
α-Synuclein at the Presynaptic Axon Terminal as a Double-Edged Sword

 ,
,
Abstract
:1. Introduction
2. The α-Syn Protein
2.1. α-Syn Splice Variants and Isoforms
2.2. Dynamic Alterations to 3D Structures of α-Syn-140
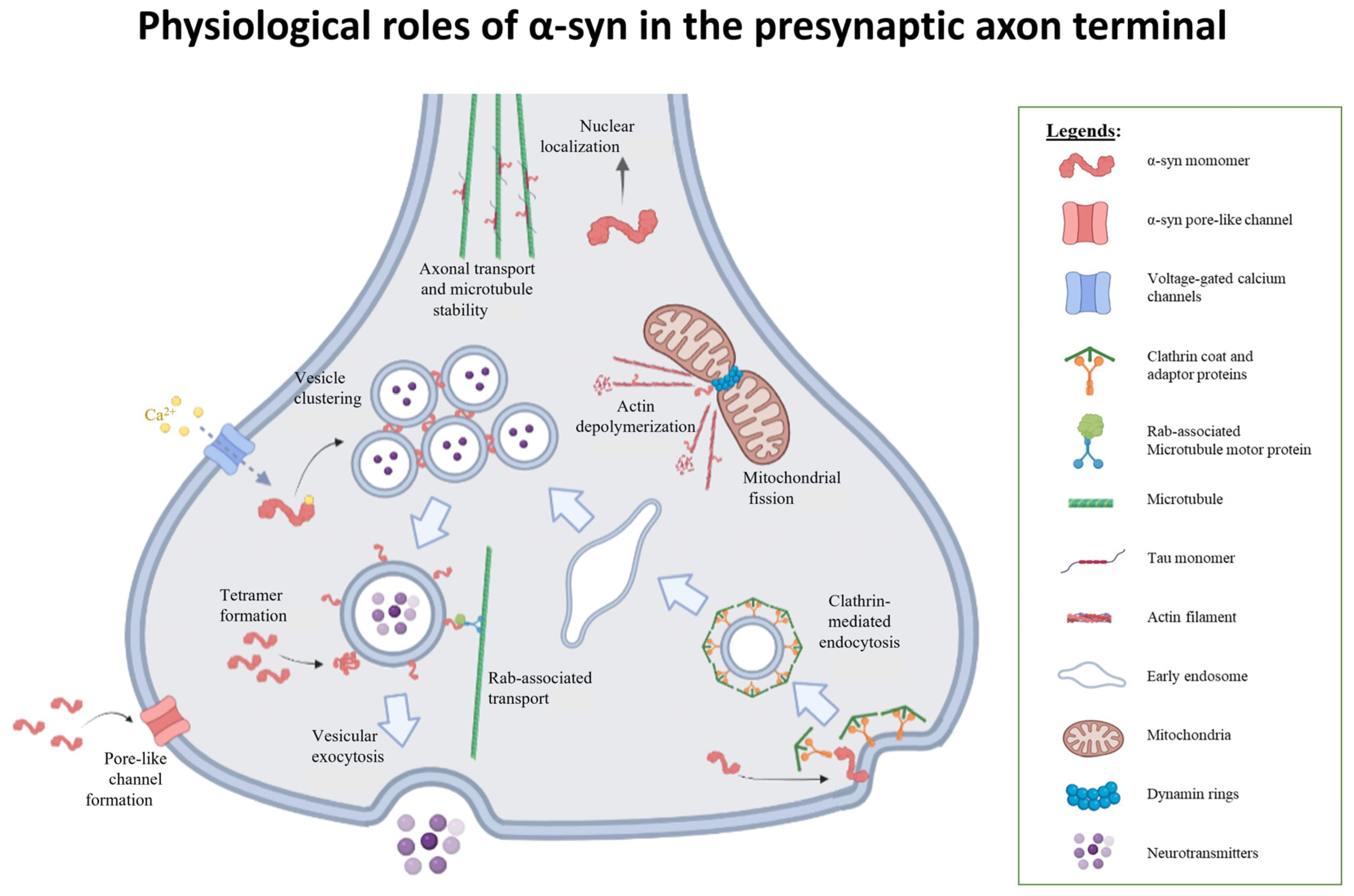
2.3. Involvement in Synaptic Vesicle Exocytosis
2.4. Involvement in Synaptic Recycling and Endocytosis
3. Pathological Impact of α-Syn Oligomers
3.1. Prevalence of α-Syn Mutations in Human Patients
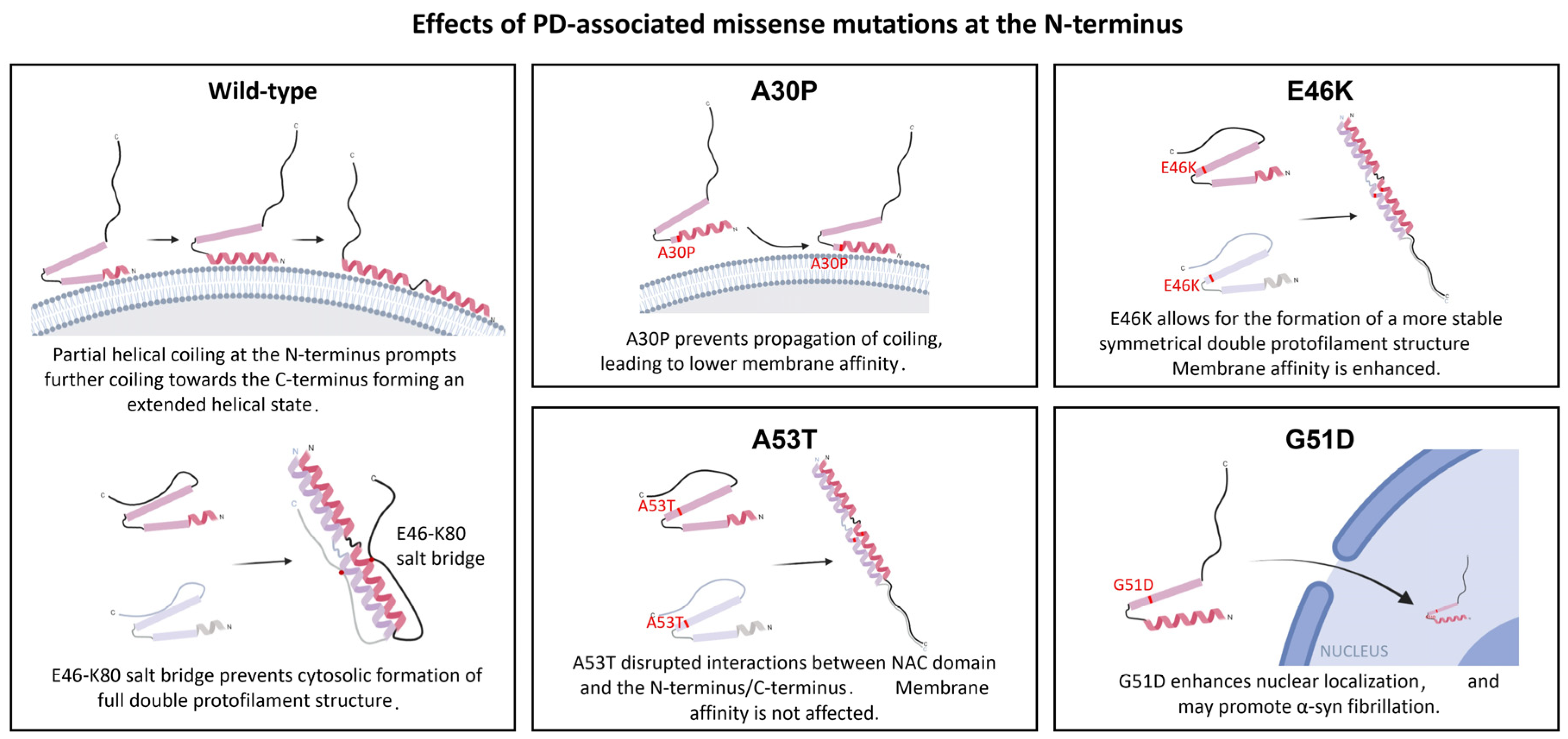
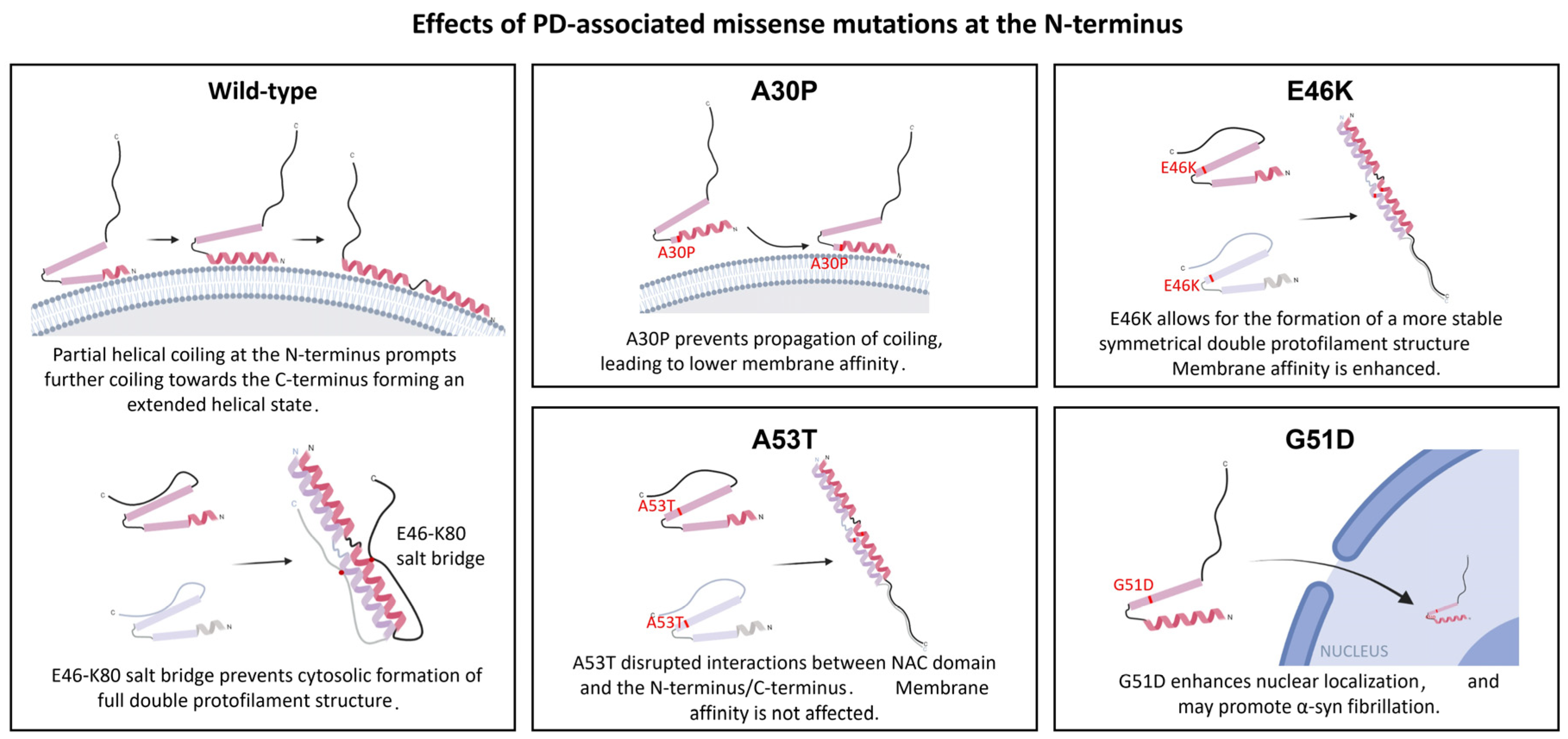
3.2. Effects of Mutations at the N-Terminus Domain of α-Syn
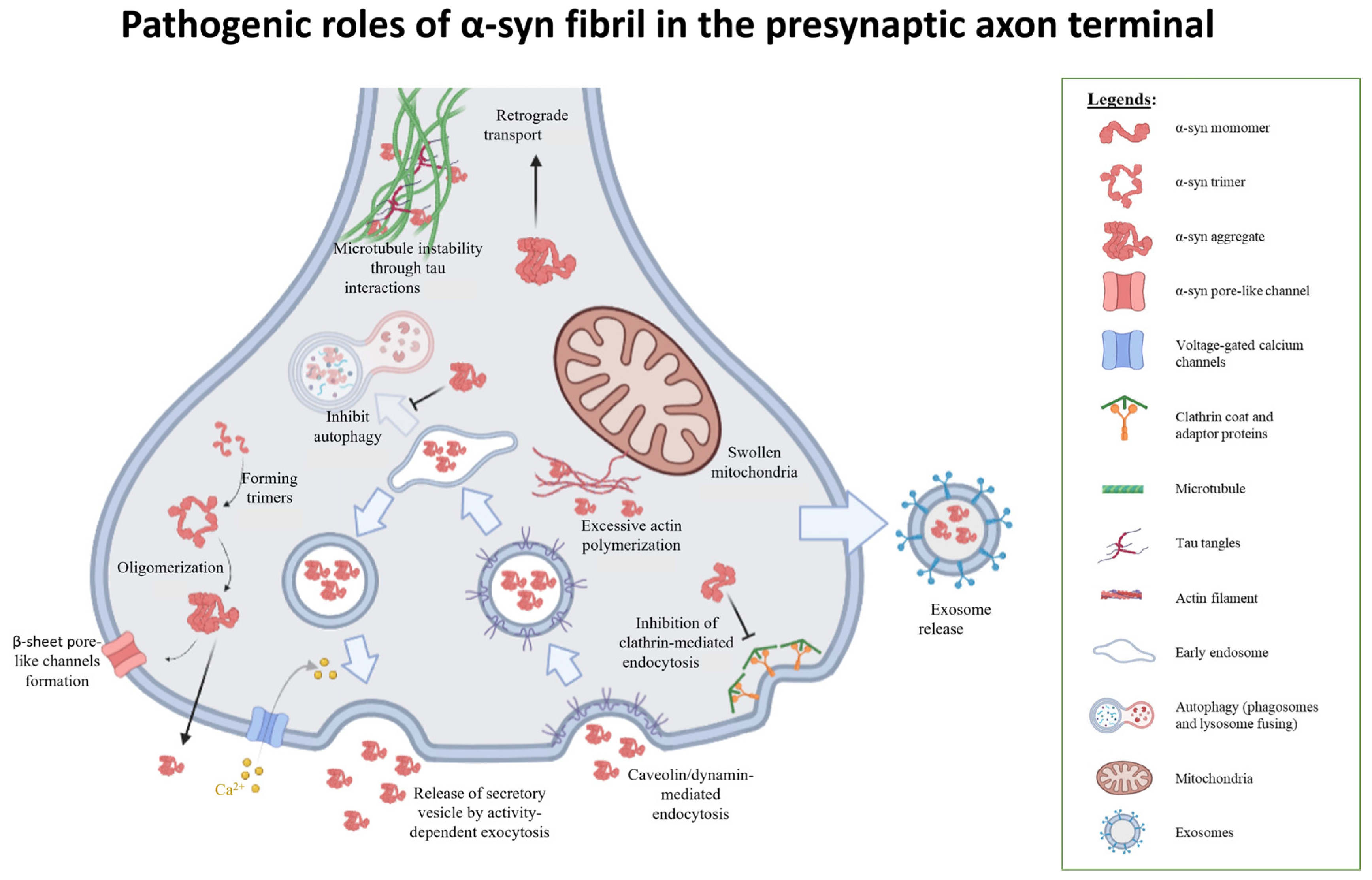
3.3. Evidence of α-Syn Multimers Influencing Membrane Permeability
3.4. Misfolded α-Syn Affects Normal Protein Recycling and Degradation
3.5. α-Syn Nuclear Localization May Alter Transcriptomic Profiles and May Be Neuroprotective
3.6. Fibrillar α-Syn Interacts with Cytoskeletal Structures
3.7. α-Syn and Mitochondrial Defects
3.8. Mode of Oligomeric α-Syn Entry into Neurons
3.9. Mechanisms of α-Syn Secretion from the Presynaptic Axon Terminals
4. α-Syn-Based Treatment Techniques
5. Limitations and Future Directions
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konno, T.; Ross, O.; Puschmann, A.; Dickson, D.W.; Wszolek, Z.K. Autosomal dominant Parkinson’s disease caused by SNCA duplications. Park. Relat. Disord. 2016, 22 (Suppl. 1), S1–S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kun-Rodrigues, C.; Orme, T.; Carmona, S.; Hernandez, D.G.; Ross, O.A.; Eicher, J.D.; Shepherd, C.; Parkkinen, L.; Darwent, L.; Heckman, M.G.; et al. A comprehensive screening of copy number variability in dementia with Lewy bodies. Neurobiol. Aging 2019, 75, 223.e1–223.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Book, A.; Guella, I.; Candido, T.; Brice, A.; Hattori, N.; Jeon, B.; Farrer, M.J.; SNCA Multiplication Investigators of the GEoPD Consortium. A Meta-Analysis of α-Synuclein Multiplication in Familial Parkinsonism. Front. Neurol. 2018, 9, 1021. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.R.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Lücking, C.B.; Brice, A. Alpha-synuclein and Parkinson’s disease. Exp. 2000, 57, 1894–1908. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Chandra, R.; Hiniker, A.; Kuo, Y.-M.; Nussbaum, R.L.; Liddle, R.A. α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2017, 2, 92295. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Ornelas, L.; Viennet, T.; Rovere, M.; Jiang, H.; Liu, L.; Nuber, S.; Ericsson, M.; Arthanari, H.; Selkoe, D.J. Altered conformation of α-synuclein drives dysfunction of synaptic vesicles in a synaptosomal model of Parkinson’s disease. Cell Rep. 2021, 36, 109333. [Google Scholar] [CrossRef]
- Emanuele, M.; Chieregatti, E. Mechanisms of Alpha-Synuclein Action on Neurotransmission: Cell-Autonomous and Non-Cell Autonomous Role. Biomolecules 2015, 5, 865–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Stroehl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, W.K.; Tahirbegi, B.; Vrettas, M.D.; Preet, S.; Ying, L.; Vendruscolo, M.; De Simone, A.; Fusco, G. The docking of synaptic vesicles on the presynaptic membrane induced by α-synuclein is modulated by lipid composition. Nat. Commun. 2021, 12, 927. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-Terminus of the Intrinsically Disordered Protein α-Synuclein Triggers Membrane Binding and Helix Folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.N.; Hirpa, D.; Zheng, K.H.; Banerjee, R.; Gunawardena, S. The Non-amyloidal Component Region of α-Synuclein Is Important for α-Synuclein Transport Within Axons. Front. Cell. Neurosci. 2020, 13, 540. [Google Scholar] [CrossRef]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Schierle, G.S.K.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef] [Green Version]
- Trexler, A.J.; Rhoades, E. N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-Terminal Acetylation of α-Synuclein on Its Random Coil and Lipid Binding Properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef]
- Kiechle, M.; Grozdanov, V.; Danzer, K.M. The Role of Lipids in the Initiation of α-Synuclein Misfolding. Front. Cell Dev. Biol. 2020, 8, 562241. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; de Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; von Saucken, V.; Bartels, T.; Selkoe, D. KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.D.; Paik, S.R.; Yang, C.-H. Structural and Functional Implications of C-Terminal Regions of α-Synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Pountney, D.L.; Jensen, P.H.; Gai, W.P.; Voelcker, N.H. Calcium(II) selectively induces α-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004, 13, 3245–3252. [Google Scholar] [CrossRef] [Green Version]
- McLean, J.R.; Hallett, P.J.; Cooper, O.; Stanley, M.; Isacson, O. Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol. Cell. Neurosci. 2012, 49, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Soll, L.G.; Eisen, J.N.; Vargas, K.J.; Medeiros, A.T.; Hammar, K.M.; Morgan, J.R. α-Synuclein-112 Impairs Synaptic Vesicle Recycling Consistent with Its Enhanced Membrane Binding Properties. Front. Cell Dev. Biol. 2020, 8, 405. [Google Scholar] [CrossRef]
- Du, T.; Wu, Z.; Luo, H.; Lu, S.; Ma, K. Injection of α-syn-98 Aggregates into the Brain Triggers α-Synuclein Pathology and an Inflammatory Response. Front. Mol. Neurosci. 2019, 12, 189. [Google Scholar] [CrossRef] [Green Version]
- Vinnakota, R.L.; Yedlapudi, D.; Manda, K.M.; Bhamidipati, K.; Bommakanti, K.T.; Rangalakshmi, G.S.; Kalivendi, S.V. Identification of an Alternatively Spliced α-Synuclein Isoform That Generates a 41-Amino Acid N-Terminal Truncated Peptide, 41-syn: Role in Dopamine Homeostasis. ACS Chem. Neurosci. 2018, 9, 2948–2958. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Kim, J. Evidence that the precursor protein of non-A beta component of Alzheimer’s disease amyloid (NACP) has an extended structure primarily composed of random-coil. Mol. Cells 1997, 7, 78–83. [Google Scholar] [PubMed]
- Jacob, R.S.; Eichmann, C.; Dema, A.; Mercadante, D.; Selenko, P. α-Synuclein plasma membrane localization correlates with cellular phosphatidylinositol polyphosphate levels. eLife 2021, 10, e61951. [Google Scholar] [CrossRef] [PubMed]
- Drescher, M.; Veldhuis, G.; van Rooijen, B.D.; Milikisyants, S.; Subramaniam, V.; Huber, M. Antiparallel Arrangement of the Helices of Vesicle-Bound α-Synuclein. J. Am. Chem. Soc. 2008, 130, 7796–7797. [Google Scholar] [CrossRef] [PubMed]
- Viennet, T.; Wördehoff, M.M.; Uluca, B.; Poojari, C.; Shaykhalishahi, H.; Willbold, D.; Strodel, B.; Heise, H.; Buell, A.K.; Hoyer, W.; et al. Structural insights from lipid-bilayer nanodiscs link α-Synuclein membrane-binding modes to amyloid fibril formation. Commun. Biol. 2018, 1, 44. [Google Scholar] [CrossRef] [Green Version]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, C.; Aguayo, L.G.; Opazo, C. An extracellular mechanism that can explain the neurotoxic effects of α-synuclein aggregates in the brain. Front. Physiol. 2012, 3, 297. [Google Scholar] [CrossRef] [Green Version]
- Tosatto, L.; Andrighetti, A.O.; Plotegher, N.; Antonini, V.; Tessari, I.; Ricci, L.; Bubacco, L.; Dalla Serra, M. Alpha-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 2876–2883. [Google Scholar] [CrossRef]
- Hoffmann, A.-C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Miranda, H.V.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep. 2019, 9, 544. [Google Scholar] [CrossRef] [Green Version]
- Dasari, A.K.R.; Kayed, R.; Wi, S.; Lim, K.H. Tau Interacts with the C-Terminal Region of α-Synuclein, Promoting Formation of Toxic Aggregates with Distinct Molecular Conformations. Biochemistry 2019, 58, 2814–2821. [Google Scholar] [CrossRef]
- Davidi, D.; Schechter, M.; Elhadi, S.A.; Matatov, A.; Nathanson, L.; Sharon, R. α-Synuclein Translocates to the Nucleus to Activate Retinoic-Acid-Dependent Gene Transcription. iScience 2020, 23, 100910. [Google Scholar] [CrossRef] [Green Version]
- Salveson, P.J.; Spencer, R.K.; Nowick, J.S. X-ray Crystallographic Structure of Oligomers Formed by a Toxic β-Hairpin Derived from α-Synuclein: Trimers and Higher-Order Oligomers. J. Am. Chem. Soc. 2016, 138, 4458–4467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L. The Consortium for Top-Down Proteomics the Human Proteoform Project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef] [PubMed]
- Bellani, S.; Sousa, V.L.; Ronzitti, G.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. The Regulation of Synaptic Function by Alpha-Synuclein. Commun. Integr. Biol. 2010, 3, 106–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular Localization and Exocytosis of α-Synuclein and its Aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, B.; Li, X.; Fu, C.; Wang, C.; Kang, X. α-Synuclein: A Multifunctional Player in Exocytosis, Endocytosis, and Vesicle Recycling. Front. Neurosci. 2019, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Khounlo, R.; Hawk, B.J.D.; Khu, T.-M.; Yoo, G.; Lee, N.K.; Pierson, J.; Shin, Y.-K. Membrane Binding of α-Synuclein Stimulates Expansion of SNARE-Dependent Fusion Pore. Front. Cell Dev. Biol. 2021, 9, 663431. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.-C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic Vesicle Depletion Correlates with Attenuated Synaptic Responses to Prolonged Repetitive Stimulation in Mice Lacking α-Synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef] [Green Version]
- Schechter, M.; Atias, M.; Abd Elhadi, S.; Davidi, D.; Gitler, D.; Sharon, R. α-Synuclein facilitates endocytosis by elevating the steady-state levels of phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2020, 295, 18076–18090. [Google Scholar] [CrossRef]
- Fakhree, M.A.; Konings, I.B.; Kole, J.; Cambi, A.; Blum, C.; Claessens, M.M. The Localization of Alpha-synuclein in the Endocytic Pathway. Neuroscience 2021, 457, 186–195. [Google Scholar] [CrossRef]
- Medeiros, A.T.; Soll, L.G.; Tessari, I.; Bubacco, L.; Morgan, J.R. α-Synuclein Dimers Impair Vesicle Fission during Clathrin-Mediated Synaptic Vesicle Recycling. Front. Cell. Neurosci. 2017, 11, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puschmann, A.; Jiménez-Ferrer, I.; Lundblad-Andersson, E.; Mårtensson, E.; Hansson, O.; Odin, P.; Widner, H.; Brolin, K.; Mzezewa, R.; Kristensen, J.; et al. Low prevalence of known pathogenic mutations in dominant PD genes: A Swedish multicenter study. Park. Relat. Disord. 2019, 66, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.; Lee, C.; Oh, E.; Park, J.; Kim, J.S.; Kim, H.-T.; Cho, J.W.; Park, W.-Y.; Jang, W.; Ki, C.-S. Genetic variants of PARK genes in Korean patients with early-onset Parkinson’s disease. Neurobiol. Aging 2019, 75, 224.e9–224.e15. [Google Scholar] [CrossRef] [PubMed]
- Keyser, R.J.; Lombard, D.; Veikondis, R.; Carr, J.; Bardien, S. Analysis of exon dosage using MLPA in South African Parkinson’s disease patients. Neurogenetics 2009, 11, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.B.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; Mackay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [Green Version]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovicic, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef]
- Bozi, M.; Papadimitriou, D.; Antonellou, R.; Moraitou, M.; Maniati, M.; Vassilatis, D.K.; Papageorgiou, S.G.; Leonardos, A.; Tagaris, G.; Malamis, G.; et al. Genetic assessment of familial and early-onset Parkinson’s disease in a Greek population. Eur. J. Neurol. 2014, 21, 963–968. [Google Scholar] [CrossRef]
- Choi, J.M.; Woo, M.S.; Ma, H.-I.; Kang, S.Y.; Sung, Y.-H.; Yong, S.W.; Chung, S.J.; Kim, J.-S.; Shin, H.-W.; Lyoo, C.H.; et al. Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 2008, 9, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, M.P.C.; Raftopoulou, A.; Gögele, M.; Borsche, M.; Emmert, D.; Fuchsberger, C.; Hantikainen, E.M.; Vukovic, V.; Klein, C.; Pramstaller, P.P.; et al. Frequency of Heterozygous Parkin (PRKN) Variants and Penetrance of Parkinson’s Disease Risk Markers in the Population-Based CHRIS Cohort. Front. Neurol. 2021, 12, 706145. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, K.; Djarmati, A.; Schafer, N.; Hering, R.; Wellenbrock, C.; Weiss, P.H.; Hilker, R.; Vieregge, P.; Ozelius, L.J.; Heutink, P.; et al. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology 2004, 62, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Tan, A.H.; Lohmann, K.; Tay, Y.W.; Lim, J.L.; Ahmad-Annuar, A.; Ramli, N.; Chin, Y.T.; Mawardi, A.S.; Azmi, K.; Aziz, Z.A.; et al. PINK1 p.Leu347Pro mutations in Malays: Prevalence and illustrative cases. Park. Relat. Disord. 2020, 79, 34–39. [Google Scholar] [CrossRef]
- Ishihara-Paul, L.; Hulihan, M.M.; Kachergus, J.; Upmanyu, R.; Warren, L.; Amouri, R.; Elango, R.; Prinjha, R.K.; Soto, A.; Kefi, M.; et al. PINK1 mutations and parkinsonism. Neurology 2008, 71, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Darvish, H.; Movafagh, A.; Omrani, M.D.; Firouzabadi, S.G.; Azargashb, E.; Jamshidi, J.; Khaligh, A.; Haghnejad, L.; Naeini, N.S.; Talebi, A.; et al. Detection of copy number changes in genes associated with Parkinson’s disease in Iranian patients. Neurosci. Lett. 2013, 551, 75–78. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Healy, D.G.; Quinn, N.; Lees, A.J.; Wood, N.W. The role of pathogenicDJ-1 mutations in Parkinson’s disease. Ann. Neurol. 2003, 54, 283–286. [Google Scholar] [CrossRef]
- Goldwurm, S.; di Fonzo, A.; Simons, E.J.; Rohé, C.F.; Zini, M.; Canesi, M.; Tesei, S.; Zecchinelli, A.; Antonini, A.; Mariani, C.; et al. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J. Med. Genet. 2005, 42, e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautier, C.; Goldwurm, S.; Dürr, A.; Giovannone, B.; Tsiaras, W.; Pezzoli, G.; Brice, A.; Smith, R.J. Mutations in the GIGYF2 (TNRC15) Gene at the PARK11 Locus in Familial Parkinson Disease. Am. J. Hum. Genet. 2008, 82, 822–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Y.P.; Kanai, K.; Tomiyama, H.; Li, Y.; Funayama, M.; Yoshino, H.; Sato, S.; Asahina, M.; Kuwabara, S.; Takeda, A.; et al. Park9-Linked Parkinsonism in Eastern Asia: Mutation Detection in Atp13a2 and Clinical Phenotype. Neurology 2008, 70, 1491–1493. [Google Scholar] [CrossRef]
- Mokretar, K.; Pease, D.; Taanman, J.-W.; Soenmez, A.; Ejaz, A.; Lashley, T.; Ling, H.; Gentleman, S.; Houlden, H.; Holton, J.L.; et al. Somatic copy number gains of α-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 2018, 141, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Björklund, T.; Petit, G.H.; Lundblad, M.; Murmu, R.P.; Brundin, P.; Li, J.-Y. A novel α-synuclein-GFP mouse model displays progressive motor impairment, olfactory dysfunction and accumulation of α-synuclein-GFP. Neurobiol. Dis. 2013, 56, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Cuvelier, E.; Méquinion, M.; Leghay, C.; Sibran, W.; Stievenard, A.; Sarchione, A.; Bonte, M.-A.; Vanbesien-Mailliot, C.; Viltart, O.; Saitoski, K.; et al. Overexpression of Wild-Type Human Alpha-Synuclein Causes Metabolism Abnormalities in Thy1-aSYN Transgenic Mice. Front. Mol. Neurosci. 2018, 11, 321. [Google Scholar] [CrossRef]
- Henrich, M.; Geibl, F.F.; Lee, B.; Chiu, W.-H.; Koprich, J.B.; Brotchie, J.M.; Timmermann, L.; Decher, N.; Matschke, L.A.; Oertel, W.H. A53T-α-synuclein overexpression in murine locus coeruleus induces Parkinson’s disease-like pathology in neurons and glia. Acta Neuropathol. Commun. 2018, 6, 39. [Google Scholar] [CrossRef]
- Perlmutter, J.; Braun, A.; Sachs, J.N. Curvature Dynamics of α-Synuclein Familial Parkinson Disease Mutants. J. Biol. Chem. 2009, 284, 7177–7189. [Google Scholar] [CrossRef] [Green Version]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.-I.; Anthony, M.D.; Edwards, R.H. Lipid Rafts Mediate the Synaptic Localization of -Synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [Green Version]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Zhou, K.; Hughes, M.P.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. USA 2020, 117, 3592–3602. [Google Scholar] [CrossRef]
- Emmer, K.L.; Waxman, E.; Covy, J.P.; Giasson, B.I. E46K Human α-Synuclein Transgenic Mice Develop Lewy-like and Tau Pathology Associated with Age-dependent, Detrimental Motor Impairment. J. Biol. Chem. 2011, 286, 35104–35118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskuner, O.; Wise-Scira, O. Structures and Free Energy Landscapes of the A53T Mutant-Type α-Synuclein Protein and Impact of A53T Mutation on the Structures of the Wild-Type α-Synuclein Protein with Dynamics. ACS Chem. Neurosci. 2013, 4, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussell, R.; Eliezer, D. Residual Structure and Dynamics in Parkinson’s Disease-associated Mutants of α-Synuclein. J. Biol. Chem. 2001, 276, 45996–46003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Hou, S.; Zhao, K.; Long, H.; Liu, Z.; Gao, J.; Zhang, Y.; Su, X.-D.; Li, D.; Liu, C. Cryo-EM structure of full-length α-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Res. 2020, 30, 360–362. [Google Scholar] [CrossRef]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic Changes in Presynaptic and Axonal Transport Proteins Combined with Striatal Neuroinflammation Precede Dopaminergic Neuronal Loss in a Rat Model of AAV -Synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [Green Version]
- Zakharov, S.D.; Hulleman, J.D.; Dutseva, E.A.; Antonenko, Y.N.; Rochet, J.-C.; Cramer, W.A. Helical α-Synuclein Forms Highly Conductive Ion Channels. Biochemistry 2007, 46, 14369–14379. [Google Scholar] [CrossRef]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by α-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.-S. Ubiquitination of -synuclein by Siah-1 promotes -synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2007, 17, 906–917. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a New Form of α-Synuclein by Parkin from Human Brain: Implications for Parkinson’s Disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Gao, P.; Arzberger, T.; Höllerhage, M.; Herms, J.; Höglinger, G.; Koeglsperger, T. Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell Death Dis. 2021, 12, 854. [Google Scholar] [CrossRef] [PubMed]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear Localization of alpha-Synuclein and Its Interaction with Histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- Schaser, A.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, O.S.; Li, S.; Sullivan, L.F.; Chen, W.; Kondrikova, G.; Manfredsson, F.P.; Mandel, R.J.; Muzyczka, N. The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2008, 105, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-Y.; Kim, H.; Jo, A.; Khang, R.; Park, C.-H.; Park, S.-J.; Kwag, E.; Shin, J.-H. α-Synuclein A53T Binds to Transcriptional Adapter 2-Alpha and Blocks Histone H3 Acetylation. Int. J. Mol. Sci. 2021, 22, 5392. [Google Scholar] [CrossRef]
- Duke, D.C.; Moran, L.B.; Kalaitzakis, M.E.; Deprez, M.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Transcriptome analysis reveals link between proteasomal and mitochondrial pathways in Parkinson’s disease. Neurogenetics 2006, 7, 139–148. [Google Scholar] [CrossRef]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. α-Synuclein and Its A30P Mutant Affect Actin Cytoskeletal Structure and Dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef] [Green Version]
- Teravskis, P.J.; Covelo, A.; Miller, E.C.; Singh, B.; Martell-Martínez, H.A.; Benneyworth, M.A.; Gallardo, C.; Oxnard, B.R.; Araque, A.; Lee, M.K.; et al. A53T Mutant Alpha-Synuclein Induces Tau-Dependent Postsynaptic Impairment Independently of Neurodegenerative Changes. J. Neurosci. 2018, 38, 9754–9767. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.C.; Stavoe, A.K.; Colón-Ramos, D.A. The actin cytoskeleton in presynaptic assembly. Cell Adhes. Migr. 2013, 7, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Morales, M.; Colicos, M.A.; Goda, Y. Actin-Dependent Regulation of Neurotransmitter Release at Central Synapses. Neuron 2000, 27, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, D.G.; Lee, M.K.; Feany, M.B. α-synuclein Induces Mitochondrial Dysfunction through Spectrin and the Actin Cytoskeleton. Neuron 2018, 97, 108–124.e6. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Peng, H.B. The Function of Mitochondria in Presynaptic Development at the Neuromuscular Junction. Mol. Biol. Cell 2008, 19, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, S.; Adi, N.; Ellul, J.; Argyriou, A.A.; Chroni, E. A Prospective Study of Familial versus Sporadic Parkinson’s Disease. Neurodegener. Dis. 2007, 4, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Kobayashi, J.; Hasegawa, T.; Sugeno, N.; Yoshida, S.; Akiyama, T.; Fujimori, K.; Hatakeyama, H.; Miki, Y.; Tomiyama, A.; Kawata, Y.; et al. Extracellular α-synuclein enters dopaminergic cells by modulating flotillin-1–assisted dopamine transporter endocytosis. FASEB J. 2019, 33, 10240–10256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xu, Y.; Lee, J.; Jarnik, M.; Wu, X.; Bonifacino, J.S.; Shen, J.; Ye, Y. A myosin-7B–dependent endocytosis pathway mediates cellular entry of α-synuclein fibrils and polycation-bearing cargos. Proc. Natl. Acad. Sci. USA 2020, 117, 10865–10875. [Google Scholar] [CrossRef]
- La, T.M.; Tachibana, H.; Li, S.; Abe, T.; Seiriki, S.; Nagaoka, H.; Takashima, E.; Takeda, T.; Ogawa, D.; Makino, S.; et al. Dynamin 1 is important for microtubule organization and stabilization in glomerular podocytes. FASEB J. 2020, 34, 16449–16463. [Google Scholar] [CrossRef]
- Masaracchia, C.; Hnida, M.; Gerhardt, E.; Lopes da Fonseca, T.; Villar-Pique, A.; Branco, T.; Stahlberg, M.A.; Dean, C.; Fernández, C.O.; Milosevic, I.; et al. Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol. Commun. 2018, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-H.; Choi, Y.R.; Heo, C.-H.; Kang, S.-J.; Joe, E.-H.; Jou, I.; Kim, H.-M.; Park, S.M. Loss of parkin promotes lipid rafts-dependent endocytosis through accumulating caveolin-1: Implications for Parkinson’s disease. Mol. Neurodegener. 2015, 10, 63. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Grasso, L.; Sibilla, C.; Stevens, T.J.; Barry, N.; Bertolotti, A. Prion-like protein aggregates exploit the RHO GTPase to cofilin-1 signaling pathway to enter cells. EMBO J. 2018, 37, e97822. [Google Scholar] [CrossRef]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length α-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- Guan, Y.; Zhao, X.; Liu, F.; Yan, S.; Wang, Y.; Du, C.; Cui, X.; Li, R.; Zhang, C.X. Pathogenic Mutations Differentially Regulate Cell-to-Cell Transmission of α-Synuclein. Front. Cell. Neurosci. 2020, 14, 159. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Crump, C.; Thomas, G. Trans-Golgi network sorting. Experientia 2001, 58, 1067–1084. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Iwatsubo, T. Extracellular α-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 2018, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Dieriks, B.V.; Park, T.I.-H.; Fourie, C.; Faull, R.L.M.; Dragunow, M.; Curtis, M.A. α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 2017, 7, srep42984. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stykel, M.G.; Humphries, K.M.; Kamski-Hennekam, E.; Buchner-Duby, B.; Porte-Trachsel, N.; Ryan, T.; Coackley, C.L.; Bamm, V.V.; Harauz, G.; Ryan, S.D. α-Synuclein mutation impairs processing of endomembrane compartments and promotes exocytosis and seeding of α-synuclein pathology. Cell Rep. 2021, 35, 109099. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced alpha-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, D.D.; Mortimer, J.A. Failure of L-Dopa to relieve activated rigidity in Parkinson’s disease. In Parkinson’s Disease; Messiha, F.S., Kenny, A.D., Eds.; Springer: New York, NY, USA, 1977; Volume 90, pp. 297–313. ISBN 978-1-4684-2513-0. [Google Scholar]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson’s Disease-Like Models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Park, S.M.; Lee, C.-H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.-T.; Paik, S.R.; Chung, K.C. Proteolytic Cleavage of Extracellular Secreted α-Synuclein via Matrix Metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef] [Green Version]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Egoz-Matia, N.; Frydman-Marom, A.; Shalev, D.E.; Segal, D.; Gazit, E. Inhibiting α-Synuclein Oligomerization by Stable Cell-Penetrating β-Synuclein Fragments Recovers Phenotype of Parkinson’s Disease Model Flies. PLoS ONE 2010, 5, e13863. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Kang, H.R.; Kang, H.; Zhang, Y.; Lee, Y.; Kim, Y.; Han, K. Unexpected Compensatory Increase in Shank3 Transcripts in Shank3 Knock-Out Mice Having Partial Deletions of Exons. Front. Mol. Neurosci. 2019, 12, 228. [Google Scholar] [CrossRef]
- Sharma, K.; Mehra, S.; Sawner, A.S.; Markam, P.S.; Panigrahi, R.; Navalkar, A.; Chatterjee, D.; Kumar, R.; Kadu, P.; Patel, K.; et al. Effect of Disease-Associated P123H and V70M Mutations on β-Synuclein Fibrillation. ACS Chem. Neurosci. 2020, 11, 2836–2848. [Google Scholar] [CrossRef]
- Jain, M.K.; Singh, P.; Roy, S.; Bhat, R. Comparative Analysis of the Conformation, Aggregation, Interaction, and Fibril Morphologies of Human α-, β-, and γ-Synuclein Proteins. Biochemistry 2018, 57, 3830–3848. [Google Scholar] [CrossRef]
- Moore, K.S.; Wehrli, S.; Roder, H.; Rogers, M.; Forrest, J.N., Jr.; McCrimmon, D.; Zasloff, M. Squalamine: An aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. USA 1993, 90, 1354–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Marshall, J.L.; Dahut, W.; Rizvi, N.; Trocky, N.; Williams, J.I.; Hait, H.; Song, S.; Holroyd, K.J.; Hawkins, M.J. A phase I and pharmacokinetic study of squalamine, a novel antiangiogenic agent, in patients with advanced cancers. Clin. Cancer Res. 2001, 7, 3912–3919. [Google Scholar] [PubMed]
- Herbst, R.S.; Hammond, L.A.; Carbone, D.P.; Tran, H.T.; Holroyd, K.J.; Desai, A.; Williams, J.I.; Bekele, B.N.; Hait, H.; Allgood, V.; et al. A phase I/IIA trial of continuous five-day infusion of squalamine lactate (MSI-1256F) plus carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 4108–4115. [Google Scholar]
- Limbocker, R.; Errico, S.; Barbut, D.; Knowles, T.P.J.; Vendruscolo, M.; Chiti, F.; Zasloff, M. Squalamine and trodusquemine: Two natural products for neurodegenerative diseases, from physical chemistry to the clinic. Nat. Prod. Rep. 2022. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [Green Version]
- Tóth, G.; Neumann, T.; Berthet, A.; Masliah, E.; Spencer, B.; Tao, J.; Jobling, M.F.; Gardai, S.J.; Bertoncini, C.W.; Cremades, N.; et al. Novel Small Molecules Targeting the Intrinsically Disordered Structural Ensemble of α-Synuclein Protect Against Diverse α-Synuclein Mediated Dysfunctions. Sci. Rep. 2019, 9, 16947. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Bu, J.; Kubiatowicz, L.J.; Chen, S.S.; Kim, Y.; Hong, S. Peptide–nanoparticle conjugates: A next generation of diagnostic and therapeutic platforms? Nano Converg. 2018, 5, 38. [Google Scholar] [CrossRef]
- Ray, R.M.; Hansen, A.H.; Taskova, M.; Jandl, B.; Hansen, J.; Soemardy, C.; Morris, K.V.; Astakhova, K. Enhanced target cell specificity and uptake of lipid nanoparticles using RNA aptamers and peptides. Beilstein J. Org. Chem. 2021, 17, 891–907. [Google Scholar] [CrossRef]
- Fouka, M.; Mavroeidi, P.; Tsaka, G.; Xilouri, M. In Search of Effective Treatments Targeting α-Synuclein Toxicity in Synucleinopathies: Pros and Cons. Front. Cell Dev. Biol. 2020, 8, 559791. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Zhang, F.; Yue, L.; Fang, X.; Wang, G.; Li, C.; Sun, X.; Jia, X.; Yang, J.; Song, J.; Zhang, Y.; et al. Altered gut microbiota in Parkinson’s disease patients/healthy spouses and its association with clinical features. Park. Relat. Disord. 2020, 81, 84–88. [Google Scholar] [CrossRef]
- Shams, R.; Banik, N.L.; Haque, A. Calpain in the cleavage of alpha-synuclein and the pathogenesis of Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2019, 167, 107–124. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef]
- Brichta, L.; Greengard, P.; Flajolet, M. Advances in the pharmacological treatment of Parkinson’s disease: Targeting neurotransmitter systems. Trends Neurosci. 2013, 36, 543–554. [Google Scholar] [CrossRef]
- Leentjens, A.F.G.; Scholtissen, B.; Vreeling, F.W.; Verhey, F.R.J. The Serotonergic Hypothesis for Depression in Parkinson’s Disease: An Experimental Approach. Neuropsychopharmacology 2006, 31, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Kano, O.; Ikeda, K.; Cridebring, D.; Takazawa, T.; Yoshii, Y.; Iwasaki, Y. Neurobiology of Depression and Anxiety in Parkinson’s Disease. Park. Dis. 2011, 2011, 143547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muslimović, D.; Post, B.; Speelman, J.D.; Schmand, B. Motor procedural learning in Parkinson’s disease. Brain 2007, 130, 2887–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Lv, G.; Lee, J.S.; Jung, B.C.; Masuda-Suzukake, M.; Hong, C.-S.; Valera, E.; Lee, H.-J.; Paik, S.R.; Hasegawa, M.; et al. Exposure to bacterial endotoxin generates a distinct strain of α-synuclein fibril. Sci. Rep. 2016, 6, 30891. [Google Scholar] [CrossRef] [Green Version]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambarè, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Lee, E.-J.; Woo, M.-S.; Moon, P.-G.; Baek, M.-C.; Choi, I.-Y.; Kim, W.-K.; Junn, E.; Kim, H.-S. α-Synuclein Activates Microglia by Inducing the Expressions of Matrix Metalloproteinases and the Subsequent Activation of Protease-Activated Receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.; Anthony, D.C. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J. Neuroinflammation 2011, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 69, 337–342. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Le, W. Profiling Non-motor Symptoms in Monogenic Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 591183. [Google Scholar] [CrossRef]
- Candini, M.; Battaglia, S.; Benassi, M.; di Pellegrino, G.; Frassinetti, F. The physiological correlates of interpersonal space. Sci. Rep. 2021, 11, 2611. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, J.S.A.M.; Ehrt, U.; Weber, W.E.J.; Aarsland, D.; Leentjens, A.F.G. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov. Disord. 2007, 23, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Yang, F.; Zheng, J.; Wang, X.; Huang, Q. Aberrant Structure MRI in Parkinson’s Disease and Comorbidity with Depression Based on Multinomial Tensor Regression Analysis. J. Pers. Med. 2022, 12, 89. [Google Scholar] [CrossRef]
- Davidsdottir, S.; Cronin-Golomb, A.; Lee, A. Visual and spatial symptoms in Parkinson’s disease. Vis. Res. 2005, 45, 1285–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellena, G.; Battaglia, S.; Làdavas, E. The spatial effect of fearful faces in the autonomic response. Exp. Brain Res. 2020, 238, 2009–2018. [Google Scholar] [CrossRef]
- Anders, S.; Sack, B.; Pohl, A.; Munte, T.F.; Pramstaller, P.P.; Klein, C.; Binkofski, F. Compensatory premotor activity during affective face processing in subclinical carriers of a single mutant Parkin allele. Brain 2012, 135, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Borgomaneri, S.; Vitale, F.; Battaglia, S.; Avenanti, A. Early Right Motor Cortex Response to Happy and Fearful Facial Expressions: A TMS Motor-Evoked Potential Study. Brain Sci. 2021, 11, 1203. [Google Scholar] [CrossRef]
- Scarpina, F.; Cau, N.; Cimolin, V.; Galli, M.; Priano, L.; Mauro, A. Defective Tool Embodiment in Body Representation of Individuals Affected by Parkinson’s Disease: A Preliminary Study. Front. Psychol. 2019, 9, 2489. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Gene | Mutations/Modifications | Population | Prevalence in PD/DLB Population | Reference |
|---|---|---|---|---|
| SNCA (PARK1) | Gene duplication | Sweden | 1/2206 (0.045%) | [53] |
| Gene triplication | Korea | 2/70 (2.86%) | [54] | |
| South Africa | 1/88 (1.14%) | [55] | ||
| A30P (N-terminus) | Germany | 5/199 (2.51%) | [56] | |
| E46K (N-terminus) | Spain | 10/337 (2.97%) | [57] | |
| H50Q (N-terminus) | British | 1/451 (0.22%) | [58] | |
| G51D (N-terminus) | France | 4/443 (0.90%) | [59] | |
| A53T (N-terminus) | Greece | 5/111 (4.5%) | [60] | |
| Korea | 1/72 (1.39%) | [61] | ||
| Y125 phosphorylation (C-terminus) | British | 3/4503 (0.067%) | [62] | |
| Parkin (PARK2) | Copy number variations, small deletions/insertions | Northern Italy | 164/3603 (4.55%) | [63] |
| Point mutations/small deletions, exon rearrangement | Mixed | 17/100 (17%) | [64] | |
| Omi/HtrA2 (PARK3) | Point mutation | Germany | 4/892 (0.45%) | [65] |
| UCHL1 (PARK5) | Point mutation | Germany | 4/507 (0.79%) | [66] |
| PINK1 (PARK6) | Point mutations (e.g., M129L, P153R) | Malaysia | 6.9% | [67] |
| Tunisia | 2.5% | [68] | ||
| Exon deletion | Iran | Familial: 6/130 (4.6%) | [69] | |
| DJ-1 (PARK7) | Exon deletion | Iran | Familial: 3/130 (2.3%) | [69] |
| Point mutation | Mixed | EOPD: 2/185 (1.1%) | [70] | |
| Exon deletion, splice site changes | Mixed | 2/100 (2%) | [64] | |
| LRRK2 (PARK8) | Point mutations | Sweden | 0.54% | [53] |
| Italy | Overall: 13/629 (2.1%) Familial: 9/177 (5.1%) Sporadic: 4/452 (0.9%) | [71] | ||
| Italy, France | Italian (familial): 6/123 (4.8%) French (familial): 2/126 (1.6%) | [72] | ||
| ATP13A2 (PARK9) | Point mutations | Japan | 28/117 (23.9%) | [73] |
| Exon deletion, gene duplication, gene triplication | Iran | Familial: 10/130 (7.7%) | [69] | |
| GIGYF2 (PARK11) | Point mutation | Italy, France | 4.8% | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, L.Y.; Tang, K.H.; Lim, L.Y.Y.; Ong, J.X.; Park, H.; Jung, S. α-Synuclein at the Presynaptic Axon Terminal as a Double-Edged Sword. Biomolecules 2022, 12, 507. https://doi.org/10.3390/biom12040507
Tan LY, Tang KH, Lim LYY, Ong JX, Park H, Jung S. α-Synuclein at the Presynaptic Axon Terminal as a Double-Edged Sword. Biomolecules. 2022; 12(4):507. https://doi.org/10.3390/biom12040507
Chicago/Turabian StyleTan, Li Yang, Kwan Hou Tang, Lynette Yu You Lim, Jia Xin Ong, Hyokeun Park, and Sangyong Jung. 2022. "α-Synuclein at the Presynaptic Axon Terminal as a Double-Edged Sword" Biomolecules 12, no. 4: 507. https://doi.org/10.3390/biom12040507
APA StyleTan, L. Y., Tang, K. H., Lim, L. Y. Y., Ong, J. X., Park, H., & Jung, S. (2022). α-Synuclein at the Presynaptic Axon Terminal as a Double-Edged Sword. Biomolecules, 12(4), 507. https://doi.org/10.3390/biom12040507