
In Silico Discovery and Optimisation of a Novel Structural Class of Hsp90 C-Terminal Domain Inhibitors



Abstract
1. Introduction
2. Materials and Methods
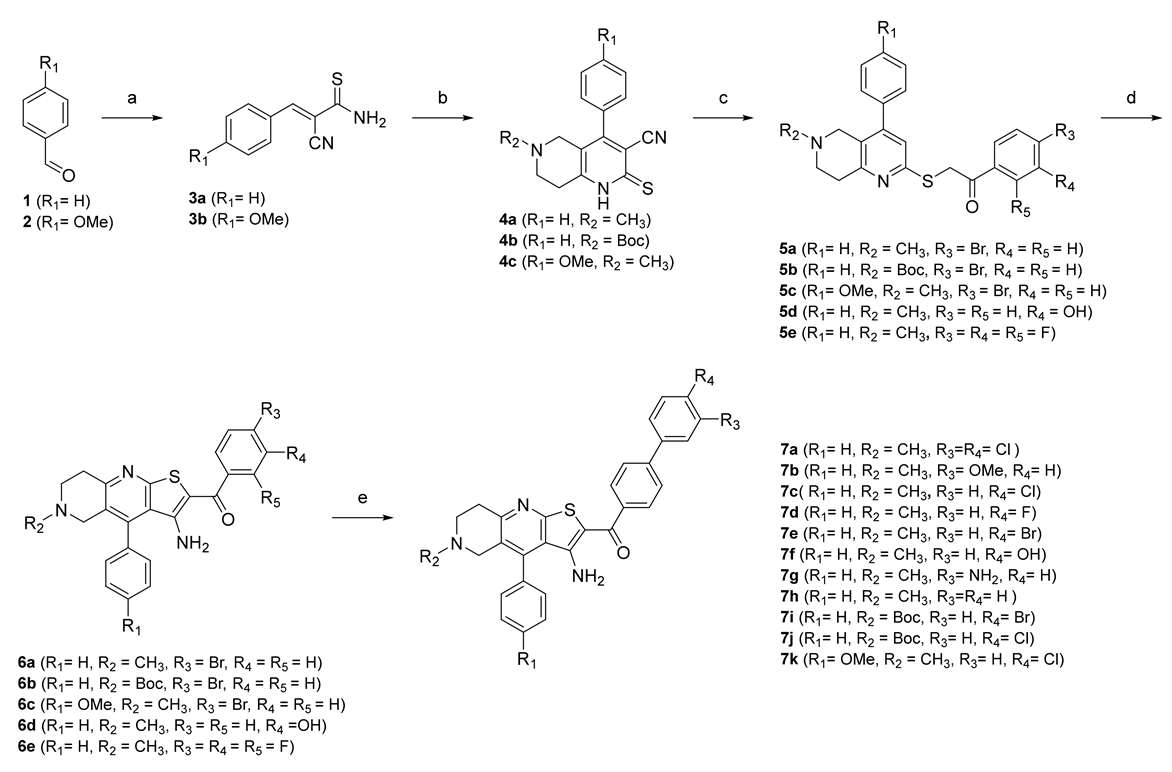
2.1. Chemistry
2.2. Virtual Screening
2.2.1. Preparation of the Compound Library
2.2.2. Virtual Screening
2.3. Molecular Dynamics Simulations
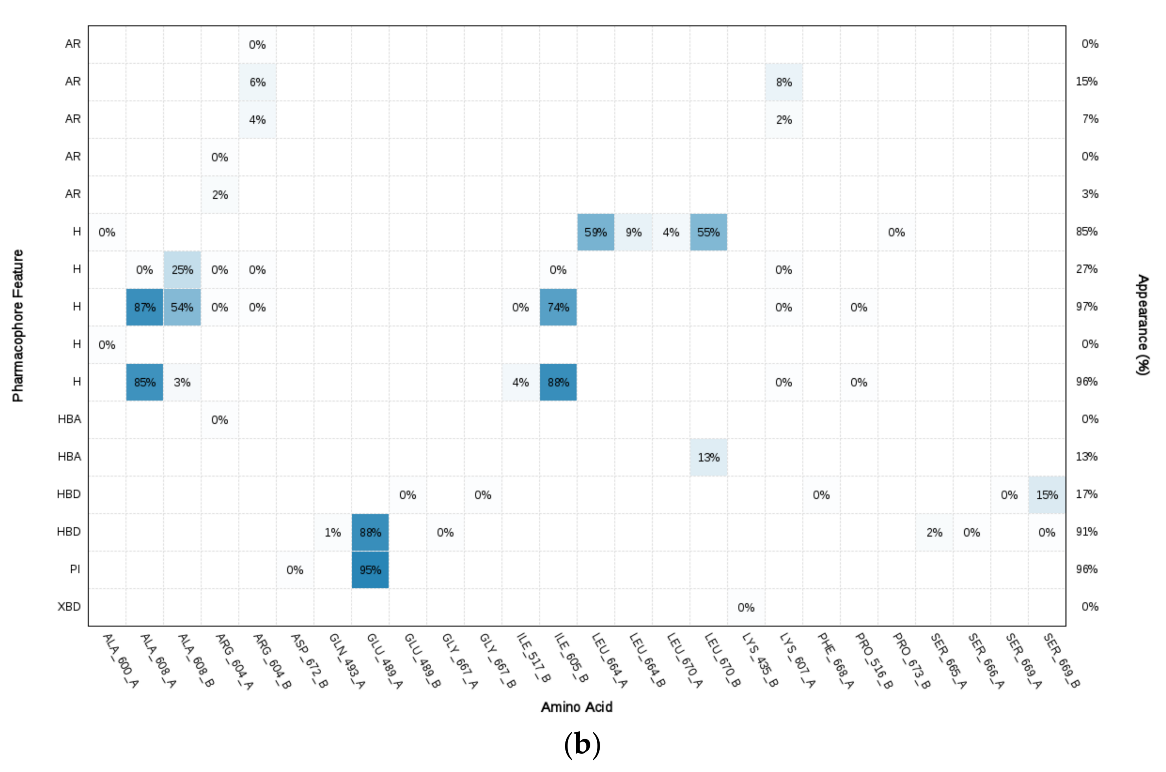
2.4. Structure-Based Pharmacophore Modeling
2.5. MTS Assay
Luciferase Refolding Assay
3. Results and Discussion
3.1. Virtual Screening
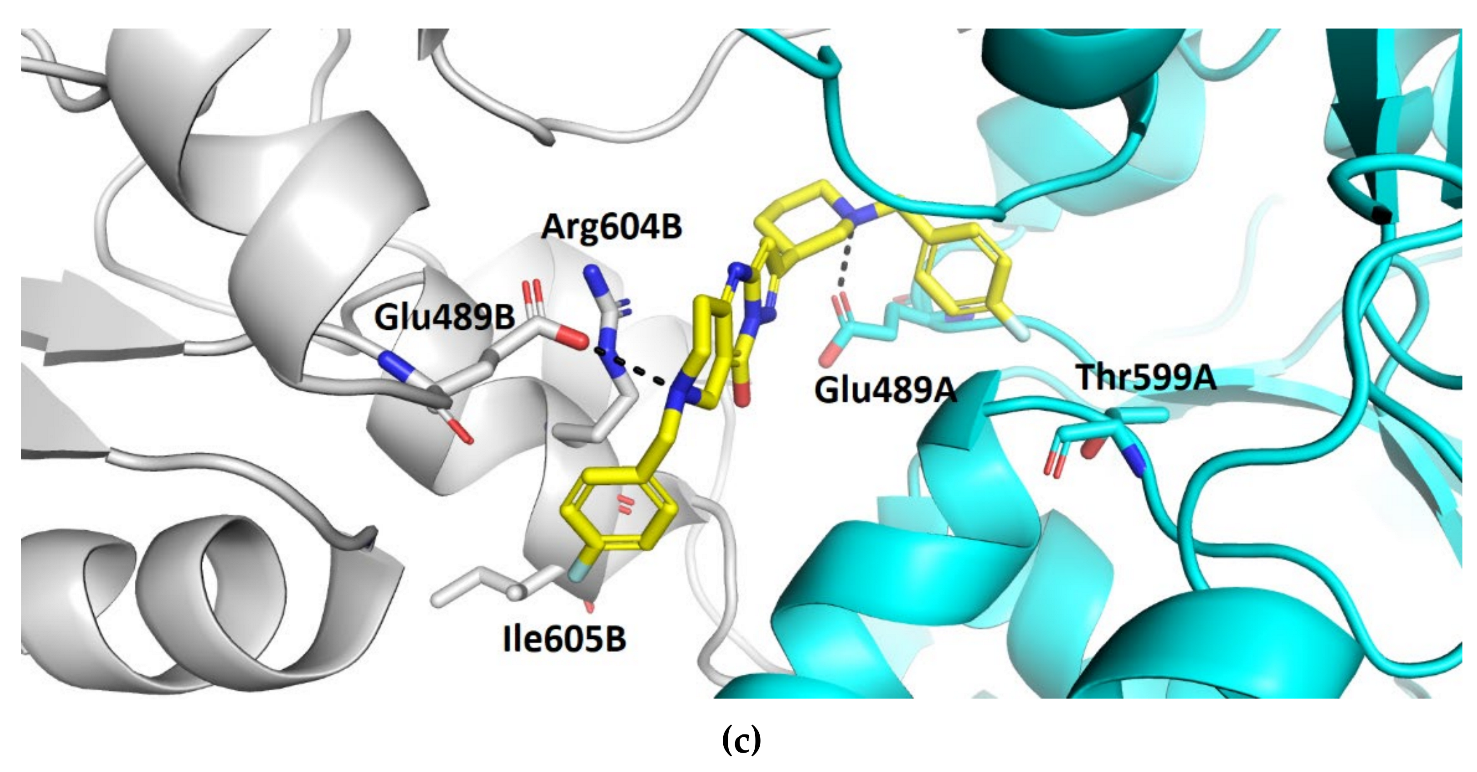
3.2. Molecular Dynamics Simulation
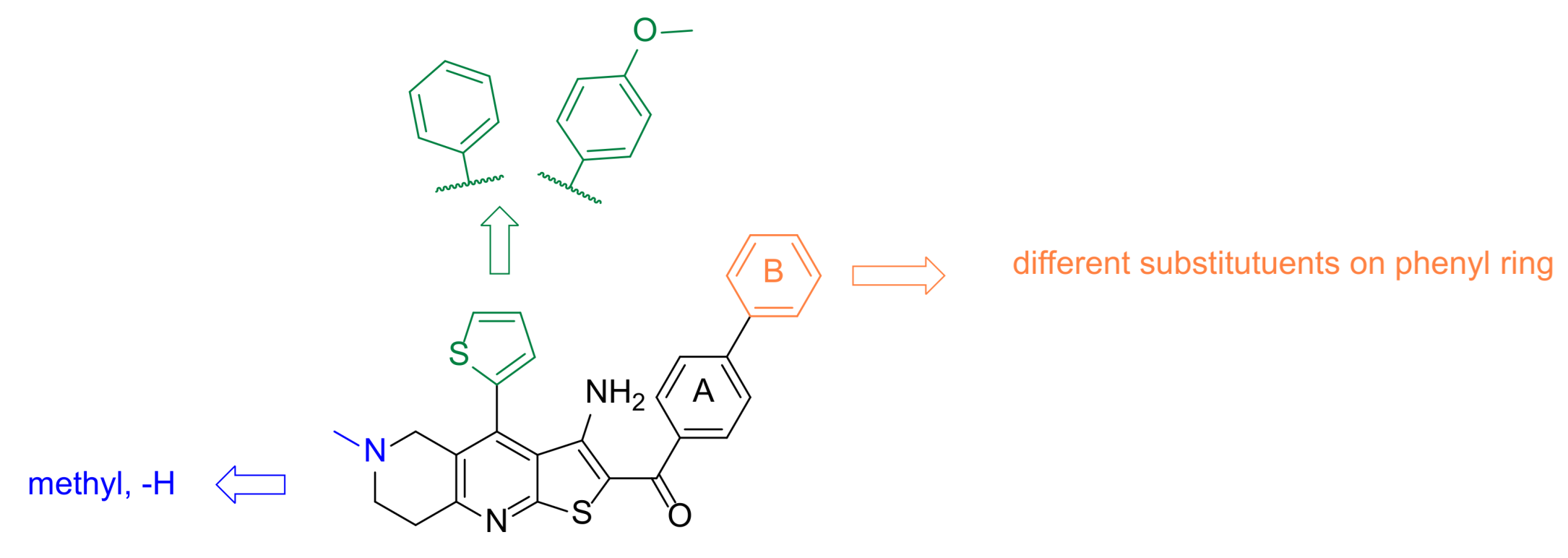
3.3. Design and Synthesis
3.4. Biological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the Hub of Protein Homeostasis: Emerging Mechanistic Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the Chaperoning of Cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat Shock Protein 90 as a Molecular Target for Cancer Therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Rüdiger, S.G.D. Hsp90 Interaction with Clients. Trends Biochem. Sci. 2015, 40, 117–125. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Kamal, A.; Boehm, M.F.; Burrows, F.J. Therapeutic and Diagnostic Implications of Hsp90 Activation. Trends Mol. Med. 2004, 10, 283–290. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.; Naim, H. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Prodromou, C.; Pearl, L. Structure and Functional Relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar] [CrossRef]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef]
- Subbarao, S.A.; Kalmár, É.; Csermely, P.; Shen, Y.-F. Hsp90 Isoforms: Functions, Expression and Clinical Importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Whitesell, L.; Neckers, L.M. Inhibition of Heat Shock Protein HSP90-Pp6Ov-Src Heteroprotein Complex Formation by Benzoquinone Ansamycins: Essential Role for Stress Proteins in Oncogenic Transformation. Cell Biol. 1994, 91, 8324–8328. [Google Scholar]
- Yuno, A.; Lee, M.-J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. In Chaperones, Methods in Molecular Biology; Calderwood, S.K., Prince, T.L., Eds.; Springer: New York, NY, USA, 2018; Volume 1709, pp. 423–441. ISBN 978-1-4939-7476-4. [Google Scholar]
- Khandelwal, A.; Crowley, V.M.; Blagg, B.S.J. Natural Product Inspired N-Terminal Hsp90 Inhibitors: From Bench to Bedside?: N-Terminal Hsp90 Inhibitors. Med. Res. Rev. 2016, 36, 92–118. [Google Scholar] [CrossRef]
- Bagatell, R.; Paine-Murrieta, G.D.; Taylor, C.W.; Pulcini, E.J.; Akinaga, S.; Benjamin, I.J.; Whitesell, L. Induction of a Heat Shock Factor 1-Dependent Stress Response Alters the Cytotoxic Activity of Hsp90-Binding Agents. Clin. Cancer Res. 2000, 6, 3312–3318. [Google Scholar]
- Kim, H.R.; Kang, H.S.; Kim, H.D. Geldanamycin Induces Heat Shock Protein Expression Through Activation of HSF1 in K562 Erythroleukemic Cells. IUBMB Life 1999, 48, 429–433. [Google Scholar] [CrossRef]
- Whitesell, L.; Bagatell, R.; Falsey, R. The Stress Response: Implications for the Clinical Development of Hsp90 Inhibitors. Curr. Cancer Drug Targets 2003, 3, 349–358. [Google Scholar] [CrossRef]
- McCollum, A.K.; TenEyck, C.J.; Sauer, B.M.; Toft, D.O.; Erlichman, C. Up-Regulation of Heat Shock Protein 27 Induces Resistance to 17-Allylamino-Demethoxygeldanamycin through a Glutathione-Mediated Mechanism. Cancer Res. 2006, 66, 10967–10975. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the Clinical Development of Heat Shock Protein 90 (Hsp90) Inhibitors in Cancers. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef]
- Koren, J.; Blagg, B.S.J. The Right Tool for the Job: An Overview of Hsp90 Inhibitors. In HSF1 and Molecular Chaperones in Biology and Cancer; Advances in Experimental Medicine and Biology; Mendillo, M.L., Pincus, D., Scherz, -S.R., Eds.; Springer International Publishing: Cham, Switzerland, 2020; Volume 1243, pp. 135–146. ISBN 978-3-030-40203-7. [Google Scholar]
- Brandt, G.; Blagg, B. Alternate Strategies of Hsp90 Modulation for the Treatment of Cancer and Other Diseases. Curr. Top. Med. Chem. 2009, 9, 1447–1461. [Google Scholar] [CrossRef]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Marcu, M.G.; Chadli, A.; Bouhouche, I.; Catelli, M.; Neckers, L.M. The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-Binding Domain in the Carboxyl Terminus of the Chaperone. J. Biol. Chem. 2000, 275, 37181–37186. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and Related Coumarins and Depletion of Heat Shock Protein 90-Dependent Signaling Proteins. JNCI J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef]
- Donnelly, A.; Blagg, B. Novobiocin and Additional Inhibitors of the Hsp90 C-Terminal Nucleotide- Binding Pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [Google Scholar] [CrossRef]
- Yu, X.M.; Shen, G.; Neckers, L.; Blake, H.; Holzbeierlein, J.; Cronk, B.; Blagg, B.S.J. Hsp90 Inhibitors Identified from a Library of Novobiocin Analogues. J. Am. Chem. Soc. 2005, 127, 12778–12779. [Google Scholar] [CrossRef]
- Zhao, H.; Moroni, E.; Colombo, G.; Blagg, B.S.J. Identification of a New Scaffold for Hsp90 C-Terminal Inhibition. ACS Med. Chem. Lett. 2014, 5, 84–88. [Google Scholar] [CrossRef]
- Garg, G.; Forsberg, L.K.; Zhao, H.; Blagg, B.S.J. Development of Phenyl Cyclohexylcarboxamides as a Novel Class of Hsp90 C-Terminal Inhibitors. Chem. Eur. J. 2017, 23, 16574–16585. [Google Scholar] [CrossRef]
- Byrd, K.M.; Subramanian, C.; Sanchez, J.; Motiwala, H.F.; Liu, W.; Cohen, M.S.; Holzbeierlein, J.; Blagg, B.S.J. Synthesis and Biological Evaluation of Novobiocin Core Analogues as Hsp90 Inhibitors. Chem. Eur. J. 2016, 22, 6921–6931. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, Y.-J.; Park, S.; Park, M.; Farrand, L.; Nguyen, C.-T.; Ann, J.; Nam, G.; Park, H.-J.; Lee, J.; et al. A Novel HSP90 Inhibitor Targeting the C-Terminal Domain Attenuates Trastuzumab Resistance in HER2-Positive Breast Cancer. Mol. Cancer 2020, 19, 161. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.-J.; Park, J.M.; Park, M.; Nam, K.D.; Farrand, L.; Nguyen, C.-T.; La, M.T.; Ann, J.; Lee, J.; et al. The C-Terminal HSP90 Inhibitor NCT-58 Kills Trastuzumab-Resistant Breast Cancer Stem-like Cells. Cell Death Discov. 2021, 7, 354. [Google Scholar] [CrossRef]
- Nguyen, C.-T.; La, M.T.; Ann, J.; Nam, G.; Park, H.-J.; Min Park, J.; Kim, Y.-J.; Young Kim, J.; Hong Seo, J.; Lee, J. Discovery of a Simplified Deguelin Analog as an HSP90 C-Terminal Inhibitor for HER2-Positive Breast Cancer. Bioorg. Med. Chem. Lett. 2021, 45, 128134. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Micol, V.; Joven, J.; Bosch, B.J.; Encinar, J.A.; Menendez, J.A. Revisiting Silibinin as a Novobiocin-like Hsp90 C-Terminal Inhibitor: Computational Modeling and Experimental Validation. Food Chem. Toxicol. 2019, 132, 110645. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Henry, E.C.; Gasiewicz, T.A. (−)-Epigallocatechin-3-Gallate Is a Novel Hsp90 Inhibitor. Biochemistry 2009, 48, 336–345. [Google Scholar] [CrossRef]
- Pugh, K.W.; Zhang, Z.; Wang, J.; Xu, X.; Munthali, V.; Zuo, A.; Blagg, B.S.J. From Bacteria to Cancer: A Benzothiazole-Based DNA Gyrase B Inhibitor Redesigned for Hsp90 C-Terminal Inhibition. ACS Med. Chem. Lett. 2020, 11, 1535–1538. [Google Scholar] [CrossRef]
- Dernovšek, J.; Zajec, Ž.; Durcik, M.; Mašič, L.P.; Gobec, M.; Zidar, N.; Tomašič, T. Structure-Activity Relationships of Benzothiazole-Based Hsp90 C-Terminal-Domain Inhibitors. Pharmaceutics 2021, 13, 1283. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic Structure of Hsp90-Cdc37-Cdk4 Reveals That Hsp90 Traps and Stabilizes an Unfolded Kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Noddings, C.M.; Wang, R.Y.-R.; Johnson, J.L.; Agard, D.A. Structure of Hsp90–P23–GR Reveals the Hsp90 Client-Remodelling Mechanism. Nature 2022, 601, 465–469. [Google Scholar] [CrossRef]
- Tomašič, T.; Durcik, M.; Keegan, B.M.; Skledar, D.G.; Zajec, Ž.; Blagg, B.S.J.; Bryant, S.D. Discovery of Novel Hsp90 C-Terminal Inhibitors Using 3D-Pharmacophores Derived from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2020, 21, 6898. [Google Scholar] [CrossRef]
- Jiang, F.; Guo, A.; Xu, J.; Wang, H.-J.; Mo, X.; You, Q.-D.; Xu, X.-L. Identification and Optimization of Novel 6-Acylamino-2-Aminoquinolines as Potent Hsp90 C-Terminal Inhibitors. Eur. J. Med. Chem. 2017, 141, 1–14. [Google Scholar] [CrossRef]
- Moroni, E.; Zhao, H.; Blagg, B.S.J.; Colombo, G. Exploiting Conformational Dynamics in Drug Discovery: Design of C-Terminal Inhibitors of Hsp90 with Improved Activities. J. Chem. Inf. Model. 2014, 54, 195–208. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Sadikot, T.; Swink, M.; Eskew, J.D.; Brown, D.; Zhao, H.; Kusuma, B.R.; Rajewski, R.A.; Blagg, B.S.J.; Matts, R.L.; Holzbeierlein, J.M.; et al. Development of a High-Throughput Screening Cancer Cell-Based Luciferase Refolding Assay for Identifying Hsp90 Inhibitors. ASSAY Drug Dev. Technol. 2013, 11, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Tomašič, T.; Zubrienė, A.; Skok, Ž.; Martini, R.; Pajk, S.; Sosič, I.; Ilaš, J.; Matulis, D.; Bryant, S.D. Selective DNA Gyrase Inhibitors: Multi-Target in Silico Profiling with 3D-Pharmacophores. Pharmaceuticals 2021, 14, 789. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | IC50 (µM) | |
|---|---|---|---|
| MCF-7 | SK-N-MC | ||
| TVS-23 | 26.4 ± 1.1 | 24.3 ± 4.0 | |
| 17-DMAG * | 0.9 ± 1 | 0.01 ± 0.007 | |
| 6e |  | >50 | >50 |
| 6d |  | >50 | >50 |
| Compound |  | IC50 (µM) | |
|---|---|---|---|
| MCF-7 | SK-N-MC | ||
| 7a |  | 20.0 ± 3.1 | 16.9 ± 2.9 |
| 7b |  | 32.4 ± 5.4 | 21.8 ± 4.8 |
| 7c |  | 10.6 ± 0.3 | 5.5 ± 0.6 |
| 7d |  | 15.7 ± 2.2 | 9.1 ± 1.4 |
| 7e |  | 7.7 ± 0.8 | 3.6 ± 0.5 |
| 7f |  | 27.4 ± 0.5 | 16.4 ± 1.7 |
| 7g |  | 14.4 ± 3.9 | 14.4 ± 1.3 |
| 7h |  | 27.3 ± 2.2 | 20.5 ± 2.0 |
| Compound |  | IC50 (µM) | |
|---|---|---|---|
| MCF-7 | SK-N-MC | ||
| 7l |  | 1.4 ± 0.4 | 2.8 ± 0.4 |
| 7m |  | 6.4 ± 0.1 | 2.6 ± 0.2 |
| 7k |  | 46.7 ± 4.8 | 15.4 ± 4.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajec, Ž.; Dernovšek, J.; Gobec, M.; Tomašič, T. In Silico Discovery and Optimisation of a Novel Structural Class of Hsp90 C-Terminal Domain Inhibitors. Biomolecules 2022, 12, 884. https://doi.org/10.3390/biom12070884
Zajec Ž, Dernovšek J, Gobec M, Tomašič T. In Silico Discovery and Optimisation of a Novel Structural Class of Hsp90 C-Terminal Domain Inhibitors. Biomolecules. 2022; 12(7):884. https://doi.org/10.3390/biom12070884
Chicago/Turabian StyleZajec, Živa, Jaka Dernovšek, Martina Gobec, and Tihomir Tomašič. 2022. "In Silico Discovery and Optimisation of a Novel Structural Class of Hsp90 C-Terminal Domain Inhibitors" Biomolecules 12, no. 7: 884. https://doi.org/10.3390/biom12070884
APA StyleZajec, Ž., Dernovšek, J., Gobec, M., & Tomašič, T. (2022). In Silico Discovery and Optimisation of a Novel Structural Class of Hsp90 C-Terminal Domain Inhibitors. Biomolecules, 12(7), 884. https://doi.org/10.3390/biom12070884