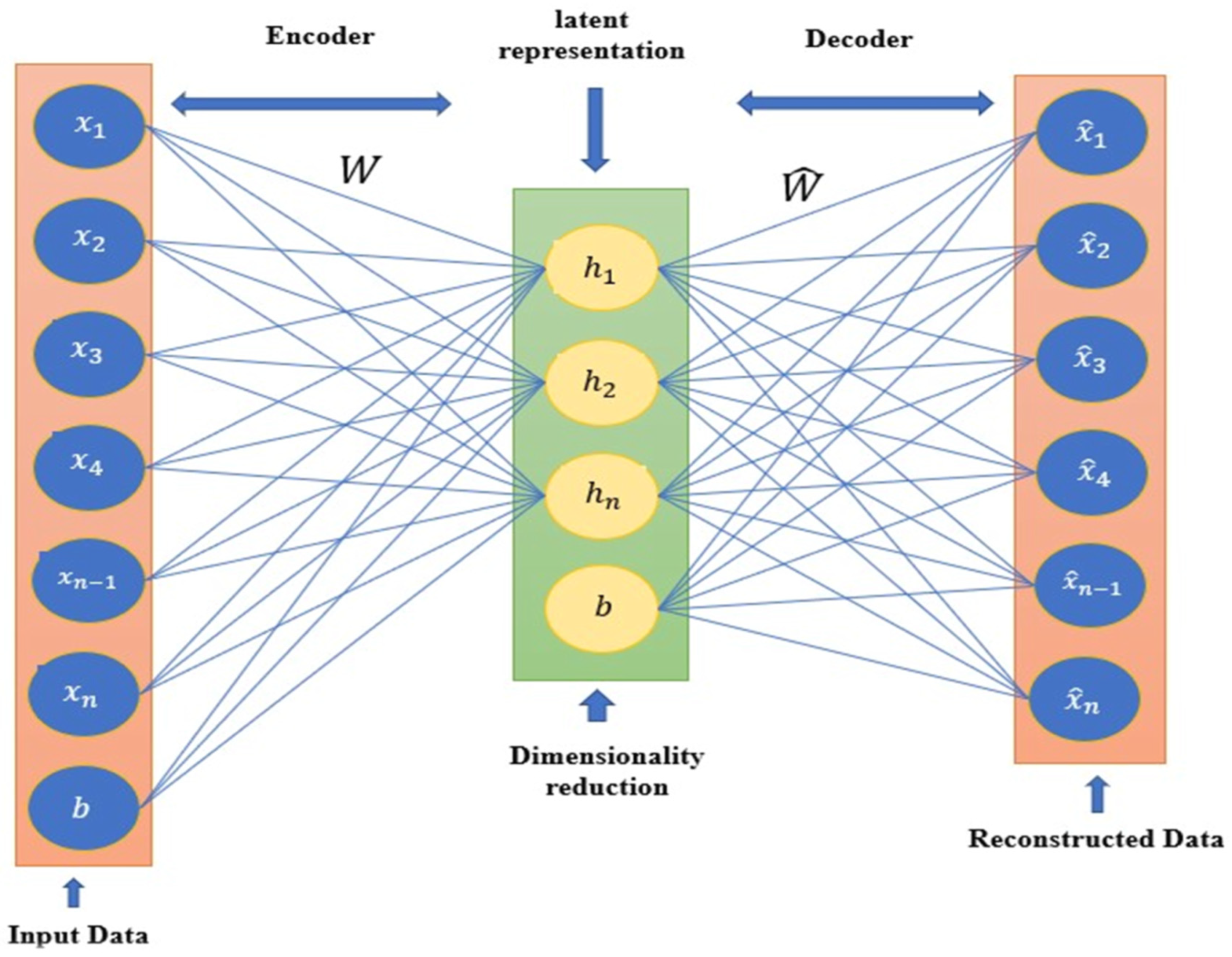
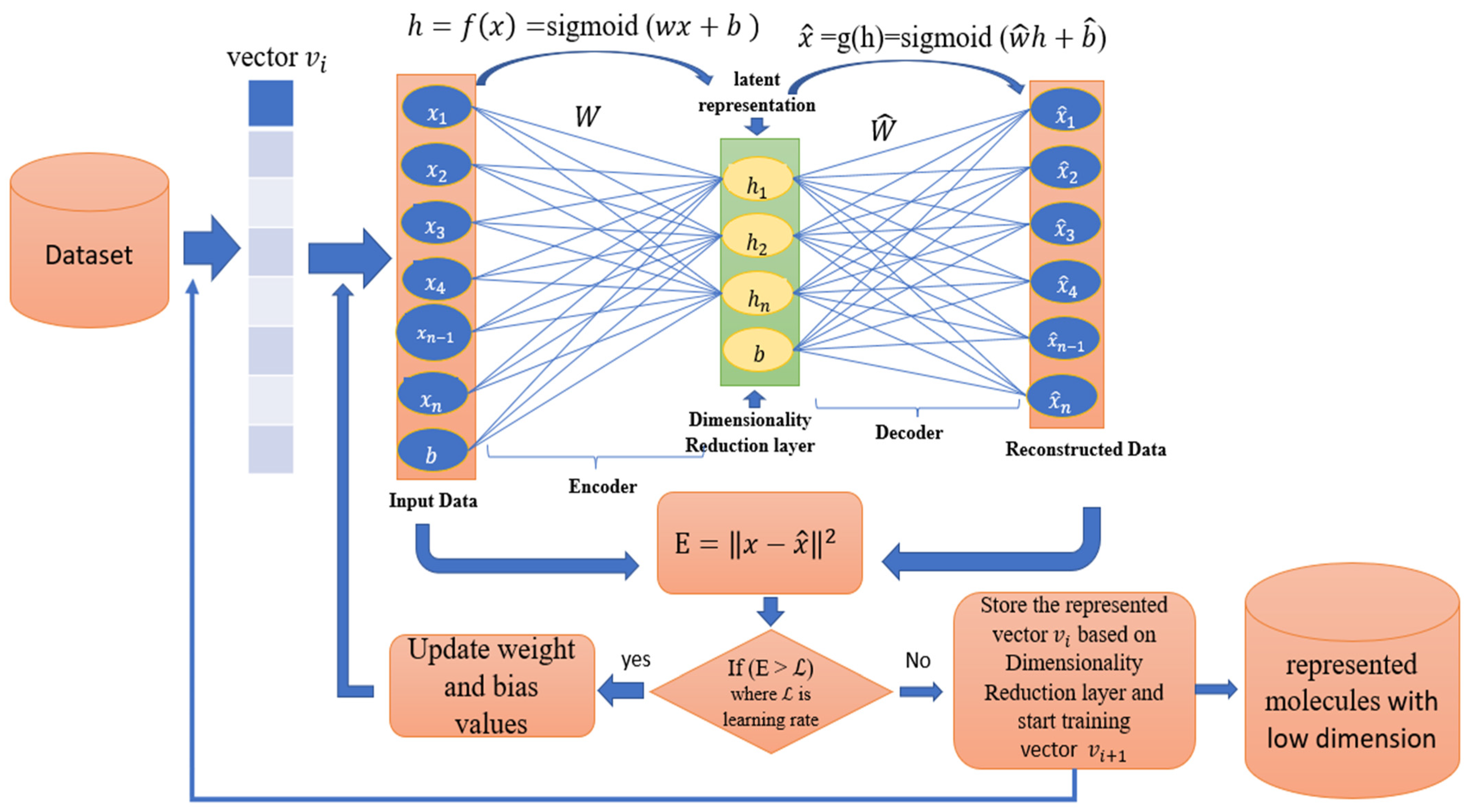
3.1. Dimensionality Reduction Based Autoencoder
3.2. Ability of Autoencoder for Molecular Dimensionality Reduction
| Algorithm 1: Autoencoder Algorithm. The Pseudocode of the proposed Autoencoder algorithm for 2D molecular fingerprints. |
| 1: Mols = 2 D figerprints dataset descriptor |
| 2: M = number of database molecules.// 102516 |
| 3: N = number of hidden layers. |
| 4: α = learning rate value. |
| 5: Epoch = 0; |
| 6: For k = 1:M // for all dataset molecules |
| 7: Input = Mlos(k) // input data |
| 8: x = mols(k); // initial the encoder input layer with molecule k. |
| 9: AE(x) // Autoencoder function |
| 10: For i = 1 until N //start the encoder phase |
| 11: If epoch = 0 do // first time training |
| 12: wi = random (0,1) // initial the weigh matrix for the first time training |
| 13: bi = random (0,1) // initial the bias vector for the first time training |
| 14: Else |
| 15: // update the weigh matrix based on the error value |
| 16: // update the bias vector based on the error value |
| 17: //calculate the hidden layers values based on Equation (1) |
| 18: x = hi // make the hidden layer values to be an input to the next hidden layer. |
| End // end encoder phase |
| 19: Encoded date = x //keep the last encoder layer which the new represented molecule. |
| 20: h = x //keep last encoder layer be an input encoder layer. |
| 21: n = N |
| 22: For j = 1 until N //start decoder phase |
| 23: = wnT //male the weight matrix of the decoder layer j be the transpose of n encoder weight matrix layer |
| 24: // make the value of the bias vector of the decoder layer j be the transpose of bias vector of n encoder layer. |
| 25: // calculate the zj reconstructed decoder layer values |
| 26: h = zj; // keep the hidden decoder layer values to be an input to the next hidden layer. |
| 27: n = n−1 |
| End // end decoder phase |
| 28: output = h // Reconstructed data |
| 29: // calculate the error value based on Equation (3) |
| 30: If () // if the error value is greater than the learning rate. |
| 31: Epoch = epoch+1 // need more training to reduce the error value |
| 32: Go to 9 // call the AE function again for new training—fine tune. |
| Else |
| 33: New_Rep_mols(k) = Encoded date;// |
3.3. Autoencoder Proposed Cases for Molecular Dimensionality Reduction
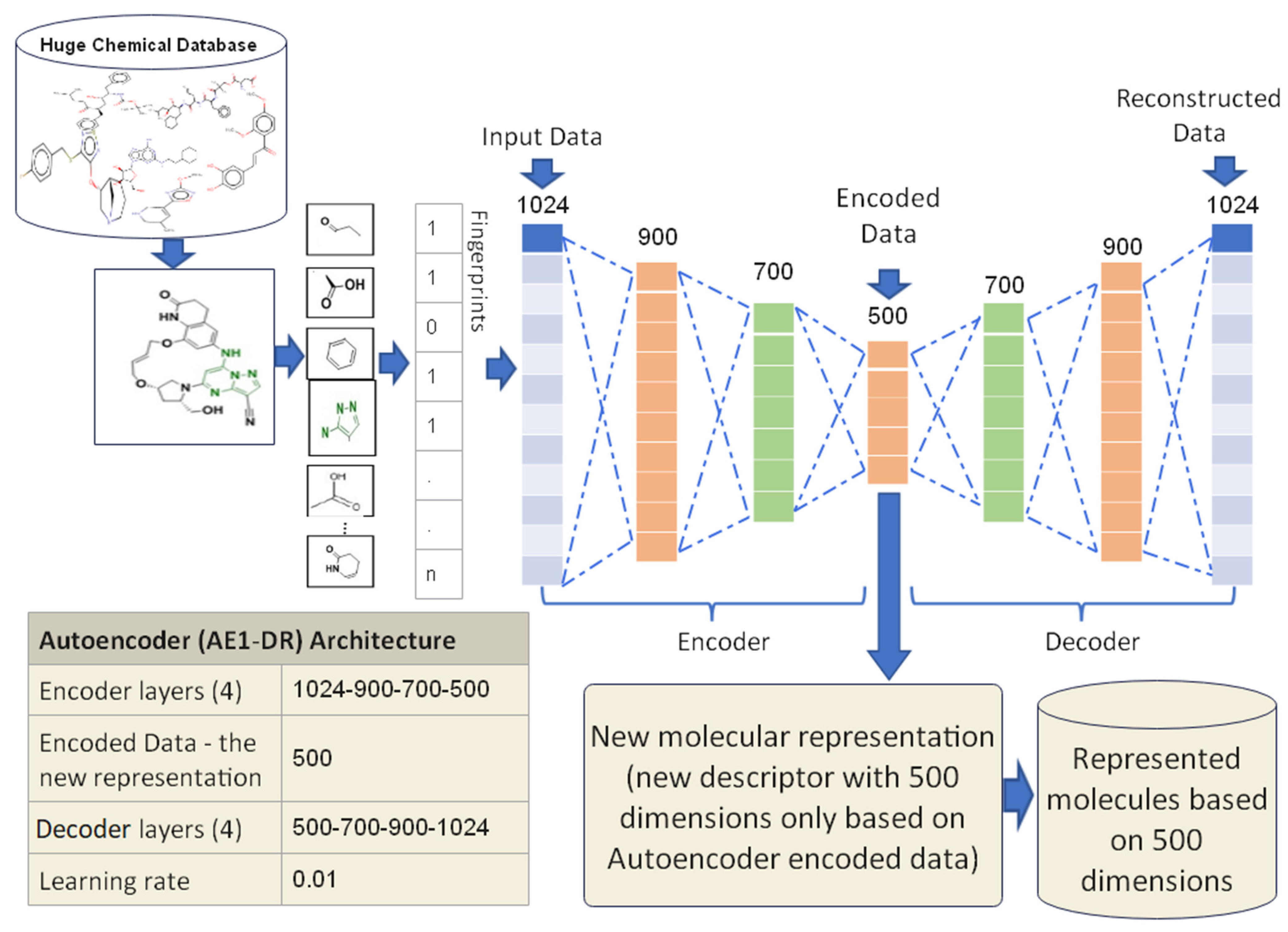
3.3.1. Proposed Autoencoder Case 1 (AE1-DR)
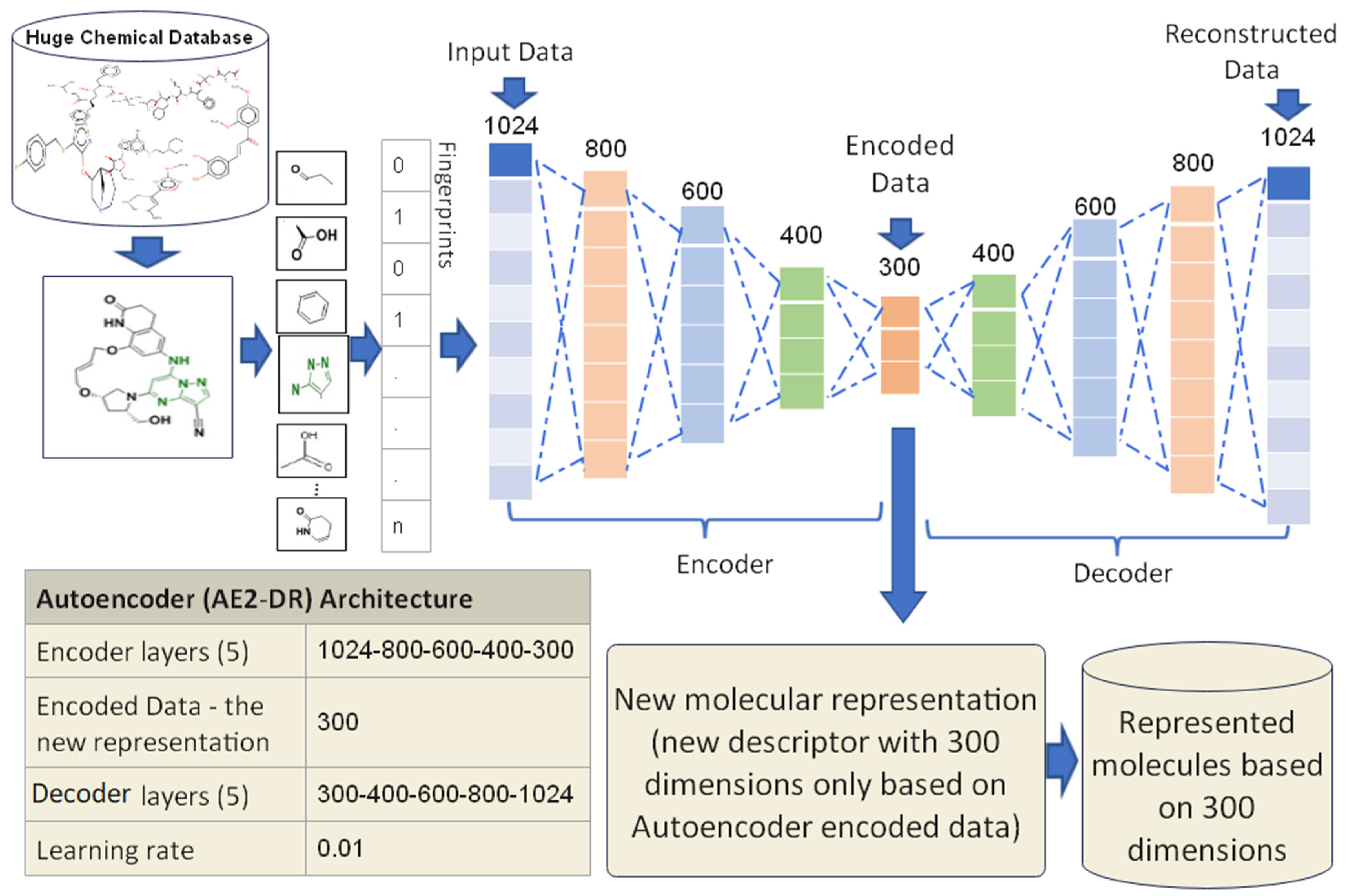
3.3.2. Proposed Autoencoder Case 2 (AE2-DR)
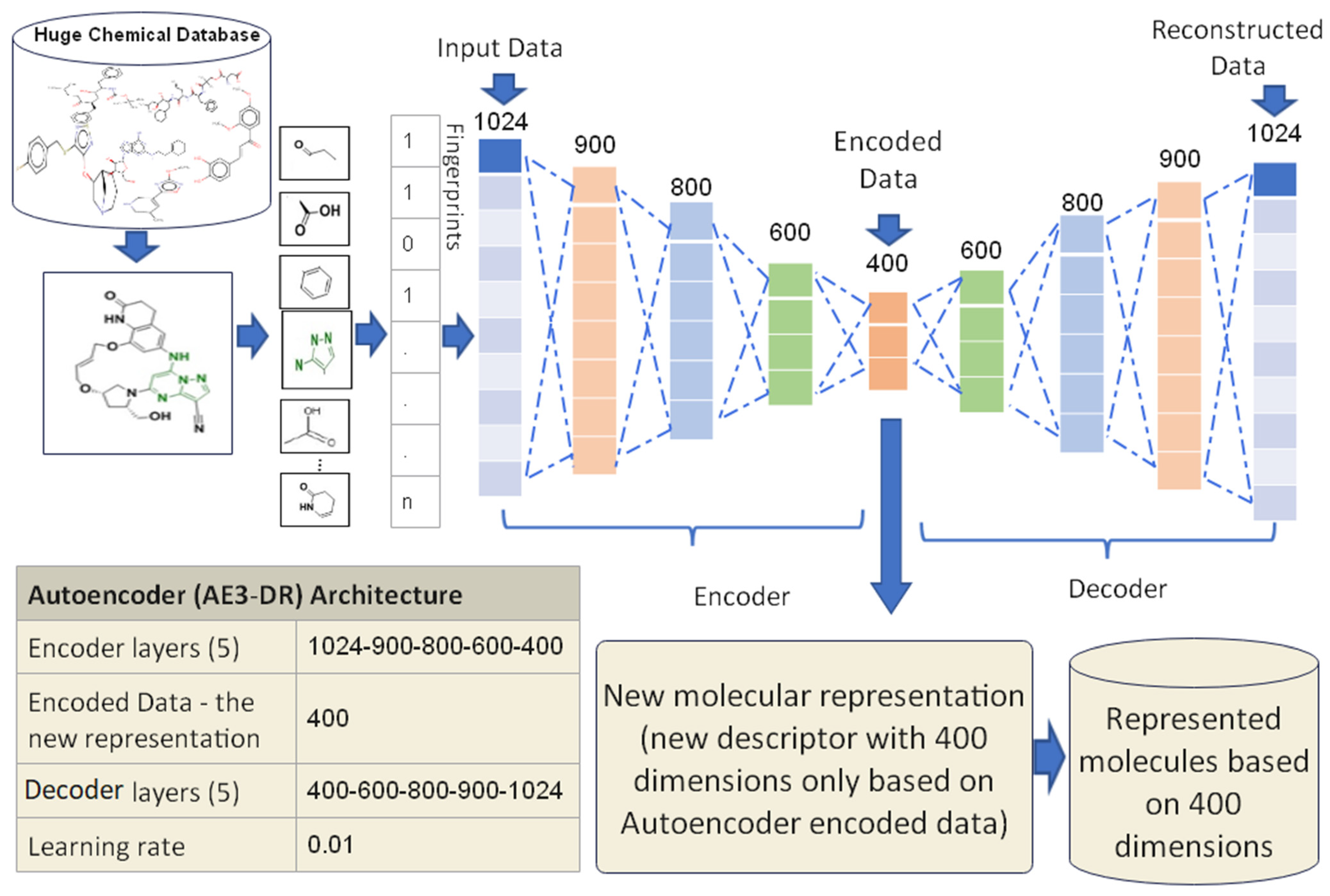
3.3.3. Proposed Autoencoder Case 3 (AE3-DR)
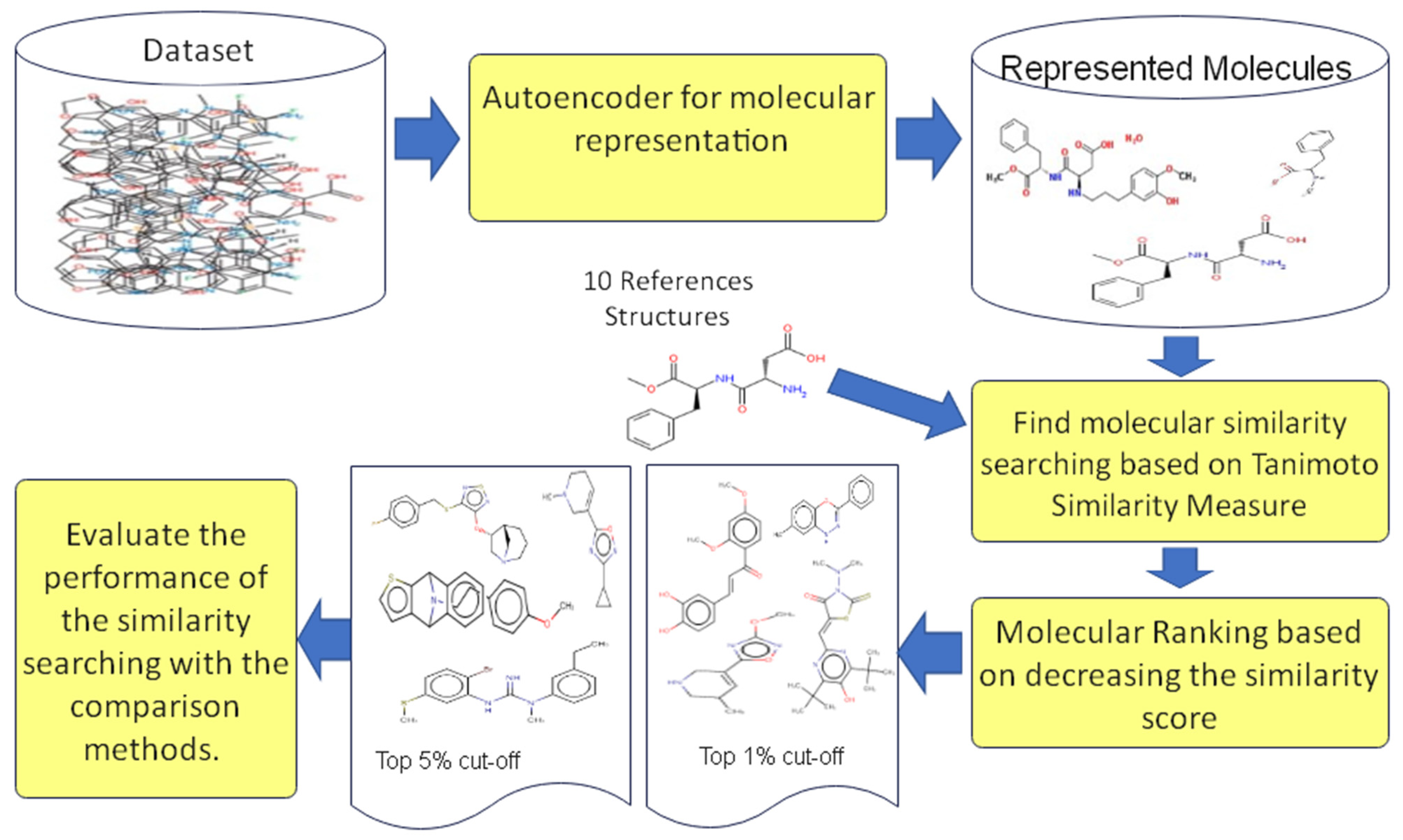
3.3.4. Similarity Searching Based Autoencoder Molecular Representation Using Tanimoto Similarity Measure
4. Experimental Design
4.1. Datasets
4.2. Evaluation Measures of the Performance
- Tanimoto Similarity Method (TAN) [63]: is used to calculate both binary and distance similarity coefficients.
- Adapted Similarity Measure of Text Processing (ASMTP) [39] is a similarity measure based on ligand-based virtual screening. It has been generated to utilize the process of chemical structure databases for a textual database.
- Quantum-Based Similarity Method (SQB) [38] is a method for determining molecular similarity based on quantum mechanics. To improve the model’s performance, the approach concentrates on the complex pure Hilbert space of molecules.
5. Experimental Results and Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rollinger, J.M.; Stuppner, H.; Langer, T. Virtual screening for the discovery of bioactive natural products. In Natural Compounds as Drugs; Springer: New York, NY, USA, 2008; pp. 211–249. [Google Scholar]
- Gonczarek, A.; Tomczak, J.M.; Zaręba, S.; Kaczmar, J.; Dąbrowski, P.; Walczak, M.J. Learning Deep Architectures for Interaction Prediction in Structure-based Virtual Screening. arXiv 2016, arXiv:1610.07187. [Google Scholar]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 2017, 3, 1237–1245. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent advances in scaffold hopping. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef]
- Vogt, M.; Bajorath, J. Chemoinformatics: A view of the field and current trends in method development. Bioorganic Med. Chem. 2012, 20, 5317–5323. [Google Scholar] [CrossRef]
- Bajorath, J. Molecular Similarity Concepts for Informatics Applications. In Bioinformatics: Volume II: Structure, Function, and Applications; Humana Press: New York, NY, USA, 2017; pp. 231–245. [Google Scholar]
- Maggiora, G.M. Introduction to molecular similarity and chemical space. In Foodinformatics; Springer: New York, NY, USA, 2014; pp. 1–81. [Google Scholar]
- Willett, P. Similarity searching using 2D structural fingerprints. In Chemoinformatics and Computational Chemical Biology; Springer: New York, NY, USA , 2010; pp. 133–158. [Google Scholar]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminformatics 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Vogt, M.; Wassermann, A.M.; Bajorath, J. Application of information—Theoretic concepts in chemoinformatics. Information 2010, 1, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Motoda, H. Computational Methods of Feature Selection; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Solorio-Fernández, S.; Carrasco-Ochoa, J.A.; Martínez-Trinidad, J.F. A review of unsupervised feature selection methods. Artif. Intell. Rev. 2020, 53, 907–948. [Google Scholar] [CrossRef]
- Le, Q.V. Building high-level features using large scale unsupervised learning. In Proceedings of the 2013 IEEE International Conference on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, 26–31 May 2013; pp. 8595–8598. [Google Scholar]
- Jia, Y.; Shelhamer, E.; Donahue, J.; Karayev, S.; Long, J.; Girshick, R.; Guadarrama, S.; Darrell, T. Caffe: Convolutional architecture for fast feature embedding. In Proceedings of the 22nd ACM International Conference on Multimedia, Orlando, FL, USA, 3–7 November 2014; pp. 675–678. [Google Scholar]
- Noda, K.; Yamaguchi, Y.; Nakadai, K.; Okuno, H.G.; Ogata, T. Audio-visual speech recognition using deep learning. Appl. Intell. 2015, 42, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Yu, D.; Dahl, G.E. Deep Belief Network for Large Vocabulary Continuous Speech Recognition. U.S. Patent 8,972,253, 3 March 2015. [Google Scholar]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed representations of words and phrases and their compositionality. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–10 December 2013; pp. 3111–3119. [Google Scholar]
- Gao, J.; He, X.; Deng, L. Deep Learning for Web Search and Natural Language Processing; Microsoft Technical Report; Microsoft Corporation: Redmond, WA, USA, 2015; MSR-TR-2015-7. [Google Scholar]
- Hamza, H.; Nasser, M.; Salim, N.; Saeed, F. Bioactivity prediction using convolutional neural network. In Proceedings of the International Conference of Reliable Information and Communication Technology, Johor, Malaysia, 22–23 September 2019; pp. 341–351. [Google Scholar]
- Hinton, G.; Deng, L.; Yu, D.; Dahl, G.E.; Mohamed, A.-R.; Jaitly, N.; Senior, A.; Vanhoucke, V.; Nguyen, P.; Sainath, T.N. Deep neural networks for acoustic modeling in speech recognition: The shared views of four research groups. IEEE Signal Processing Mag. 2012, 29, 82–97. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Tang, X. Deep convolutional network cascade for facial point detection. In Proceedings of the IEEE conference on Computer Vision and Pattern Recognition, Portland, OR, USA, 23–28 June 2013; pp. 3476–3483. [Google Scholar]
- Tompson, J.J.; Jain, A.; LeCun, Y.; Bregler, C. Joint training of a convolutional network and a graphical model for human pose estimation. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 8–13 December 2014; pp. 1799–1807. [Google Scholar]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Alsoufi, M.A.; Razak, S.; Siraj, M.M.; Al-Rimy, B.A.; Ali, A.; Nasser, M.; Abdo, S. A Review of Anomaly Intrusion Detection Systems in IoT using Deep Learning Techniques. Adv. Data Sci. Adapt. Anal. 2021, 72, 2143001. [Google Scholar] [CrossRef]
- Alsoufi, M.A.; Razak, S.; Siraj, M.M.; Nafea, I.; Ghaleb, F.A.; Saeed, F.; Nasser, M. Anomaly-based intrusion detection systems in iot using deep learning: A systematic literature review. Appl. Sci. 2021, 11, 8383. [Google Scholar] [CrossRef]
- Alsoufi, M.; Razak, S.; Siraj, M.M.; Ali, A.; Nasser, M.; Abdo, S. Anomaly Intrusion Detection Systems in IoT Using Deep Learning Techniques: A Survey. In Proceedings of the International Conference of Reliable Information and Communication Technology, Langkawi, Malaysia, 21–22 December 2020; pp. 659–675. [Google Scholar]
- Wang, H.; Raj, B. On the origin of deep learning. arXiv 2017, arXiv:1702.07800. [Google Scholar]
- Wang, H.; Yeung, D.-Y. Towards Bayesian deep learning: A survey. arXiv 2016, arXiv:1604.01662. [Google Scholar]
- Strub, F.; Mary, J. Collaborative filtering with stacked denoising autoencoders and sparse inputs. In Proceedings of the NIPS Workshop on Machine Learning for eCommerce, Montreal, QC, Canada, 27 December 2015. [Google Scholar]
- Liu, W.; Wang, Z.; Liu, X.; Zeng, N.; Liu, Y.; Alsaadi, F.E. A survey of deep neural network architectures and their applications. Neurocomputing 2017, 234, 11–26. [Google Scholar] [CrossRef]
- Zhang, C.; Patras, P.; Haddadi, H. Deep learning in mobile and wireless networking: A survey. IEEE Commun. Surv. Tutor. 2019, 21, 2224–2287. [Google Scholar] [CrossRef] [Green Version]
- Lemke, T.; Peter, C. EncoderMap: Dimensionality reduction and generation of molecule conformations. J. Chem. Theory Comput. 2019, 15, 1209–1215. [Google Scholar] [CrossRef]
- Willett, P. Textual and chemical information processing: Different domains but similar algorithms. Inf. Res. 2000, 5, XIII–XIV. [Google Scholar]
- Abdo, A.; Saeed, F.; Hamza, H.; Ahmed, A.; Salim, N. Ligand expansion in ligand-based virtual screening using relevance feedback. J. Comput.-Aided Mol. Des. 2012, 26, 279–287. [Google Scholar] [CrossRef]
- Abdo, A.; Salim, N.; Ahmed, A. Implementing relevance feedback in ligand-based virtual screening using Bayesian inference network. J. Biomol. Screen. 2011, 16, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Abdo, A.; Chen, B.; Mueller, C.; Salim, N.; Willett, P. Ligand-based virtual screening using bayesian networks. J. Chem. Inf. Modeling 2010, 50, 1012–1020. [Google Scholar] [CrossRef]
- Abdo, A.; Leclère, V.; Jacques, P.; Salim, N.; Pupin, M. Prediction of new bioactive molecules using a bayesian belief network. J. Chem. Inf. Modeling 2014, 54, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Al-Dabbagh, M.M.; Salim, N.; Himmat, M.; Ahmed, A.; Saeed, F. A quantum-based similarity method in virtual screening. Molecules 2015, 20, 18107–18127. [Google Scholar] [CrossRef]
- Himmat, M.; Salim, N.; Al-Dabbagh, M.M.; Saeed, F.; Ahmed, A. Adapting document similarity measures for ligand-based virtual screening. Molecules 2016, 21, 476. [Google Scholar] [CrossRef] [Green Version]
- Abdo, A.; Salim, N. New fragment weighting scheme for the bayesian inference network in ligand-based virtual screening. J. Chem. Inf. Modeling 2010, 51, 25–32. [Google Scholar] [CrossRef]
- Himmat, M.H.I. New Similarity Measures for Ligand-Based Virtual Screening; Universiti Teknologi Malaysia: Johor, Malaysia, 2017. [Google Scholar]
- Arif, S.M.; Holliday, J.D.; Willett, P. The Use of Weighted 2D Fingerprints in Similarity-Based Virtual Screening. In Advances in Mathematical Chemistry and Applications; Bentham Science Publishers: Oak Par, IL, USA, 2015. [Google Scholar]
- Ahmed, A.; Abdo, A.; Salim, N. Ligand-based Virtual screening using Bayesian inference network and reweighted fragments. Sci. World J. 2012, 2012, 410914. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.; Salim, N.; Abdo, A. Fragment Reweighting in Ligand-Based Virtual Screening. Adv. Sci. Lett. 2013, 19, 2782–2786. [Google Scholar] [CrossRef]
- Ahmed, A.; Abdo, A.; Salim, N. An enhancement of Bayesian inference network for ligand-based virtual screening using minifingerprints. In Proceedings of the Fourth International Conference on Machine Vision (ICMV 11), Singapore, 9 December 2011; p. 83502U-83502U-83505. [Google Scholar]
- Ahmed, A.; Abdo, A.; Salim, N. Ligand-based virtual screening using fuzzy correlation coefficient. Int. J. Comput. Appl. 2011, 19, 38–43. [Google Scholar]
- Willett, P. Combination of similarity rankings using data fusion. J. Chem. Inf. Modeling 2013, 53, 1–10. [Google Scholar] [CrossRef]
- Ahmed, A.; Saeed, F.; Salim, N.; Abdo, A. Condorcet and borda count fusion method for ligand-based virtual screening. J. Cheminform. 2014, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.L.; McMullen, S.A. Mathematical Techniques in Multisensor Data Fusion; Artech House: Norwood, MA, USA, 2004. [Google Scholar]
- Liggins II, M.; Hall, D.; Llinas, J. Handbook of Multisensor Data Fusion: Theory and Practice; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Salim, N.; Holliday, J.; Willett, P. Combination of fingerprint-based similarity coefficients using data fusion. J. Chem. Inf. Comput. Sci. 2003, 43, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Saeed, F.; Salim, N.; Abdo, A. Combining multiple clusterings of chemical structures using cluster-based similarity partitioning algorithm. Int. J. Comput. Biol. Drug Des. 2014, 7, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; O’Hagan, S.; Swainston, N.; Roberts, T.J.; Kell, D.B. VAE-Sim: A novel molecular similarity measure based on a variational autoencoder. Molecules 2020, 25, 3446. [Google Scholar] [CrossRef]
- Nasser, M.; Salim, N.; Hamza, H.; Saeed, F.; Rabiu, I. Features Reweighting and Selection in ligand-based Virtual Screening for Molecular Similarity Searching Based on Deep Belief Networks. Adv. Data Sci. Adapt. Anal. 2020, 12, 2050009. [Google Scholar] [CrossRef]
- Nasser, M.; Salim, N.; Hamza, H.; Saeed, F. Deep Belief Network for Molecular Feature Selection in Ligand-Based Virtual Screening. In Proceedings of the International Conference of Reliable Information and Communication Technology, Kuala Lumpur, Malaysia, 23–24 July 2018; pp. 3–14. [Google Scholar]
- Nasser, M.; Salim, N.; Hamza, H.; Saeed, F.; Rabiu, I. Improved deep learning based method for molecular similarity searching using stack of deep belief networks. Molecules 2021, 26, 128. [Google Scholar] [CrossRef]
- Nasser, M.; Salim, N.; Hamza, H. Molecular Similarity Searching Based on Deep Belief Networks with Different Molecular Descriptors. In Proceedings of the 2020 2nd International Conference on Big Data Engineering and Technology, Singapore, 3–5 January 2020; pp. 18–24. [Google Scholar]
- Ponti, M.A.; Ribeiro, L.S.F.; Nazare, T.S.; Bui, T.; Collomosse, J. Everything you wanted to know about deep learning for computer vision but were afraid to ask. In Proceedings of the 2017 30th SIBGRAPI Conference on Graphics, Patterns and Images Tutorials (SIBGRAPI-T), Niteroi, Brazil, 17–20 October 2017; pp. 17–41. [Google Scholar]
- Coates, A.; Ng, A.; Lee, H. An analysis of single-layer networks in unsupervised feature learning. In Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, Fort Lauderdale, FL, USA, 11–13 April 2011; pp. 215–223. [Google Scholar]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of generative autoencoder in de novo molecular design. Mol. Inform. 2018, 37, 1700123. [Google Scholar] [CrossRef] [Green Version]
- Pathirage, C.S.N.; Li, J.; Li, L.; Hao, H.; Liu, W.; Wang, R. Development and application of a deep learning–based sparse autoencoder framework for structural damage identification. Struct. Health Monit. 2019, 18, 103–122. [Google Scholar] [CrossRef] [Green Version]
- Konda, K.R. Unsupervised Relational Feature Learning for Vision. Ph.D. Thesis, Johann Wolfgang Goethe-University, Frankfurt am Main, Germany, 2016. [Google Scholar]
- Ellis, D.; Furner-Hines, J.; Willett, P. Measuring the degree of similarity between objects in text retrieval systems. Perspect. Inf. Manag. Annu. Rev. 1993, 3, 61108641. [Google Scholar]
- MDL Drug Data Report (MDDR). Available online: http://www.accelrys.com (accessed on 15 January 2020).
- San Diego Accelrys Inc. Pipeline Pilot Software: SciTegic Accelrys Inc; San Diego Accelrys Inc.: San Diego, CA, USA, 2008. [Google Scholar]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of fingerprint-based methods for virtual screening using multiple bioactive reference structures. J. Chem. Inf. Comput. Sci. 2004, 44, 1177–1185. [Google Scholar] [CrossRef]
- Legendre, P. Species associations: The Kendall coefficient of concordance revisited. J. Agric. Biol. Environ. Stat. 2005, 10, 226. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity Class | Active Molecules | Activity Index | Pairwise Similarity |
|---|---|---|---|
| Renin inhibitors | 1130 | 31,420 | 0.290 |
| HIV protease inhibitors | 750 | 71,523 | 0.198 |
| Thrombin inhibitors | 803 | 37,110 | 0.180 |
| Angiotensin II AT1 antagonists | 943 | 31,432 | 0.229 |
| Substance P antagonists | 1246 | 42,731 | 0.149 |
| 5HT3 antagonist | 752 | 06233 | 0.140 |
| 5HT reuptake inhibitors | 359 | 06245 | 0.122 |
| D2 antagonists | 395 | 07701 | 0.138 |
| 5HT1A agonists | 827 | 06235 | 0.133 |
| Protein kinase C inhibitors | 453 | 78,374 | 0.120 |
| Cyclooxygenase inhibitors | 636 | 78,331 | 0.108 |
| Activity Class | Active Molecules | Activity Index | Pairwise Similarity |
|---|---|---|---|
| Adenosine (A1) agonists | 207 | 07707 | 0.229 |
| Adenosine (A2) agonists | 156 | 07708 | 0.305 |
| Renin inhibitors | 1130 | 31,420 | 0.290 |
| CCK agonists | 111 | 42,710 | 0.361 |
| Monocyclic -lactams | 1346 | 64,100 | 0.336 |
| Cephalosporins | 113 | 64,200 | 0.322 |
| Carbacephems | 1051 | 64,220 | 0.269 |
| Carbapenems | 126 | 64,500 | 0.260 |
| Tribactams | 388 | 64,350 | 0.305 |
| Activity Class | Active Molecules | Activity Index | Pairwise Similarity |
|---|---|---|---|
| Muscarinic (M1) agonists | 900 | 09249 | 0.111 |
| NMDA receptor antagonists | 1400 | 12,455 | 0.098 |
| Nitric oxide synthase inhibitors | 505 | 12,464 | 0.102 |
| Dopamine -hydroxylase inhibitors | 106 | 31,281 | 0.125 |
| Aldose reductase inhibitors | 957 | 43,210 | 0.119 |
| Reverse transcriptase inhibitors | 700 | 71,522 | 0.103 |
| Aromatase inhibitors | 636 | 75,721 | 0.110 |
| Cyclooxygenase inhibitors | 636 | 78,331 | 0.108 |
| Phospholipase A2 inhibitors | 617 | 78,348 | 0.123 |
| Lipoxygenase inhibitors | 2111 | 78,351 | 0.113 |
| Activity Index | TAN | ASMTP | SQB | AE1_DR | AE2_DR | AE3_DR |
|---|---|---|---|---|---|---|
| 31,420 | 69.69 | 73.84 | 73.73 | 71.31 | 70.43 | 70.99 |
| 71,523 | 25.94 | 15.03 | 26.84 | 28.37 | 25.37 | 25.85 |
| 37,110 | 9.63 | 20.82 | 24.73 | 21.40 | 21.90 | 20.92 |
| 31,432 | 35.82 | 37.14 | 36.66 | 41.34 | 40.71 | 41.04 |
| 42,731 | 17.77 | 19.53 | 21.17 | 19.23 | 17.67 | 22.03 |
| 06233 | 13.87 | 10.35 | 12.49 | 13.01 | 14.04 | 14.87 |
| 06245 | 6.51 | 5.50 | 6.03 | 6.03 | 7.78 | 7.08 |
| 07701 | 8.63 | 7.99 | 11.35 | 9.87 | 8.91 | 12.31 |
| 06235 | 9.71 | 9.94 | 10.15 | 10.71 | 11.07 | 10.49 |
| 78,374 | 13.69 | 13.90 | 13.08 | 11.91 | 12.04 | 13.74 |
| 78,331 | 7.17 | 6.89 | 5.92 | 7.23 | 7.07 | 8.14 |
| Mean | 19.86 | 20.08 | 22.01 | 21.86 | 21.54 | 22.50 |
| Shaded cells | 0 | 2 | 1 | 2 | 2 | 4 |
| Activity Index | TAN | ASMTP | SQB | AE1_DR | AE2_DR | AE3_DR |
|---|---|---|---|---|---|---|
| 31,420 | 83.49 | 86 | 87.75 | 85.8 | 85.03 | 87.08 |
| 71,523 | 48.92 | 51.33 | 60.16 | 55.21 | 57.22 | 56.41 |
| 37,110 | 21.01 | 23.87 | 39.81 | 43.53 | 42.17 | 41.79 |
| 31,432 | 74.29 | 76.63 | 82 | 78.72 | 80.40 | 80.12 |
| 42,731 | 29.68 | 32.9 | 28.77 | 27.04 | 26.03 | 27.04 |
| 06233 | 27.68 | 26.2 | 20.96 | 23.8 | 24.11 | 25.19 |
| 06245 | 16.54 | 15.5 | 15.39 | 19.76 | 21.17 | 21.07 |
| 07701 | 24.09 | 23.9 | 26.90 | 25.21 | 24.78 | 26.25 |
| 06235 | 20.06 | 23.6 | 22.47 | 22.08 | 21.91 | 24.17 |
| 78,374 | 20.51 | 22.26 | 20.95 | 18.19 | 19.88 | 23.74 |
| 78,331 | 16.2 | 15 | 10.31 | 11.07 | 11.9 | 13.19 |
| Mean | 34.77 | 36.11 | 37.77 | 37.31 | 37.69 | 38.73 |
| Shaded cells | 2 | 1 | 3 | 0 | 1 | 3 |
| Activity Index | TAN | ASMTP | SQB | AE1_DR | AE2_DR | AE3_DR |
|---|---|---|---|---|---|---|
| 07707 | 61.84 | 67.86 | 72.09 | 70.15 | 73.18 | 73.46 |
| 07708 | 47.03 | 97.87 | 95.68 | 95.73 | 97.57 | 98.75 |
| 31,420 | 65.10 | 73.51 | 78.56 | 73.75 | 75.17 | 74.04 |
| 42,710 | 81.27 | 81.17 | 76.82 | 80.12 | 83.03 | 82.01 |
| 64,100 | 80.31 | 86.62 | 87.80 | 86.19 | 88.17 | 87.79 |
| 64,200 | 53.84 | 69.11 | 70.18 | 67.61 | 67.02 | 69.08 |
| 64,220 | 38.64 | 66.26 | 67.58 | 67.96 | 66.74 | 67.19 |
| 64,500 | 30.56 | 46.24 | 79.20 | 74.04 | 76.02 | 79.72 |
| 64,350 | 80.18 | 68.01 | 81.68 | 81.96 | 81.77 | 83.09 |
| 75,755 | 87.56 | 93.48 | 98.02 | 97.26 | 97.08 | 98.15 |
| Mean | 62.63 | 75.01 | 80.76 | 79.48 | 80.58 | 81.33 |
| Shaded cells | 0 | 0 | 2 | 1 | 2 | 5 |
| Activity Index | TAN | ASMTP | SQB | AE1_DR | AE2_DR | AE3_DR |
|---|---|---|---|---|---|---|
| 07707 | 70.39 | 76.17 | 74.22 | 73.33 | 77.78 | 80.24 |
| 07708 | 56.58 | 99.99 | 100 | 97.9 | 98.03 | 99.28 |
| 31,420 | 88.19 | 95.75 | 95.24 | 92.08 | 94.11 | 95.22 |
| 42,710 | 88.09 | 96.73 | 93 | 91.06 | 91.27 | 92.71 |
| 64,100 | 93.75 | 98.27 | 98.94 | 98.90 | 97.41 | 97.85 |
| 64,200 | 77.68 | 96.16 | 98.93 | 93.80 | 94.80 | 95.90 |
| 64,220 | 52.19 | 94.13 | 90.9 | 91.5 | 92.09 | 92.33 |
| 64,500 | 44.8 | 90.6 | 92.72 | 89.04 | 91.08 | 91.07 |
| 64,350 | 91.71 | 98.6 | 93.75 | 91.11 | 92.44 | 90.9 |
| 75,755 | 94.82 | 97.27 | 98.75 | 98.08 | 97.09 | 97.19 |
| Mean | 75.82 | 94.36 | 93.61 | 91.68 | 92.61 | 93.27 |
| Shaded cells | 0 | 4 | 5 | 0 | 0 | 1 |
| Activity Index | TAN | SQB | AE1_DR | AE2_DR | AE3_DR |
|---|---|---|---|---|---|
| 09249 | 12.12 | 10.99 | 15.01 | 16.03 | 17.76 |
| 12,455 | 6.57 | 7.03 | 7.88 | 9.17 | 6.77 |
| 12,464 | 8.17 | 6.92 | 11.12 | 12.50 | 12.04 |
| 31,281 | 16.95 | 18.67 | 17.66 | 17.75 | 16.5 |
| 43,210 | 6.27 | 6.83 | 9.76 | 9.07 | 10.90 |
| 71,522 | 3.75 | 6.57 | 7.19 | 9.14 | 9.02 |
| 75,721 | 17.32 | 20.38 | 22.29 | 21.66 | 23.90 |
| 78,331 | 6.31 | 6.16 | 6.09 | 5.06 | 8.98 |
| 78,348 | 10.15 | 8.99 | 9.11 | 6.89 | 6.40 |
| 78,351 | 9.84 | 12.5 | 14.02 | 15.78 | 16.06 |
| Mean | 9.75 | 10.50 | 12.01 | 12.31 | 12.83 |
| Shaded cells | 1 | 1 | 0 | 3 | 5 |
| Activity Index | TAN | SQB | AE1_DR | AE2_DR | AE3_DR |
|---|---|---|---|---|---|
| 09249 | 24.17 | 17.8 | 26.08 | 26.02 | 25.79 |
| 12,455 | 10.29 | 11.42 | 14.85 | 15.86 | 14.99 |
| 12,464 | 15.22 | 16.79 | 19.76 | 20.74 | 19.78 |
| 31,281 | 29.62 | 29.05 | 32.33 | 33.19 | 35.01 |
| 43,210 | 16.07 | 14.12 | 19.11 | 20.22 | 19.55 |
| 71,522 | 12.37 | 13.82 | 15.44 | 15.07 | 16.06 |
| 75,721 | 25.21 | 30.61 | 33.71 | 34.45 | 35.33 |
| 78,331 | 15.01 | 11.97 | 13.22 | 13.10 | 14.12 |
| 78,348 | 24.67 | 21.14 | 20.87 | 20.98 | 21.89 |
| 78,351 | 11.71 | 13.30 | 17.50 | 16.45 | 18.08 |
| Mean | 18.43 | 18.00 | 21.29 | 21.61 | 22.06 |
| Shaded cells | 2 | 0 | 1 | 3 | 4 |
| Data Set | Recall Cut-Off | W | P | Mean Rank | |||||
|---|---|---|---|---|---|---|---|---|---|
| TAN | ASMTP | SQB | AE1_DR | AE2_DR | AE3_DR | ||||
| DS1 | 1% | 0.19 | 0.00012 | 1.56 | 1.64 | 2.59 | 2.95 | 2.55 | 3.737 |
| DS1 | 5% | 0.11 | 0.03 | 1.73 | 2.55 | 2.82 | 2.05 | 2.36 | 3.5 |
| DS2 | 1% | 0.49 | 0.002 | 0.4 | 1.7 | 3.2 | 2.4 | 3.2 | 4.1 |
| DS2 | 5% | 0.61 | 0.001 | 0.2 | 4.4 | 4.4 | 1.8 | 2.6 | 3.1 |
| DS3 | 1% | 0.23 | 0.0001 | 1 | Not used | 1.4 | 2.3 | 2.6 | 2.7 |
| DS3 | 5% | 0.47 | 0.0011 | 1.1 | Not used | 0.7 | 2.2 | 2.7 | 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasser, M.; Salim, N.; Saeed, F.; Basurra, S.; Rabiu, I.; Hamza, H.; Alsoufi, M.A. Feature Reduction for Molecular Similarity Searching Based on Autoencoder Deep Learning. Biomolecules 2022, 12, 508. https://doi.org/10.3390/biom12040508
Nasser M, Salim N, Saeed F, Basurra S, Rabiu I, Hamza H, Alsoufi MA. Feature Reduction for Molecular Similarity Searching Based on Autoencoder Deep Learning. Biomolecules. 2022; 12(4):508. https://doi.org/10.3390/biom12040508
Chicago/Turabian StyleNasser, Maged, Naomie Salim, Faisal Saeed, Shadi Basurra, Idris Rabiu, Hentabli Hamza, and Muaadh A. Alsoufi. 2022. "Feature Reduction for Molecular Similarity Searching Based on Autoencoder Deep Learning" Biomolecules 12, no. 4: 508. https://doi.org/10.3390/biom12040508
APA StyleNasser, M., Salim, N., Saeed, F., Basurra, S., Rabiu, I., Hamza, H., & Alsoufi, M. A. (2022). Feature Reduction for Molecular Similarity Searching Based on Autoencoder Deep Learning. Biomolecules, 12(4), 508. https://doi.org/10.3390/biom12040508